
Identification of miRNAs Mediating Seed Storability of Maize during Germination Stage by High-Throughput Sequencing, Transcriptome and Degradome Sequencing
 , ,
, ,
Abstract
:1. Introduction
2. Results
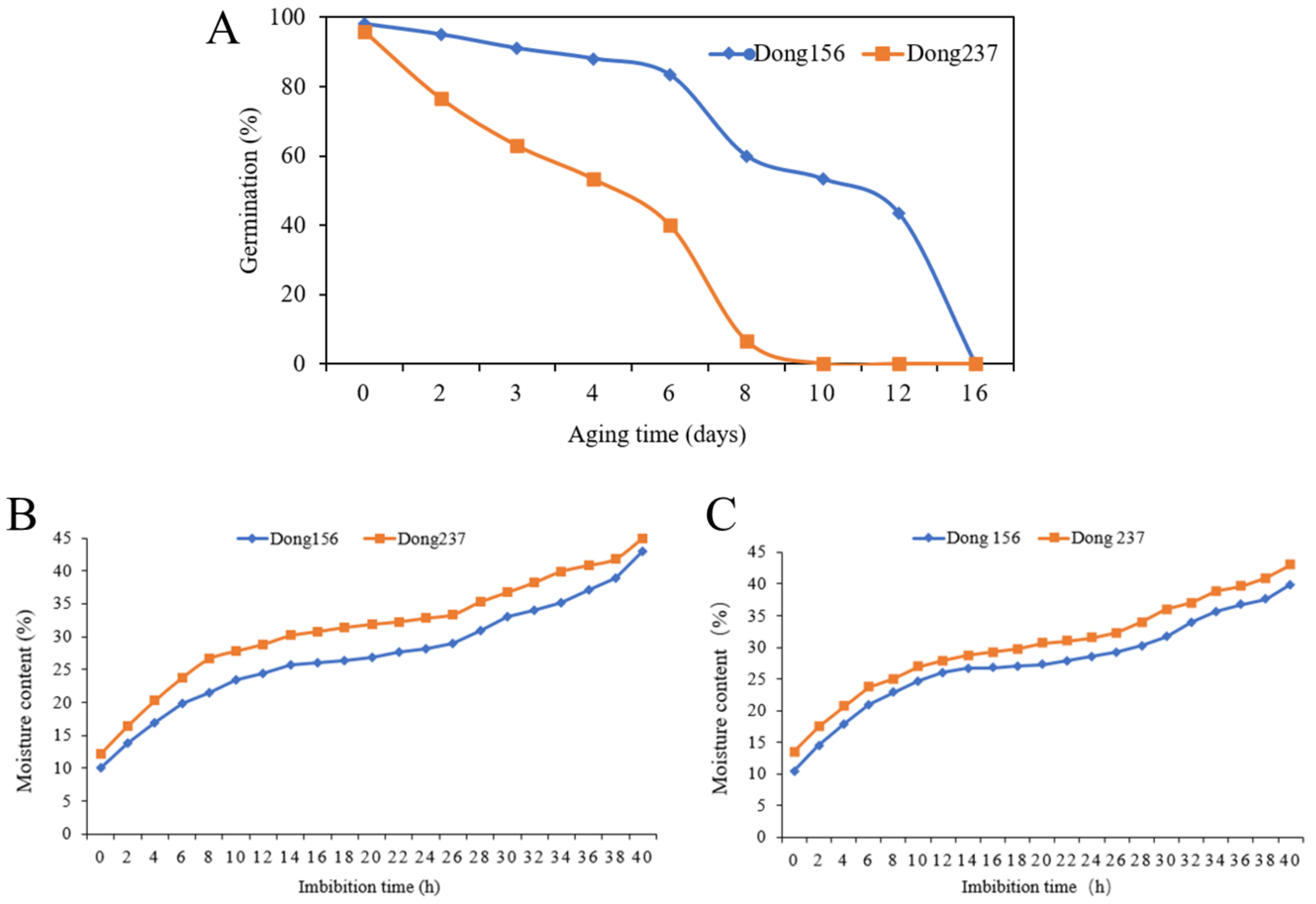
2.1. The Artificial Aging and Sampling Method of Inbred Lines
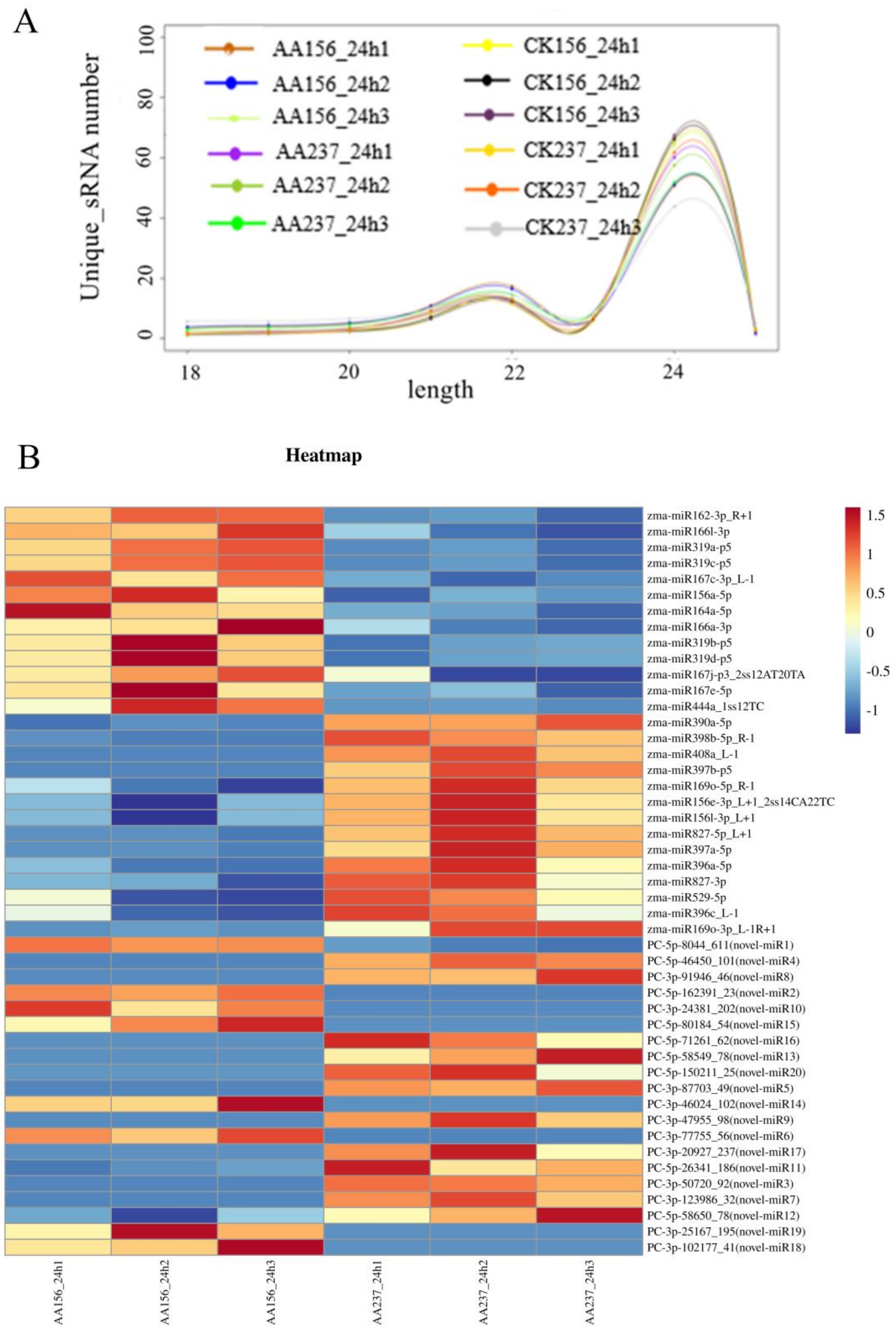
2.2. Overview and Validation of Small RNA Sequencing Data
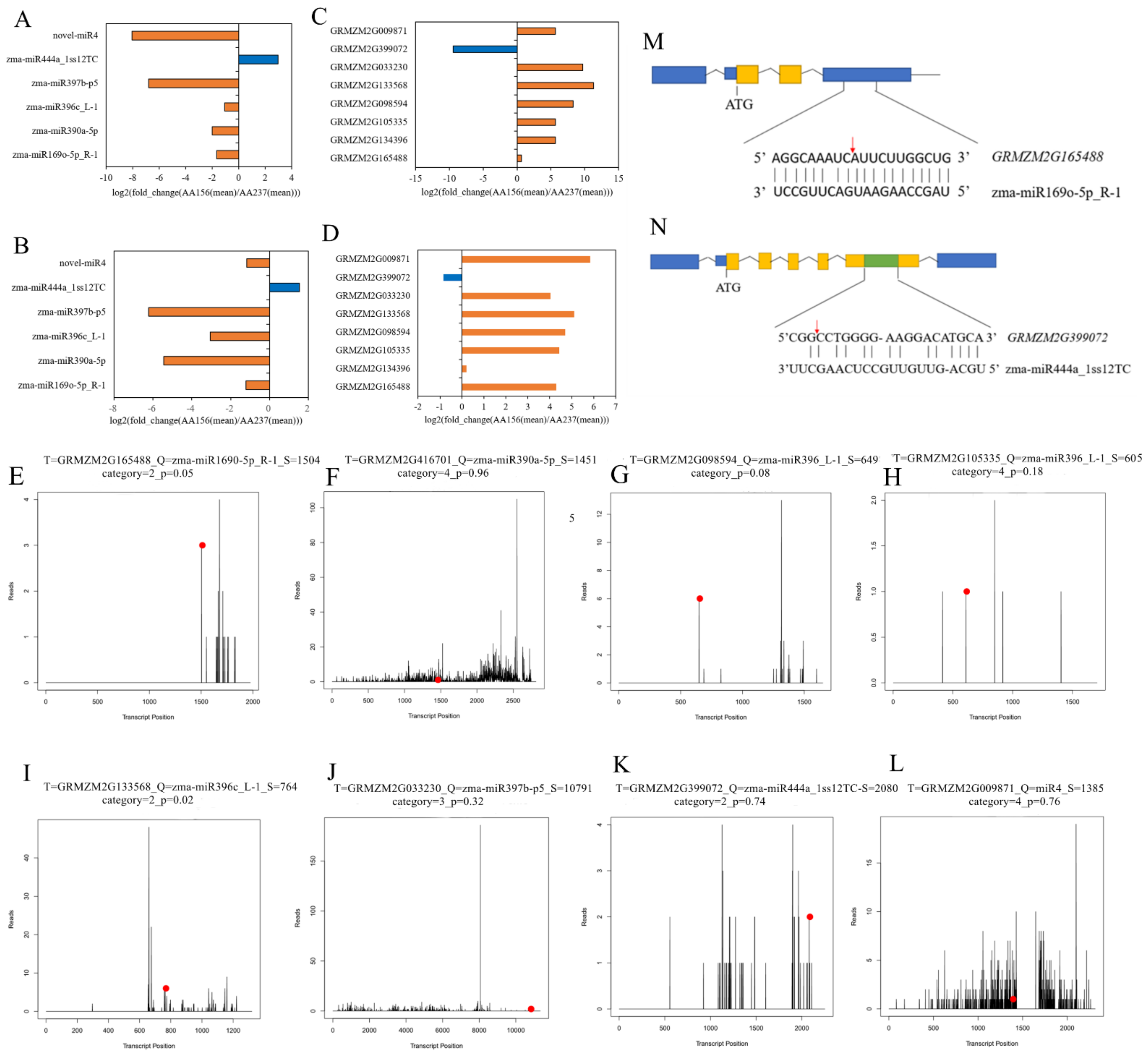
2.3. Prediction of Target Genes of miRNAs by Degradome Sequencing and Validation of miRNA Targets by 5′RLM-RACE
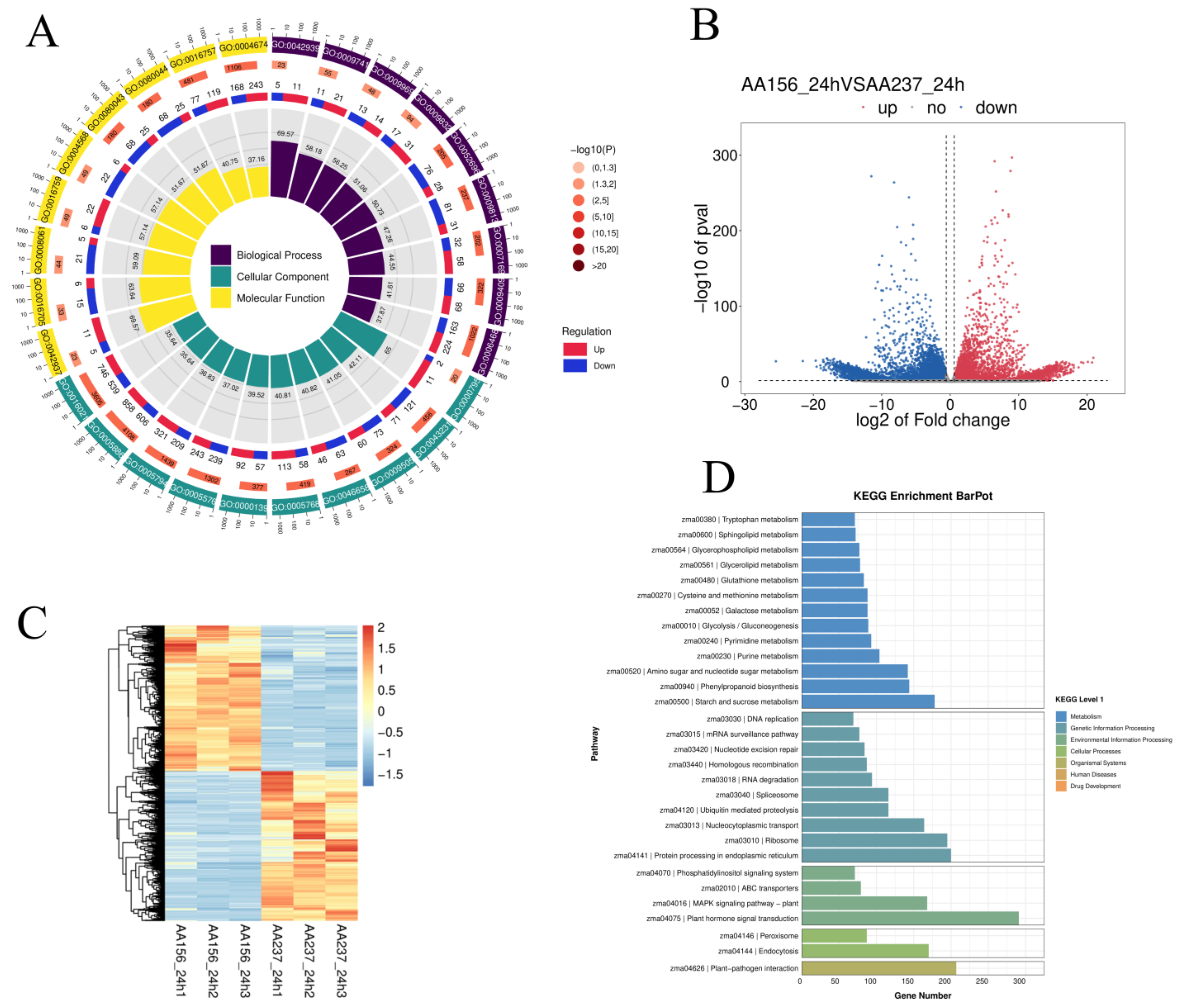
2.4. Transcriptome Analysis Explored the of DEGs Involved in Maize Seed Storability
2.5. The Combined Analysis of Transcriptome, Small RNA, and the Degradome Sequencing
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Seed-Accelerated Aging and Changes of Water Content Test during Seed Germination
4.3. RNA Extraction, Library Construction, and Sequencing
4.4. Sequence Data Analysis and Verification
4.5. Transcriptome—Small RNA—The Degradome Combined Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiang, C.; Li, S.K.; Li, Q.H.; Wang, J.Y. Research Progress in Storability of Rice. Acta Agric. Jiangxi 2011, 23, 39–43. [Google Scholar]
- Bertram, C.; Hass, R. Cellular responses to reactive oxygen species-induced DNA damage and aging. Biol. Chem. 2008, 389, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S. Aging as war between chemical and biochemical processes: Protein methylation and the recognition of age-damaged proteins for repair. Ageing. Res. Rev. 2003, 2, 263–285. [Google Scholar] [CrossRef]
- Bailly, C.; El-Maarouf-Bouteau, H.; Corbineau, F. From intracellular signaling networks to cell death: The dual role of reactive oxygen species in seed physiology. Comptes Rendus Biol. 2008, 331, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, S.; Wang, L.; Wu, D.; Cheng, H.; Du, X.; Mao, D.; Zhang, C.; Jiang, X. miR164c and miR168a regulate seed vigor in rice. J. Integr. Plant Biol. 2020, 62, 470–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdsworth, M.J.; Bentsink, L.; Soppe, W.J.J. Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 2008, 179, 33–54. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Lee, J.; Jin, Y.M.; Qiao, Y.; Piao, R.; Jang, S.M.; Woo, M.O.; Kwon, S.W.; Liu, X.; Pan, H.Y.; et al. Identification of QTLs for seed germination capability after various storage periods using two RIL populations in rice. Mol. Cells 2011, 31, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Zhang, S.Q.; Yao, Q.H.; Peng, R.H.; Xiong, A.S.; Li, X.; Zhu, W.M.; Zhu, Y.Y.; Zha, D.S. Identification of quantitative trait loci for seed storability in rice (Oryza sativa L.). Euphytica 2008, 164, 739–744. [Google Scholar] [CrossRef]
- Bettey, M.; Finch-Savage, W.E.; King, G.J.; Lynn, J.R. Quantitative genetic analysis of seed vigour and pre-emergence seedling growth traits in Brassica oleracea. New Phytol. 2000, 148, 277–286. [Google Scholar] [CrossRef]
- Rehman Arif, M.A.; Nagel, M.; Neumann, K.; Kobiljski, B.; Lohwasser, U.; Börner, A. Genetic studies of seed longevity in hexaploid wheat using segregation and association mapping approaches. Euphytica 2011, 186, 1–13. [Google Scholar] [CrossRef]
- Xie, L.; Tan, Z.; Zhou, Y.; Xu, R.; Feng, L.; Xing, Y.; Qi, X. Identification and fine mapping of quantitative trait loci for seed vigor in germination and seedling establishment in rice. J. Integr. Plant Biol. 2014, 56, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Rajjou, L.; Lovigny, Y.; Groot, S.P.; Belghazi, M.; Job, C.; Job, D. Proteome-wide characterization of seed aging in Arabidopsis: A comparison between artificial and natural aging protocols. Plant Physiol. 2008, 148, 620–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Higo, K.; Hagiwara, K.; Ohtsubo, K.; Kobayashi, A.; Nozu, Y. Production and Use of Monoclonal Antibodies against Rice Embryo Lipoxygenase-3. Biosci. Biotechnol. Biochem. 1992, 56, 678–679. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Dong, Z.Y.; Xu, Z.B.; Wang, D.W.; Wang, T. Molecular characterization of a novel type of lipoxygenase (LOX) gene from common wheat (Triticum aestivum L.). Mol. Breed. 2011, 30, 113–124. [Google Scholar] [CrossRef]
- Borrelli, G.M.; De Leonardis, A.M.; Platani, C.; Troccoli, A. Distribution along durum wheat kernel of the components involved in semolina colour. J. Cereal Sci. 2008, 48, 494–502. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, S.R.; An, G. Rice aldehyde dehydrogenase7 is needed for seed maturation and viability. Plant Physiol. 2009, 149, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, Y.; Wang, D.; Liu, Y.; Dirk, L.M.A.; Goodman, J.; Downie, A.B.; Wang, J.; Wang, G.; Zhao, T. Regulation of Seed Vigor by Manipulation of Raffinose Family Oligosaccharides in Maize and Arabidopsis thaliana. Mol. Plant 2017, 10, 1540–1555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, D.; Dirk, L.M.A.; Downie, A.B.; Zhao, T. ZmAGA1 Hydrolyzes RFOs Late during the Lag Phase of Seed Germination, Shifting Sugar Metabolism toward Seed Germination Over Seed Aging Tolerance. J. Agric. Food Chem. 2021, 69, 11606–11615. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, K.; Zhou, Y.; Hu, Y.; Xiao, Y.; Chen, T.; Yin, M.; Liu, Y.; Xu, M.; Jiang, X. Degradome sequencing reveals an integrative miRNA-mediated gene interaction network regulating rice seed vigor. BMC Plant Biol. 2022, 22, 269. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Gou, L.; Chen, D.; Wu, P.; Chen, M. High-throughput degradome sequencing can be used to gain insights into microRNA precursor metabolism. J. Exp. Bot. 2010, 61, 3833–3837. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kwak, K.J.; Jung, H.J.; Lee, H.J.; Kang, H. MicroRNA402 affects seed germination of Arabidopsis thaliana under stress conditions via targeting DEMETER-LIKE Protein3 mRNA. Plant Cell Physiol. 2010, 51, 1079–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.-M.; Qiao, Y.; Mao, K.; Hao, Y.-J. Ectopic overexpression of AtmiR398b gene in tobacco influences seed germination and seedling growth. Plant Cell Tissue Organ Cult. (PCTOC) 2010, 102, 53–59. [Google Scholar] [CrossRef]
- Willmann, M.R.; Mehalick, A.J.; Packer, R.L.; Jenik, P.D. MicroRNAs Regulate the Timing of Embryo Maturation in Arabidopsis. Plant Physiol. 2011, 155, 1871–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Gonzalez, N.; Inze, D.; Dubois, M. Emerging Connections between Small RNAs and Phytohormones. Trends Plant Sci. 2020, 25, 912–929. [Google Scholar] [CrossRef]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef]
- Sarkar, D.S.; Prakash, K.; Kumar, N.A.; Neeti, S.M. Small RNA mediated regulation of seed germination. Front. Plant Sci. 2015, 6, 828–834. [Google Scholar]
- Chen, L.; Wang, T.Z.; Zhao, M.G.; Zhang, W.H. Ethylene-responsive miRNAs in roots of Medicago truncatula identified by high-throughput sequencing at whole genome level. Plant Sci. 2012, 184, 14–19. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Zhao, Y.; Feng, Z.; Li, Q.; Yang, H.Q.; Luan, S.; Li, J.; He, Z.H. Auxin controls seed dormancy through stimulation of abscisic acid signaling by inducing ARF-mediated ABI3 activation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2013, 110, 15485–15490. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Liu, H.; Wang, K.; Liu, L.; Wang, S.; Zhao, Y.; Li, Y. Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol. 2013, 13, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Zhou, S.; Shen, K.; Zhou, Y.; Wang, F.; Jiang, X. Elucidation of the miR164c-Guided Gene/Protein Interaction Network Controlling Seed Vigor in Rice. Front. Plant Sci. 2020, 11, 589005. [Google Scholar] [CrossRef] [PubMed]
- Lujan-Soto, E.; Juarez-Gonzalez, V.T.; Reyes, J.L.; Dinkova, T.D. MicroRNA Zma-miR528 Versatile Regulation on Target mRNAs during Maize Somatic Embryogenesis. Int. J. Mol. Sci. 2021, 22, 5310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chia, J.M.; Kumari, S.; Stein, J.C.; Liu, Z.; Narechania, A.; Maher, C.A.; Guill, K.; McMullen, M.D.; Ware, D. A genome-wide characterization of microRNA genes in maize. PLoS Genet. 2009, 5, e1000716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marc, J.; Bernd, W.; Wolfgang, D.L.; Jesus, V.C.; Jens, T.; Thomas, K.; Franois, P.; Group, B. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2019, 7, 106–111. [Google Scholar]
- Hawker, N.P.; Bowman, J.L. Roles for Class III HD-Zip and KANADI genes in Arabidopsis root development. Plant Physiol. 2004, 135, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Wang, Y.; Han, M.; Fu, Z.; Li, W.; Liu, Z.; Hu, Y.; Tang, J. MicroRNA transcriptomic analysis of heterosis during maize seed germination. PLoS ONE 2012, 7, e39578. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Liu, Y.; Zhang, J.; Liu, H.; Hu, Y.; Du, H.; Li, Y.; Chen, J.; Wei, B.; Huang, Y. Identification and characterization of microRNAs in the developing maize endosperm. Genomics 2013, 102, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.Y.; Tang, C.Y.; Guo, Y.M.; Yang, M.K.; Yang, R.W.; Lu, G.H.; Yang, Y.H. Comparison of miRNAs and Their Targets in Seed Development between Two Maize Inbred Lines by High-Throughput Sequencing and Degradome Analysis. PLoS ONE 2016, 11, e0159810. [Google Scholar] [CrossRef] [Green Version]
- Gong, S.; Ding, Y.; Huang, S.; Zhu, C. Identification of miRNAs and Their Target Genes Associated with Sweet Corn Seed Vigor by Combined Small RNA and Degradome Sequencing. J. Agric. Food Chem. 2015, 63, 5485–5491. [Google Scholar] [CrossRef]
- Yue, J.; Lu, X.; Zhang, H.; Ge, J.; Gao, X.; Liu, Y. Identification of Conserved and Novel MicroRNAs in Blueberry. Front. Plant Sci. 2017, 8, 1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Lai, B.; Hu, B.; Qin, Y.; Hu, G.; Zhao, J. Identification of MicroRNAs and Their Target Genes Related to the Accumulation of Anthocyanins in Litchi chinensis by High-Throughput Sequencing and Degradome Analysis. Front. Plant Sci. 2016, 7, 2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef]
- Lima, J.C.D.; Loss-Morais, G.; Margis, R. MicroRNAs play critical roles during plant development and in response to abiotic stresses. Genet. Mol. Biol. 2012, 35, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nodine, M.D.; Bartel, D.P. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.C.; Wang, H.J.; Liang, H.H.; Gao, Z.; Luo, L.N.; He, C.; Xiang, Z.X. Identification and differential expression of miRNA related to seed dormancy of Paris polyphylla var. chinensischinensis. China J. Chin. Mater. Med. 2020, 45, 5958–5966. [Google Scholar]
- Yan, Y.; Wang, H.; Hamera, S.; Chen, X.; Fang, R. miR444a has multiple functions in the rice nitrate-signaling pathway. Plant J. 2014, 78, 44–55. [Google Scholar] [CrossRef]
- Sun, X.Z.; Wang, Z.W.; Lou, G.C.; Wang, D.J.; Qi, Y.; Yu, J.; Cang, J. Exogenous abscisic acid and methyl jasmonate regulate the expression of miR444a and target gene TaMADS57 in winter wheat under cold stress. Plant Physiol. J. 2022, 58, 708–722. [Google Scholar]
- Wang, Z.N. OsMADS27 Targeted by miR444b. 2 Functions in Seed Dormancy Regulation in Rice (Oryza sativa); University of Chinese Academy of Sciences: Beijing, China, 2015. [Google Scholar]
- Zhang, X.Y.; Tian, Y.H.; Qin, Y.Z.; Xiong, X.Y.; Hu, X.X. The Role of miR169 Family Members in the Processes of Growth, Development and Abiotic Stress Response in Planta. J. Plant Genet. Resour. 2021, 22, 900–909. [Google Scholar]
- Mu, J.; Tan, H.; Hong, S.; Liang, Y.; Zuo, J. Arabidopsis transcription factor genes NF-YA1, 5, 6, and 9 play redundant roles in male gametogenesis, embryogenesis, and seed development. Mol. Plant 2013, 6, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Jian, C.; Lv, J.; Yan, Y.; Chi, Q.; Li, Z.; Wang, Q.; Liu, X.; Zhao, H. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.). BMC Genom. 2014, 15, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Zhao, M.; Gao, W.; Sun, S.; Li, W.X. microRNA/microRNA* complementarity is important for the regulation pattern of NFYA5 by miR169 under dehydration shock in Arabidopsis. Plant J. 2017, 91, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xu, Z.; Mo, Q.; Zou, C.; Li, W.; Xu, Y.; Xie, C. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann. Bot. 2013, 112, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, M.B.; Tu, J.X.; Helliwell, C.A.; Waterhouse, P.M.; Dennis, E.S.; Fu, T.D.; Fan, Y.L. Cloning and characterization of microRNAs from Brassica napus. FEBS Lett. 2007, 581, 3848–3856. [Google Scholar] [CrossRef]
- Serivichyaswat, P.T.; Susila, H.; Ahn, J.H. Elongated Hypocotyl 5-Homolog (HYH) Negatively Regulates Expression of the Ambient Temperature-Responsive MicroRNA Gene MIR169. Front. Plant Sci. 2017, 8, 2087. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ge, L.; Liang, R.; Li, W.; Ruan, K.; Lin, H.; Jin, Y. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. 2009, 10, 29. [Google Scholar] [CrossRef]
- Liu, H.J.; Qin, C.; Chen, Z.; Zuo, T.; Pan, G.T. Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genom. 2014, 15, 25. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H.; Jiang, C.; Xu, C.; Li, P.; Li, S.; et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar]
- Li, W.X.; Oono, Y.; Zhu, J.; He, X.J.; Wu, J.M.; Iida, K.; Lu, X.Y.; Cui, X.; Jin, H.; Zhu, J.K. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 2008, 20, 2238–2251. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Jia, W.; Zhang, J. AtMKK1 mediates ABA-induced CAT1 expression and H2O2 production via AtMPK6-coupled signaling in Arabidopsis. Plant J. 2008, 54, 440–451. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, L.; Liu, Y.; Wang, Y.; Zhang, A.; Tan, M.; Jiang, M. The C2H2-type zinc finger protein ZFP182 is involved in abscisic acid-induced antioxidant defense in rice. J. Integr. Plant Biol. 2012, 54, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Wen, F.; Yao, D.; Wang, L.; Guo, J.; Ni, L.; Zhang, A.; Tan, M.; Jiang, M. A novel rice C2H2-type zinc finger protein, ZFP36, is a key player involved in abscisic acid-induced antioxidant defence and oxidative stress tolerance in rice. J. Exp. Bot. 2014, 65, 5795–5809. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, M.Z.; Jia, B.W.; Leng, Y.; Sun, X.L. Research progress regarding the function and mechanism of rice AP2/ERF transcription factor in stress response. Acta Agron. Sin. 2022, 48, 781–790. [Google Scholar] [CrossRef]
- Sun, M.; Shen, Y.; Chen, Y.; Wang, Y.; Cai, X.; Yang, J.; Jia, B.; Dong, W.; Chen, X.; Sun, X. Osa-miR1320 targets the ERF transcription factor OsERF096 to regulate cold tolerance via JA-mediated signaling. Plant Physiol. 2022, 5, 2500–2516. [Google Scholar] [CrossRef]
- Fu, J.; Zhu, C.; Wang, C.; Liu, L.; Shen, Q.; Xu, D.; Wang, Q. Maize transcription factor ZmEREB20 enhanced salt tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2021, 159, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Huang, M.; Zhang, Z.; Cheng, S. Genome-wide analysis of the AP2/ERF gene family in maize waterlogging stress response. Euphytica 2014, 198, 115–126. [Google Scholar] [CrossRef]
- Tang, J.; Chu, C. MicroRNAs in crop improvement: Fine-tuners for complex traits. Nat. Plants 2017, 3, 17077. [Google Scholar] [CrossRef]
- Liu, D.; Song, Y.; Chen, Z.; Yu, D. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant 2009, 136, 223–236. [Google Scholar] [CrossRef]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Li, Y.; Cai, H.; Ji, W.; Guo, D.; Zhu, Y. Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 2010, 231, 991–1001. [Google Scholar] [CrossRef]
- Guo, X.; Sun, X.; Liu, S.; Gong, C.; Feng, C.; Han, X.; Lv, T.; Zhou, Y.; Wang, Z.; Di, H. Screening and application of SSR markers related to seed storability traits in maize (Zea mays L.). Genet. Resour. Crop. Evol. 2021, 68, 2521–2535. [Google Scholar] [CrossRef]
- McDonough, C.M.; Floyd, C.D.; Waniska, R.D.; Rooney, L.W. Effect of accelerated aging on maize, sorghum, and sorghum meal. J. Cereal Sci. 2004, 39, 351–361. [Google Scholar] [CrossRef]
- Zhu, J.B.; Shan, C.G.; Ni, D.P.; Zhu, Y.W.; Wang, Z.F. Study on the advantages of “inverted filter paper” paper bed method in the determination of germination rate of small seeds. Seed Technol. 2016, 34, 127–128. [Google Scholar]
- Kandala, C.; Leffler, R.G.; Nelson, S.O.; Lawrence, K.C. Capacitive Sensors for Measuring Single-Kernel Moisture Content in Corn. Trans. Asae 1986, 30, 0793–0797. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Li, D.; Chen, H. Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genom. 2011, 12, 154. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR_Name | miR_Seq | Up/Down | Fold_Change (AA156_24h (Mean)/AA237_24h (Mean)) | log2 (Fold_Change) | p Value (t_Test) | AA237_24h (Mean) | AA156_24h (Mean) |
|---|---|---|---|---|---|---|---|
| zma-miR156a-5p | TGACAGAAGAGAGTGAGCAC | up | 4.13 | 2.04 | 0.0203 | 366 | 1510 |
| zma-miR156e-3p_L+1_2ss14CA22TC | TGCTCACTGCTCTATCTGTCACC | down | 0.42 | −1.24 | 0.0132 | 220 | 93 |
| zma-miR156l-3p_L+1 | TGCTCACTGCTCTATCTGTCACC | down | 0.42 | −1.24 | 0.0132 | 220 | 93 |
| zma-miR162-3p_R+1 | TCGATAAACCTCTGCATCCAG | up | 12.92 | 3.69 | 0.0039 | 3 | 44 |
| zma-miR164a-5p | TGGAGAAGCAGGGCACGTGCA | up | 4.96 | 2.31 | 0.0274 | 23 | 115 |
| zma-miR166a-3p | TCGGACCAGGCTTCATTCCCC | up | 3.83 | 1.94 | 0.0442 | 24,545 | 94,027 |
| zma-miR166l-3p | TCGGACCAGGCTTCATTCCTC | up | 3.95 | 1.98 | 0.0046 | 412 | 1625 |
| zma-miR167c-3p_L-1 | ATCATGCTGTGGCAGCCTCACT | up | 5.18 | 2.37 | 0.0095 | 28 | 146 |
| zma-miR167e-5p | TGAAGCTGCCAGCATGATCTG | up | 2.70 | 1.43 | 0.0459 | 54 | 147 |
| zma-miR167j-p3_2ss12AT20TA | TCTGAAGAAAGTGTTGGCTATC | up | 4.71 | 2.23 | 0.0451 | 2 | 7 |
| zma-miR169o-3p_L-1R+1 | GCAGGTCTTCTTGGCTAGCC | down | 0.02 | −5.96 | 0.0459 | 153 | 2 |
| zma-miR169o-5p_R-1 | TAGCCAAGAATGACTTGCCT | down | 0.32 | −1.64 | 0.0111 | 1327 | 427 |
| zma-miR319a-p5 | TAGCTGCCGACTCATCCATTCA | up | 6.62 | 2.73 | 0.0068 | 19 | 128 |
| zma-miR319b-p5 | AGCTGCCGACTCATTCACCCA | up | 2.94 | 1.56 | 0.0450 | 241 | 708 |
| zma-miR319c-p5 | TAGCTGCCGACTCATCCATTCA | up | 6.62 | 2.73 | 0.0068 | 19 | 128 |
| zma-miR319d-p5 | AGCTGCCGACTCATTCACCCA | up | 2.94 | 1.56 | 0.0450 | 241 | 708 |
| zma-miR390a-5p | AAGCTCAGGAGGGATAGCGCC | down | 0.25 | −1.99 | 0.0016 | 237 | 60 |
| zma-miR396a-5p | TTCCACAGCTTTCTTGAACTG | down | 0.21 | −2.26 | 0.0248 | 1603 | 334 |
| zma-miR396c_L-1 | TCCACAGGCTTTCTTGAACTG | down | 0.48 | −1.05 | 0.0455 | 519 | 251 |
| zma-miR397a-5p | TCATTGAGCGCAGCGTTGATG | down | 0.03 | −4.87 | 0.0233 | 42 | 1 |
| zma-miR397b-p5 | TTGAGCGCAGCGTTGATGAGC | down | 0.01 | −6.79 | 0.0103 | 40,574 | 366 |
| zma-miR398b-5p_R-1 | GGGGCGGACTGGGAACACAT | down | 0.08 | −3.56 | 0.0063 | 340 | 29 |
| zma-miR408a_L-1 | TGCACTGCCTCTTCCCTGGC | down | 0.01 | −6.51 | 0.0083 | 9270 | 102 |
| zma-miR444a_1ss12TC | TGCAGTTGTTGCCTCAAGCTT | up | 7.87 | 2.98 | 0.0484 | 820 | 6457 |
| zma-miR529-5p | AGAAGAGAGAGAGTACAGCCT | down | 0.60 | −0.73 | 0.0453 | 628 | 379 |
| zma-miR827-3p | TTAGATGACCATCAGCAAACA | down | 0.25 | −2.00 | 0.0306 | 10,114 | 2520 |
| zma-miR827-5p_L+1 | TTTTGTTGGTGGTCATTTAACC | down | 0.10 | −3.36 | 0.0154 | 2511 | 244 |
| miRNA | log2 (Fold_Change) | Up/Down | Transcript | GnenID | log2 (Fold_Change) | Description | Up/Down | Focus on GO | KEGG |
|---|---|---|---|---|---|---|---|---|---|
| zma-miR169o-5p_R-1 | −1.63707 | down | XM_008682881.3 | GRMZM2G165488 | 0.68 | Nuclear transcription factor Y subunit A-10 | up | GO:0009793 (embryo development ending in seed dormancy); GO:0048316 (seed development) | NA |
| zma-miR390a-5p | −1.98813 | down | NM_001320234.1 | GRMZM2G134396 | 5.682455 | uncharacterized protein LOC100275036 | up | GO:0003876 (AMP deaminase activity) GO:0009737 (response to abscisic acid);GO:0009793 (embryo development ending in seed dormancy) | NA |
| zma-miR396c_L-1 | −1.04713 | down | XM_008649043.2 | GRMZM2G098594 | 8.267418 | putative growth-regulating factor 14 | up | GO:0008757 (S-adenosylmethionine-dependent methyltransferase activity) | NA |
| XM_008678196.2 | GRMZM2G105335 | 5.682455 | putative growth-regulating factor 3 | up | GO:0009739 (response to gibberellin) | NA | |||
| XM_020552600.2 | GRMZM2G133568 | 11.31 | MADS-box transcription factor 27 isoform X1 | up | GO:0008134 (transcription factor binding) | NA | |||
| zma-MIR397b-p5 | -6.79 | down | XM_020541848.1 | GRMZM2G033230 | 9.73 | putative bZIP transcription factor superfamily protein | up | GO:0006970 (response to osmotic stress); | NA |
| zma-miR444a_1ss12TC | 2.976548 | up | XM_008653282.2 | GRMZM2G399072 | −9.41933 | AP2-like ethylene-responsive transcription factor BBM2 isoform X2 | down | GO:0009873 (ethylene-activated signaling pathway) | NA |
| novel-miR4 | −8.05 | down | XR_002268591.2 | GRMZM2G009871 | 5.6825732 | putative TCP-1/cpn60 chaperonin family protein | up | GO:0044183 (protein binding involved in protein folding) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Lv, Z.; Wang, Y.; Li, C.; Jia, Y.; Zhu, Y.; Cao, M.; Zhou, Y.; Zeng, X.; Wang, Z.; et al. Identification of miRNAs Mediating Seed Storability of Maize during Germination Stage by High-Throughput Sequencing, Transcriptome and Degradome Sequencing. Int. J. Mol. Sci. 2022, 23, 12339. https://doi.org/10.3390/ijms232012339
Song Y, Lv Z, Wang Y, Li C, Jia Y, Zhu Y, Cao M, Zhou Y, Zeng X, Wang Z, et al. Identification of miRNAs Mediating Seed Storability of Maize during Germination Stage by High-Throughput Sequencing, Transcriptome and Degradome Sequencing. International Journal of Molecular Sciences. 2022; 23(20):12339. https://doi.org/10.3390/ijms232012339
Chicago/Turabian StyleSong, Yongfeng, Zhichao Lv, Yue Wang, Chunxiang Li, Yue Jia, Yong Zhu, Mengna Cao, Yu Zhou, Xing Zeng, Zhenhua Wang, and et al. 2022. "Identification of miRNAs Mediating Seed Storability of Maize during Germination Stage by High-Throughput Sequencing, Transcriptome and Degradome Sequencing" International Journal of Molecular Sciences 23, no. 20: 12339. https://doi.org/10.3390/ijms232012339