
Longitudinal Serum Protein Analysis of Women with a High Risk of Developing Breast Cancer Reveals Large Interpatient Versus Small Intrapatient Variations: First Results from the TESTBREAST Study

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
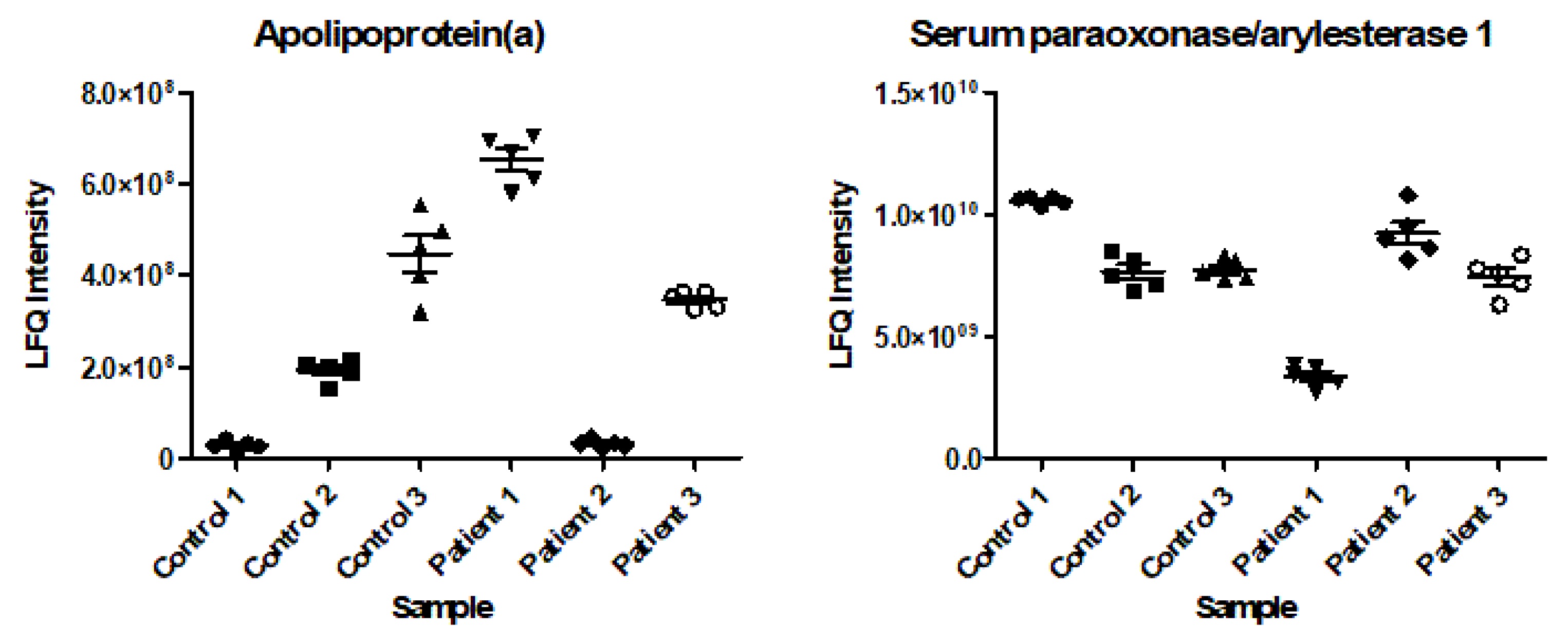
2.2. Clustering Analysis
2.3. Intrapatient and Interpatient Variability in Protein Levels
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Serum Samples
4.3. Questionnaires
4.4. Data Management
4.5. Sample Preparation
4.6. Mass Spectrometry
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| CTC | Circulating Tumor Cell |
| miRNA | microRNA |
| TESTBREAST | Trial Early Serum Test BREAST cancer |
| MS | Mass Spectrometry |
| TEAB | Triethylammonium Bicarbonate Buffer |
| ACN | Acetonitrile |
| SDC | Sodium Deoxycholate |
| DTT | Dithiothreitol |
| TFA | Trifluoroacetic Acid |
| LC-MS | Liquid Chromatography-Mass Spectrometry |
| RP-HPLC | Reverse-Phase High-Performance Liquid Chromatography |
| LC-MS/MS | Liquid Chromatography Tandem Mass Spectrometry |
| DDA | Data-Dependent Acquisition |
| CV | Compensation Voltages |
| AGC | Automatic Gain Control |
| HCD | Higher-Energy Collisional Dissociation |
| NCE | Normalized Collision Energy |
| LFQ | Label-Free Quantitation |
| TNBC | Triple-Negative Breast Cancer |
References
- Lee, T.C.; Reyna, C.; Shaughnessy, E.; Lewis, J.D. Screening of populations at high risk for breast cancer. J. Surg. Oncol. 2019, 120, 820–830. [Google Scholar] [CrossRef]
- Siu, A.L. Screening for Breast Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2016, 164, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.D.; Pappas, M.; Cantor, A.; Griffin, J.; Daeges, M.; Humphrey, L. Harms of Breast Cancer Screening: Systematic Review to Update the 2009 U.S. Preventive Services Task Force Recommendation. Ann. Intern. Med. 2016, 164, 256–267. [Google Scholar] [CrossRef]
- Marrugo-Ramírez, J.; Mir, M.; Samitier, J. Blood-Based Cancer Biomarkers in Liquid Biopsy: A Promising Non-Invasive Alternative to Tissue Biopsy. Int. J. Mol. Sci. 2018, 19, 2877. [Google Scholar] [CrossRef]
- Nassar, F.J.; Chamandi, G.; Tfaily, M.A.; Zgheib, N.K.; Nasr, R. Peripheral Blood-Based Biopsy for Breast Cancer Risk Prediction and Early Detection. Front. Med. (Lausanne) 2020, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Hayes, D.F.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Reuben, J.M.; Doyle, G.V.; Matera, J.; Allard, W.J.; Miller, M.C.; et al. Circulating tumor cells: A novel prognostic factor for newly diagnosed metastatic breast cancer. J. Clin. Oncol. 2005, 23, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guan, X.; Fan, Z.; Ching, L.M.; Li, Y.; Wang, X.; Cao, W.M.; Liu, D.X. Non-Invasive Biomarkers for Early Detection of Breast Cancer. Cancers 2020, 12, 2767. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Silva, J.; Sanchez, A.; Garcia, J.M.; Dominguez, G.; Provencio, M.; Sanfrutos, L.; Jareño, E.; Colas, A.; España, P.; et al. Tumor DNA in plasma at diagnosis of breast cancer patients is a valuable predictor of disease-free survival. Clin. Cancer Res. 2002, 8, 3761–3766. [Google Scholar] [PubMed]
