


Dysregulated Hemostasis and Immunothrombosis in Cerebral Cavernous Malformations

 , , and
, , and
Abstract
:1. Introduction
2. Endothelial Dysfunction in CCM
3. The Hemostatic System in CCM Is Dysregulated
3.1. The Hemostatic System
3.2. