

Modulating p-AMPK/mTOR Pathway of Mitochondrial Dysfunction Caused by MTERF1 Abnormal Expression in Colorectal Cancer Cells
Abstract
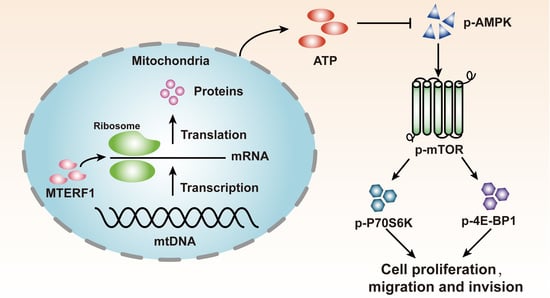
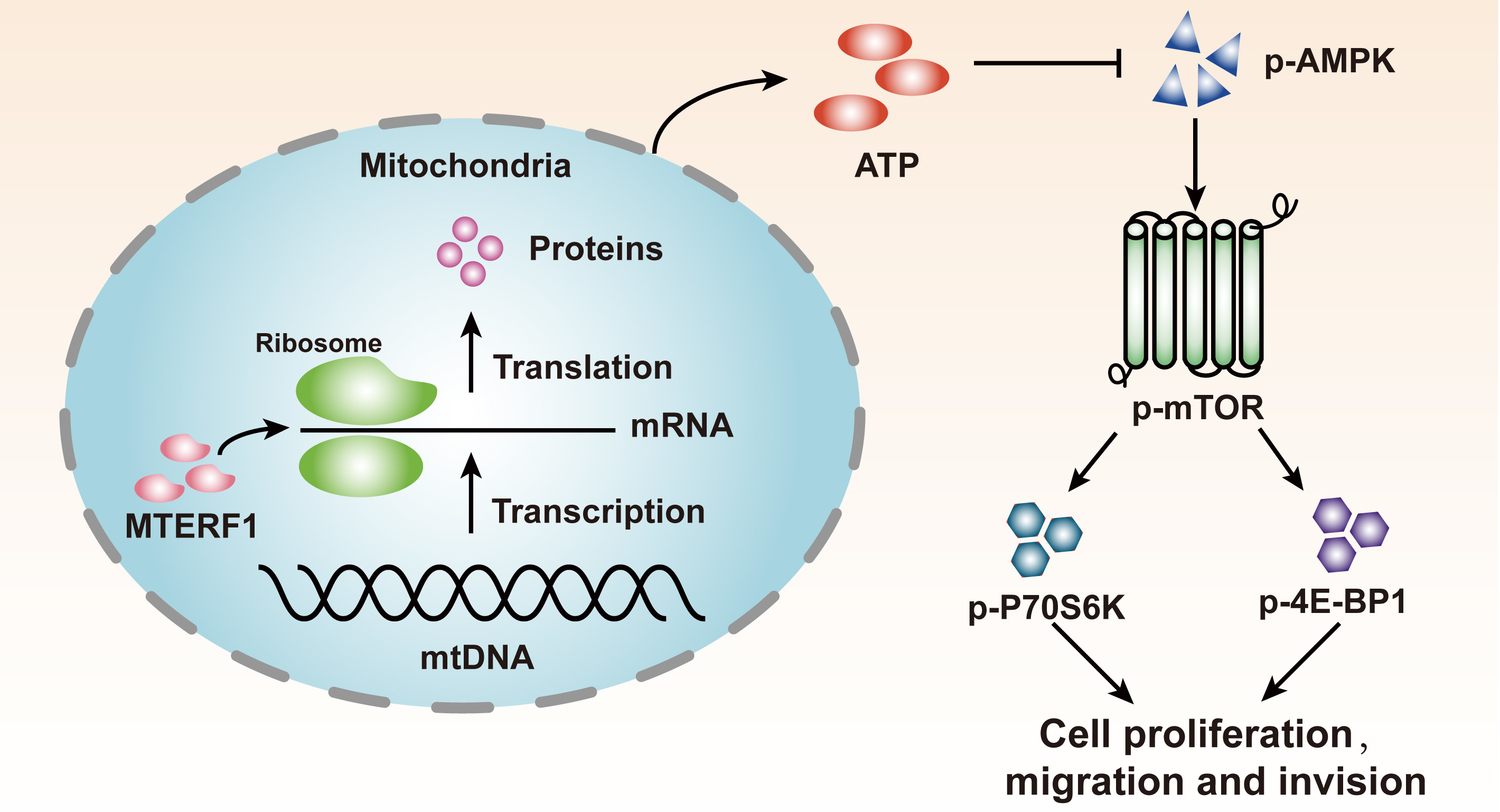{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
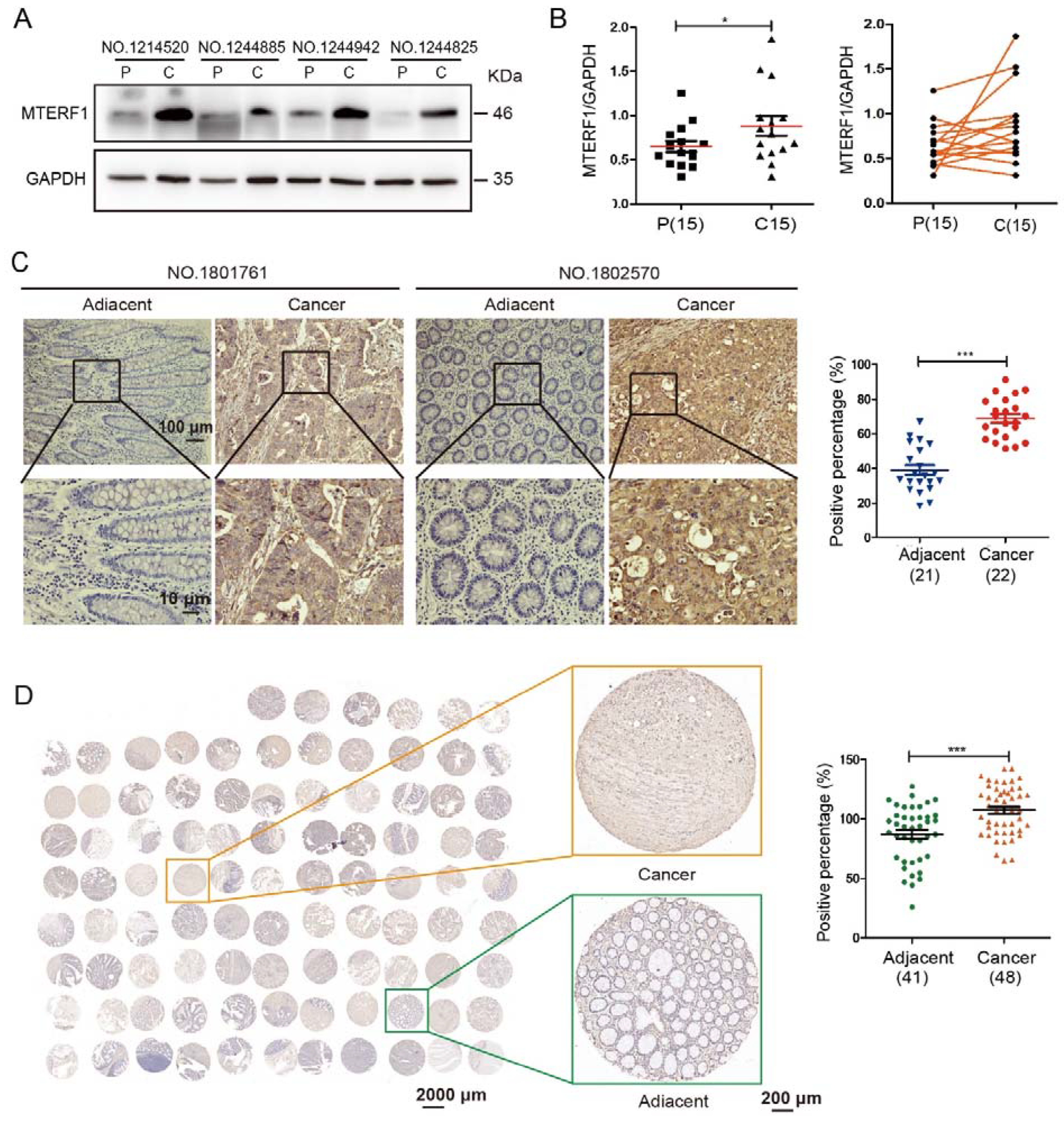
2.1. MTERF1 Is Highly Expressed in CRC Tissues
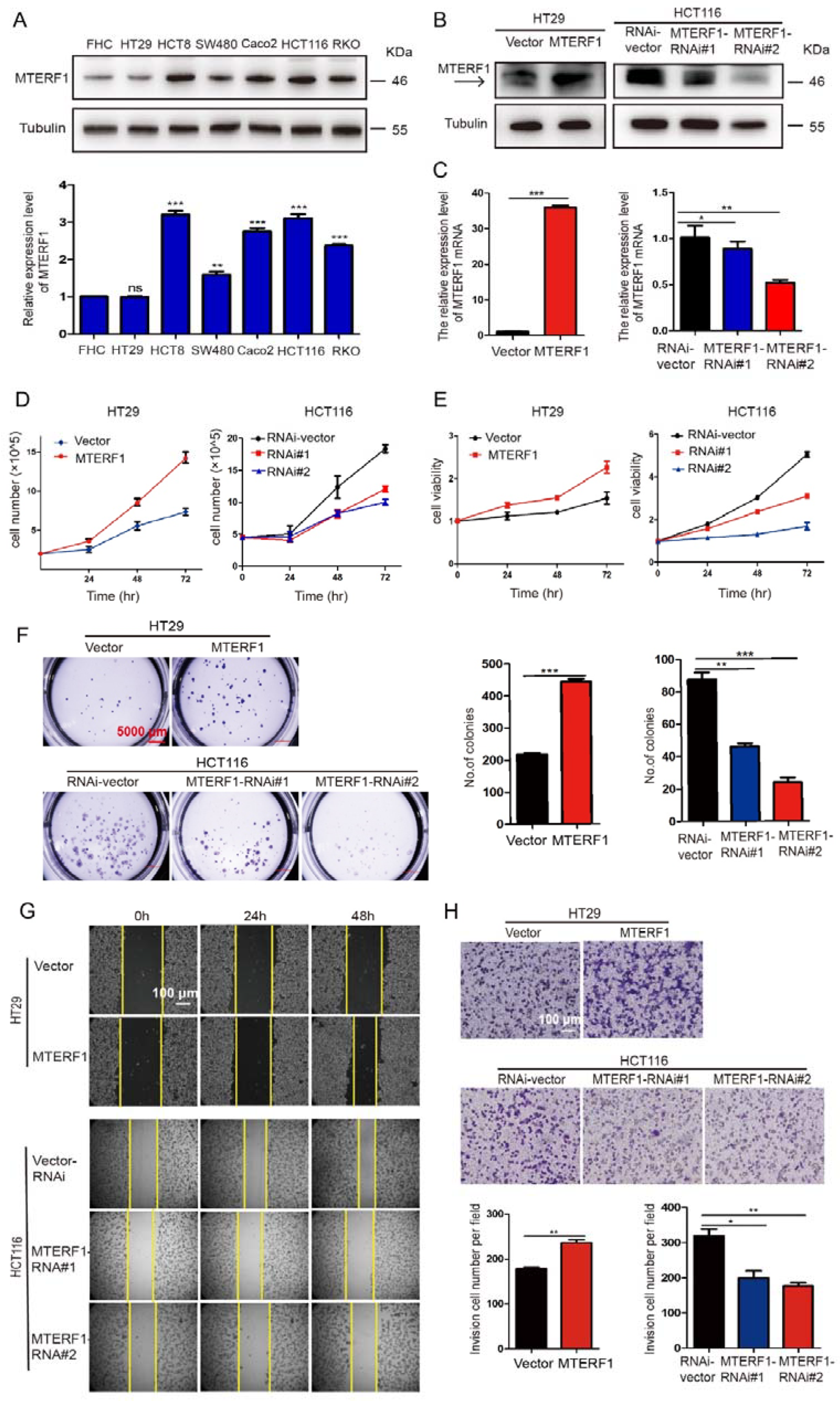
2.2. MTERF1 Promotes the Proliferation, Migration, and Invasion of CRC Cells and Affects Cell Cycle Transition and Apoptosis
2.3. MTERF1 Promotes CRC Xenograft Tumor Formation
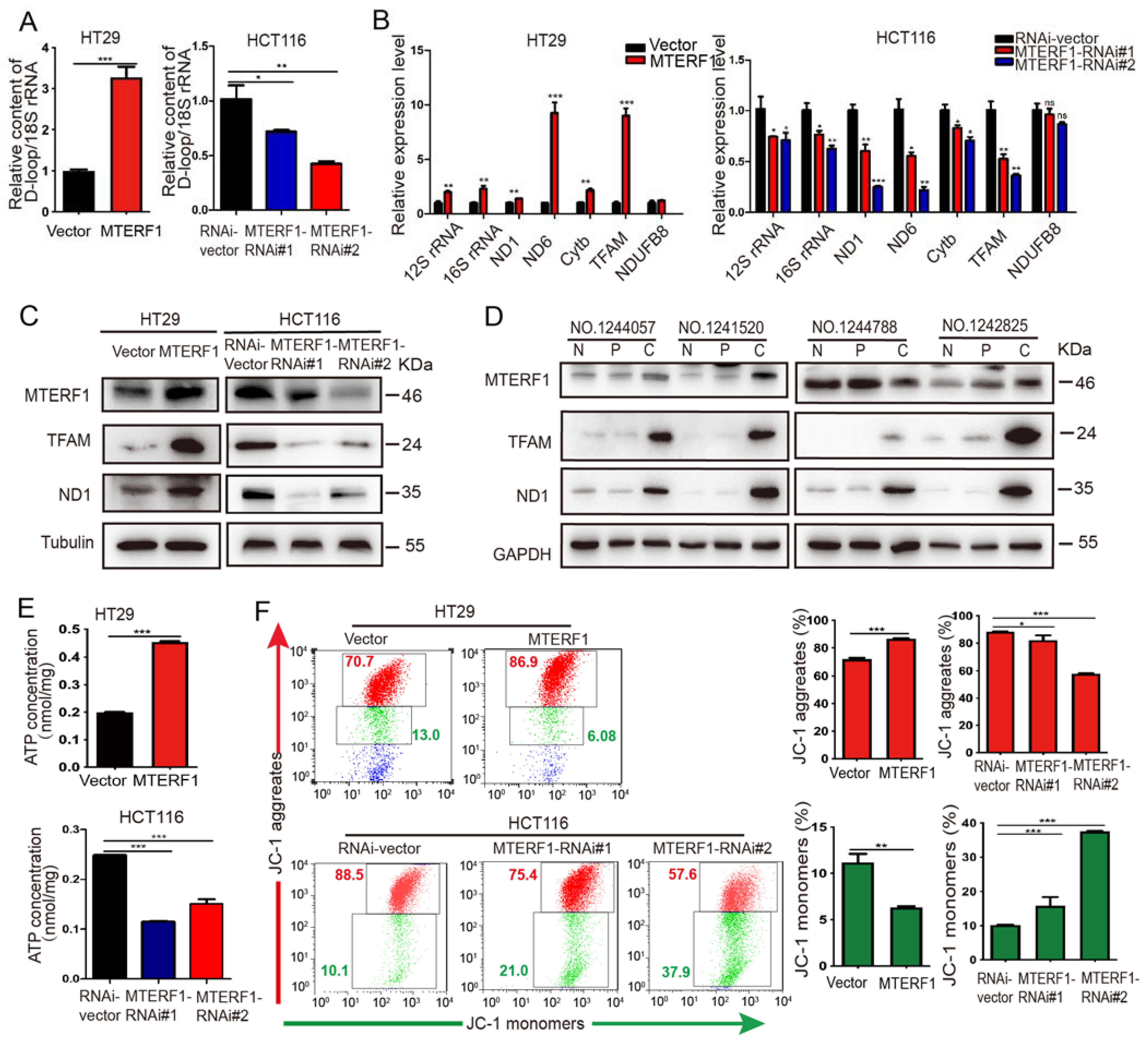
2.4. MTERF1 Positively Regulates Mitochondrial Gene Replication, Transcription, and OXPHOS
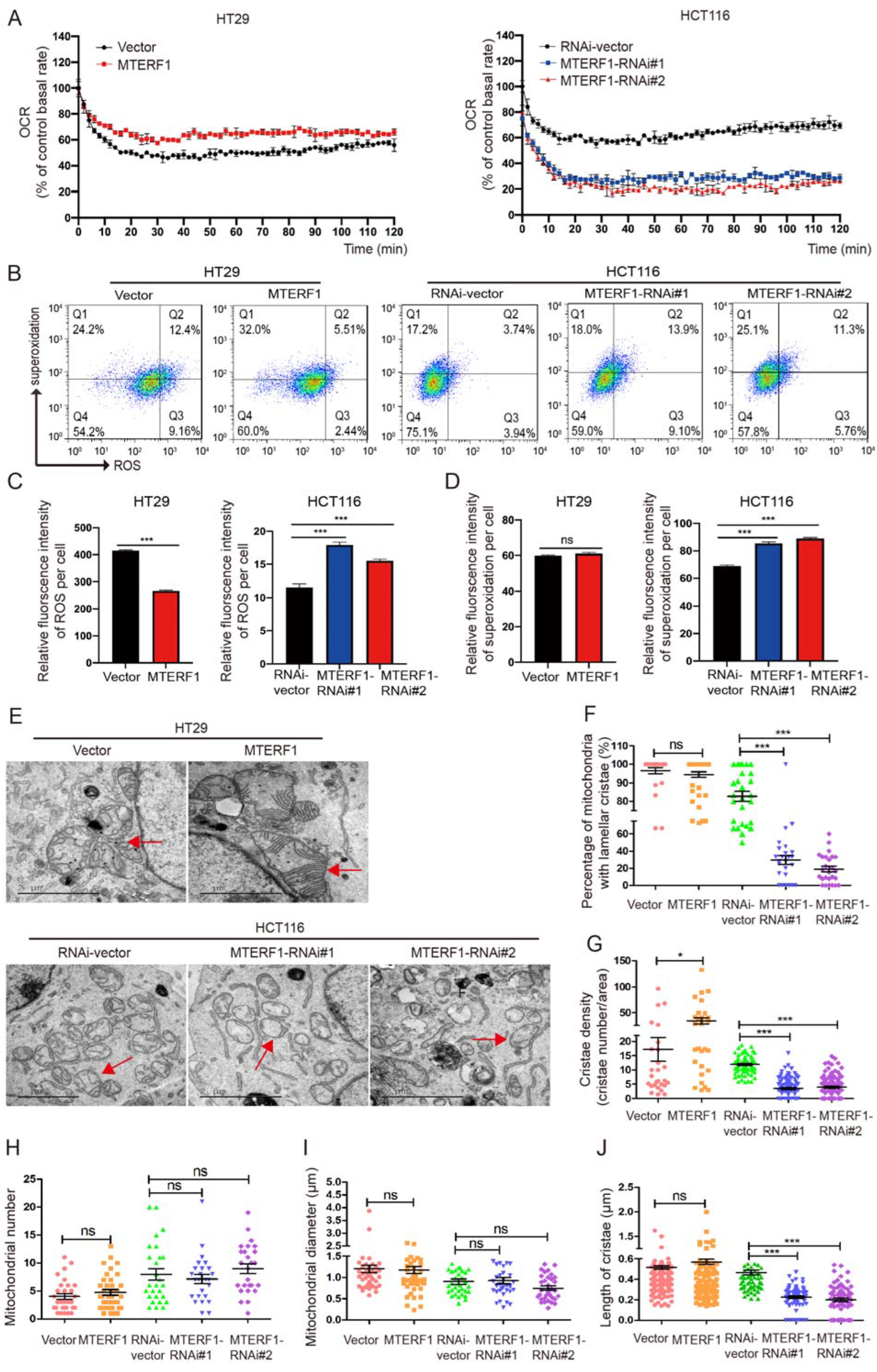
2.5. MTERF1 Expression Changes the Mitochondrial Inner Membrane Ultrastructure
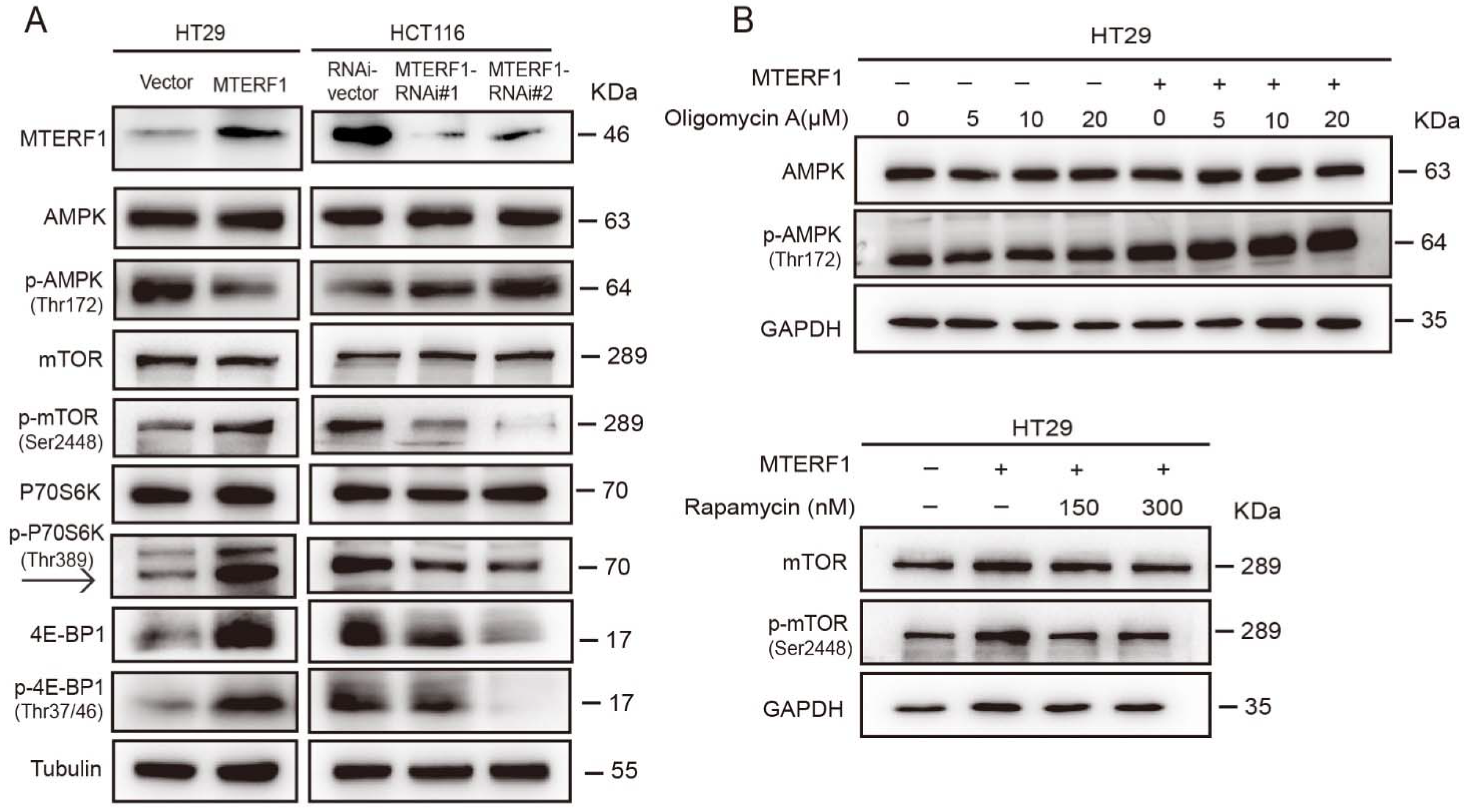
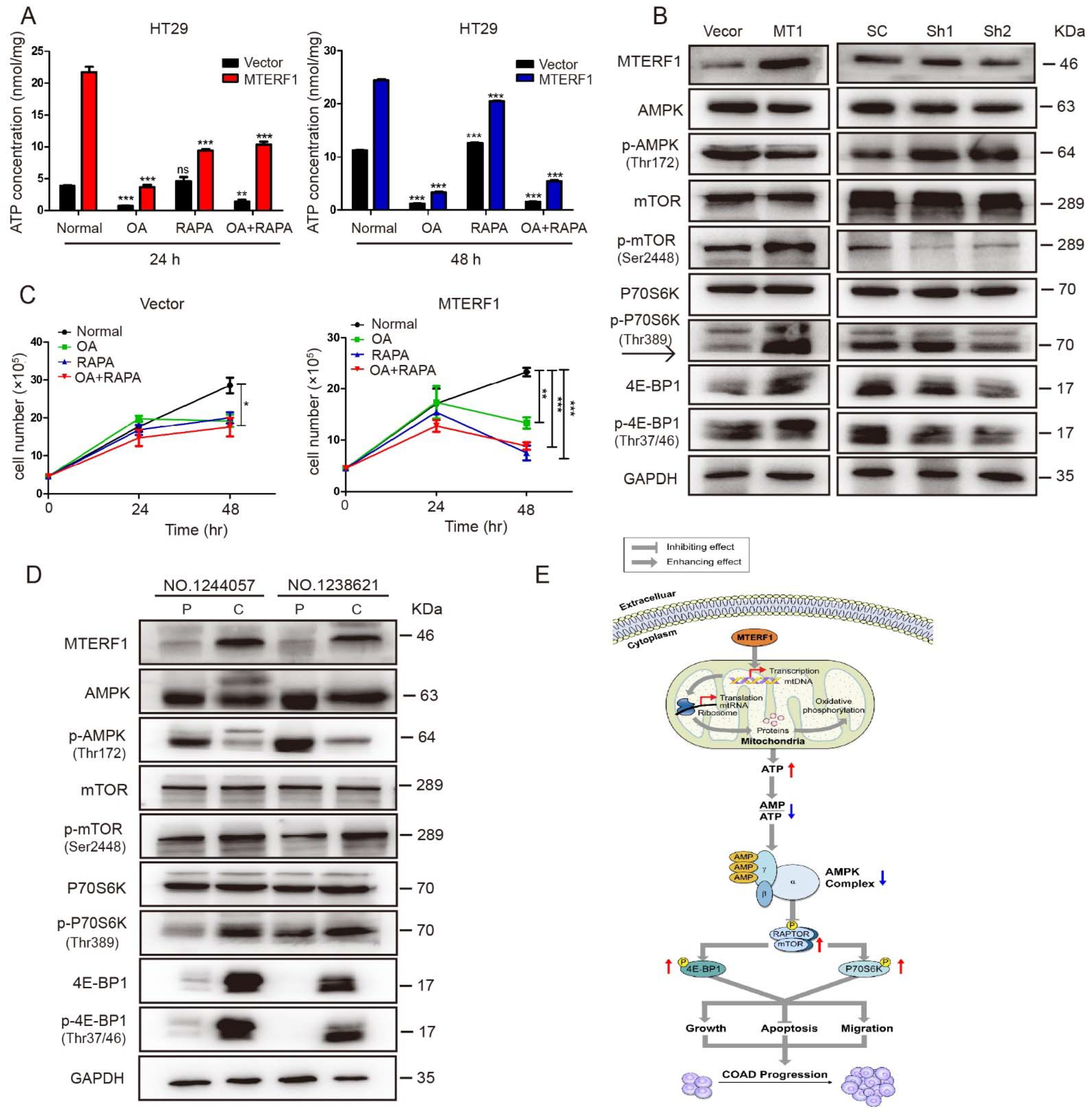
2.6. MTERF1 Expression Correlates with AMPK/mTOR Pathway Activation in Human CRC Cells
3. Discussion
4. Materials and Methods
4.1. Clinical Samples and Data
4.2. Cell Culture
4.3. Plasmids, Transfection, and Infection
4.4. RNA Extraction, DNA Extraction, and Quantitative Real-Time PCR (qPCR)
4.5. Immunohistochemical (IHC) and Immunofluorescence (IF) Staining
4.6. Cell Proliferation, Viability, and Colony Formation Assays
4.7. Wound Healing and Invasion Assay
4.8. Xenograft Tumor Formation Assay
4.9. Western Blotting Analysis
4.10. Flow Cytometric Analyses of Cell Cycle and Apoptosis
4.11. Mitochondrial Membrane Potential Measurement
4.12. Determination of ATP Levels
4.13. ROS/Superoxide Detection Assay
4.14. OCR (Rates of Oxygen consumption) Detection Assay
4.15. Transmission Electron Microscopy
4.16. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, X.; Meng, Y.; Ma, J.; Zhang, Q.; Shao, G.; Wang, L.; Cheng, X.; Hong, X.; Wang, Y.; et al. A Novel Mechanism of Endoplasmic Reticulum Stress- and c-Myc-Degradation-Mediated Therapeutic Benefits of Antineurokinin-1 Receptor Drugs in Colorectal Cancer. Adv. Sci. 2021, 8, e2101936. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lou, S.; Jiang, Z. PHLDA2 regulates EMT and autophagy in colorectal cancer via the PI3K/AKT signaling pathway. Aging 2020, 12, 7985–8000. [Google Scholar] [CrossRef]
- van der Geest, L.G.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.; de Wilt, J.H. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin. Exp. Metastasis 2015, 32, 457–465. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Clinical utility of circulating tumor DNA for colorectal cancer. Cancer Sci. 2019, 110, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Mohammadalipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef]
- Bouda, E.; Stapon, A.; Garcia-Diaz, M. Mechanisms of mammalian mitochondrial transcription. Protein Sci. 2019, 28, 1594–1605. [Google Scholar] [CrossRef]
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature 2020, 588, 712–716. [Google Scholar] [CrossRef]
- Kleine, T.; Leister, D. Emerging functions of mammalian and plant mTERFs. Biochim. Biophys. Acta 2015, 1847, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T. Arabidopsis thaliana mTERF proteins: Evolution and functional classification. Front. Plant Sci. 2012, 3, 233. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Silva, P.; Martinez-Azorin, F.; Micol, V.; Attardi, G. The human mitochondrial transcription termination factor (mTERF) is a multizipper protein but binds to DNA as a monomer, with evidence pointing to intramolecular leucine zipper interactions. EMBO J. 1997, 16, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Roberti, M.; Polosa, P.L.; Bruni, F.; Manzari, C.; Deceglie, S.; Gadaleta, M.N.; Cantatore, P. The MTERF family proteins: Mitochondrial transcription regulators and beyond. Biochim. Biophys. Acta 2009, 1787, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, M.; Ruzzenente, B.; Harmel, J.; Mourier, A.; Jemt, E.; Lopez, M.D.; Kukat, C.; Stewart, J.B.; Wibom, R.; Meharg, C.; et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013, 17, 618–626. [Google Scholar] [CrossRef]
- Hyvarinen, A.K.; Pohjoismaki, J.L.; Reyes, A.; Wanrooij, S.; Yasukawa, T.; Karhunen, P.J.; Spelbrink, J.N.; Holt, I.J.; Jacobs, H.T. The mitochondrial transcription termination factor mTERF modulates replication pausing in human mitochondrial DNA. Nucleic Acids Res. 2007, 35, 6458–6474. [Google Scholar] [CrossRef]
- Shi, Y.; Posse, V.; Zhu, X.; Hyvarinen, A.K.; Jacobs, H.T.; Falkenberg, M.; Gustafsson, C.M. Mitochondrial transcription termination factor 1 directs polar replication fork pausing. Nucleic Acids Res. 2016, 44, 5732–5742. [Google Scholar] [CrossRef]
- Chen, G.; Dai, J.; Tan, S.; Meng, S.; Liu, Z.; Li, M.; Cui, Q.; Yu, M. MTERF1 regulates the oxidative phosphorylation activity and cell proliferation in HeLa cells. Acta Biochim. Biophys. Sin. 2014, 46, 512–521. [Google Scholar] [CrossRef]
- Sun, S.; Wu, C.; Yang, C.; Chen, J.; Wang, X.; Nan, Y.; Huang, Z.; Ma, L. Prognostic roles of mitochondrial transcription termination factors in non-small cell lung cancer. Oncol. Lett. 2019, 18, 3453–3462. [Google Scholar] [CrossRef]
- Leveille, C.F.; Mikhaeil, J.S.; Turner, K.D.; Silvera, S.; Wilkinson, J.; Fajardo, V.A. Mitochondrial cristae density: A dynamic entity that is critical for energy production and metabolic power in skeletal muscle. J. Physiol. 2017, 595, 2779–2780. [Google Scholar] [CrossRef]
- Illes, P.; Burnstock, G.; Tang, Y. Astroglia-Derived ATP Modulates CNS Neuronal Circuits. Trends Neurosci. 2019, 42, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Elkholi, R.; Renault, T.T.; Serasinghe, M.N.; Chipuk, J.E. Putting the pieces together: How is the mitochondrial pathway of apoptosis regulated in cancer and chemotherapy? Cancer Metab. 2014, 2, 16. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, N.W. The impact of mitochondrial endosymbiosis on the evolution of calcium signaling. Cell Calcium 2015, 57, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, D.; Kee, S.H. Metformin-activated AMPK regulates beta-catenin to reduce cell proliferation in colon carcinoma RKO cells. Oncol. Lett. 2019, 17, 2695–2702. [Google Scholar]
- Martinez-Azorin, F. The mitochondrial ribomotor hypothesis. IUBMB Life 2005, 57, 27–30. [Google Scholar] [CrossRef]
- Hyvarinen, A.K.; Kumanto, M.K.; Marjavaara, S.K.; Jacobs, H.T. Effects on mitochondrial transcription of manipulating mTERF protein levels in cultured human HEK293 cells. BMC Mol. Biol. 2010, 11, 72. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Zhang, K.; Zhou, X.; Wang, J.; Zhou, Y.; Qi, W.; Chen, H.; Nie, S.; Xie, M. Dendrobium officinale polysaccharide triggers mitochondrial disorder to induce colon cancer cell death via ROS-AMPK-autophagy pathway. Carbohydr. Polym. 2021, 264, 118018. [Google Scholar] [CrossRef]
- Liu, G.; Kuang, S.; Cao, R.; Wang, J.; Peng, Q.; Sun, C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J. 2019, 33, 10089–10103. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhang, F.; Zhao, Y.; Shao, D.; Zheng, X.; Chen, Y.; He, K.; Li, J.; Chen, L. Berberine Enhances Chemosensitivity and Induces Apoptosis through Dose-orchestrated AMPK Signaling in Breast Cancer. J. Cancer 2017, 8, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, M.C.; Jing, L.; Liu, Z.Y.; Guo, H.; Liu, Y.; Bai, Y.Y.; Cheng, Y.Z.; Nan, K.J.; Liang, X. Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des. Dev. Ther. 2015, 9, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, L.; Guo, X.; Cheng, C.; Luo, Y.; Wang, J.; Wang, J.; Liu, Y.; Cao, Y.; Li, P.; et al. Combating multidrug resistance and metastasis of breast cancer by endoplasmic reticulum stress and cell-nucleus penetration enhanced immunochemotherapy. Theranostics 2022, 12, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.; Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur. J. Med. Chem. 2022, 239, 114542. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Rong, H.; Chen, Y.; Wang, Q.; Qian, S.; Ji, Y.; Yao, W.; Yin, J.; Gao, X. Targeted disruption of mitochondria potently reverses multidrug resistance in cancer therapy. Br. J. Pharmacol. 2022, 179, 3346–3362. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Zhang, L.; Zou, Y.; Tao, Y.; Wang, B.; Li, B.; Liu, R.; Wang, B.; Ding, L.; Cui, Q.; et al. Modulating p-AMPK/mTOR Pathway of Mitochondrial Dysfunction Caused by MTERF1 Abnormal Expression in Colorectal Cancer Cells. Int. J. Mol. Sci. 2022, 23, 12354. https://doi.org/10.3390/ijms232012354
Liu Q, Zhang L, Zou Y, Tao Y, Wang B, Li B, Liu R, Wang B, Ding L, Cui Q, et al. Modulating p-AMPK/mTOR Pathway of Mitochondrial Dysfunction Caused by MTERF1 Abnormal Expression in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2022; 23(20):12354. https://doi.org/10.3390/ijms232012354
Chicago/Turabian StyleLiu, Qianqian, Longlong Zhang, Yayan Zou, Ying Tao, Bing Wang, Bin Li, Ruai Liu, Boyong Wang, Lei Ding, Qinghua Cui, and et al. 2022. "Modulating p-AMPK/mTOR Pathway of Mitochondrial Dysfunction Caused by MTERF1 Abnormal Expression in Colorectal Cancer Cells" International Journal of Molecular Sciences 23, no. 20: 12354. https://doi.org/10.3390/ijms232012354
APA StyleLiu, Q., Zhang, L., Zou, Y., Tao, Y., Wang, B., Li, B., Liu, R., Wang, B., Ding, L., Cui, Q., Lin, J., Mao, B., Xiong, W., & Yu, M. (2022). Modulating p-AMPK/mTOR Pathway of Mitochondrial Dysfunction Caused by MTERF1 Abnormal Expression in Colorectal Cancer Cells. International Journal of Molecular Sciences, 23(20), 12354. https://doi.org/10.3390/ijms232012354