- Patuleia, S.I.S.; Hagenaars, S.C.; Moelans, C.B.; Ausems, M.; van Gils, C.H.; Tollenaar, R.; van Diest, P.J.; Mesker, W.E.; van der Wall, E. Lessons Learned from Setting Up a Prospective, Longitudinal, Multicenter Study with Women at High Risk for Breast Cancer. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 441–449. [Google Scholar] [CrossRef] [PubMed]
- de Noo, M.E.; Deelder, A.; van der Werff, M.; Ozalp, A.; Mertens, B.; Tollenaar, R. MALDI-TOF serum protein profiling for the detection of breast cancer. Onkologie 2006, 29, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Shi, T.; Petyuk, V.A.; Schepmoes, A.A.; Fillmore, T.L.; Wang, Y.T.; Cardoni, W.; Coppit, G.; Srivastava, S.; Goodman, J.F.; et al. Detection of Head and Neck Cancer Based on Longitudinal Changes in Serum Protein Abundance. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Núñez, C. Blood-based protein biomarkers in breast cancer. Clin. Chim. Acta. 2019, 490, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Stastna, M.; Van Eyk, J.E. Secreted proteins as a fundamental source for biomarker discovery. Proteomics 2012, 12, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Bodde, M.C.; Hermans, M.P.J.; Jukema, J.W.; Schalij, M.J.; Lijfering, W.M.; Rosendaal, F.R.; Romijn, F.; Ruhaak, L.R.; van der Laarse, A.; Cobbaert, C.M. Apolipoproteins A1, B, and apoB/apoA1 ratio are associated with first ST-segment elevation myocardial infarction but not with recurrent events during long-term follow-up. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2019, 108, 520–538. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.G.; Kramer, B.S.; McIntosh, M.; Patterson, B.H.; Shyr, Y.; Skates, S. Evaluating markers for the early detection of cancer: Overview of study designs and methods. Clin. Trials 2006, 3, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Brüning, T.; Johnen, G.; Casjens, S.; Bonberg, N.; Taeger, D.; Müller, A.; Weber, D.G.; Behrens, T. Biomarker research with prospective study designs for the early detection of cancer. Biochim. Biophys. Acta. 2014, 1844, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Galvão, E.R.; Martins, L.M.; Ibiapina, J.O.; Andrade, H.M.; Monte, S.J. Breast cancer proteomics: A review for clinicians. J. Cancer. Res. Clin. Oncol. 2011, 137, 915–925. [Google Scholar] [CrossRef]
- Gast, M.C.; Schellens, J.H.; Beijnen, J.H. Clinical proteomics in breast cancer: A review. Breast Cancer Res. Treat 2009, 116, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Neagu, A.N.; Whitham, D.; Buonanno, E.; Jenkins, A.; Alexa-Stratulat, T.; Tamba, B.I.; Darie, C.C. Proteomics and its applications in breast cancer. Am. J. Cancer Res. 2021, 11, 4006–4049. [Google Scholar] [PubMed]
- Bakker, M.F.; de Lange, S.V.; Pijnappel, R.M.; Mann, R.M.; Peeters, P.H.M.; Monninkhof, E.M.; Emaus, M.J.; Loo, C.E.; Bisschops, R.H.C.; Lobbes, M.B.I.; et al. Supplemental MRI Screening for Women with Extremely Dense Breast Tissue. N. Engl. J. Med. 2019, 381, 2091–2102. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Birth Year | Year Tumor Diagnosis | Histology Tumor | Hormonal Status Tumor | Mutational Status | Medical History of Cancer | Menopause | Hormonal Use | Smoking | Medicine Use | Adnexal Surgery | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | P1 | 1972 | 2014 | Ductal (NST) | TNBC | BRCA1 | No | Yes | Yes | Yes | No | Preventive adnex extirpation |
| Case | P2 | 1959 | 2014 | Ductal (NST) | TNBC | BRCA1 | No | Yes | No | No | Yes | Preventive adnex extirpation |
| Case | P3 | 1975 | 2011 | Ductal (NST) | TNBC | BRCA1 | No | Yes | No | No | Yes | Preventive adnex extirpation |
| Control | C1 | 1971 | N/A | N/A | N/A | No, high risk | No | No | Yes | Sometimes | No | No |
| Control | C2 | 1960 | N/A | N/A | N/A | No, high risk | Yes, ovarium carcinoma | Yes | Yes | No | No | Hysterosalpingo-oophorectomy |
| Control | C3 | 1969 | N/A | N/A | N/A | BRCA2 | No | No | No | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagenaars, S.C.; Dekker, L.J.M.; Ravesteijn, B.; van Vlierberghe, R.L.P.; Romijn, F.P.H.T.M.; Verhoeff, L.; Witkamp, A.J.; Schenk, K.E.; Keymeulen, K.B.I.M.; Menke-Pluijmers, M.B.E.; et al. Longitudinal Serum Protein Analysis of Women with a High Risk of Developing Breast Cancer Reveals Large Interpatient Versus Small Intrapatient Variations: First Results from the TESTBREAST Study. Int. J. Mol. Sci. 2022, 23, 12399. https://doi.org/10.3390/ijms232012399
Hagenaars SC, Dekker LJM, Ravesteijn B, van Vlierberghe RLP, Romijn FPHTM, Verhoeff L, Witkamp AJ, Schenk KE, Keymeulen KBIM, Menke-Pluijmers MBE, et al. Longitudinal Serum Protein Analysis of Women with a High Risk of Developing Breast Cancer Reveals Large Interpatient Versus Small Intrapatient Variations: First Results from the TESTBREAST Study. International Journal of Molecular Sciences. 2022; 23(20):12399. https://doi.org/10.3390/ijms232012399
Chicago/Turabian StyleHagenaars, Sophie C., Lennard J. M. Dekker, Bob Ravesteijn, Ronald L. P. van Vlierberghe, Fred P. H. T. M. Romijn, Linda Verhoeff, Arjen J. Witkamp, Karin E. Schenk, Kristien B. I. M. Keymeulen, Marian B. E. Menke-Pluijmers, and et al. 2022. "Longitudinal Serum Protein Analysis of Women with a High Risk of Developing Breast Cancer Reveals Large Interpatient Versus Small Intrapatient Variations: First Results from the TESTBREAST Study" International Journal of Molecular Sciences 23, no. 20: 12399. https://doi.org/10.3390/ijms232012399
APA StyleHagenaars, S. C., Dekker, L. J. M., Ravesteijn, B., van Vlierberghe, R. L. P., Romijn, F. P. H. T. M., Verhoeff, L., Witkamp, A. J., Schenk, K. E., Keymeulen, K. B. I. M., Menke-Pluijmers, M. B. E., Dassen, A. E., Kortmann, B. A., de Vries, J., Rutgers, E. J. T., van der Burgt, Y. E. M., Meershoek-Klein Kranenbarg, E., Cobbaert, C. M., Luider, T. M., Mesker, W. E., & Tollenaar, R. A. E. M. (2022). Longitudinal Serum Protein Analysis of Women with a High Risk of Developing Breast Cancer Reveals Large Interpatient Versus Small Intrapatient Variations: First Results from the TESTBREAST Study. International Journal of Molecular Sciences, 23(20), 12399. https://doi.org/10.3390/ijms232012399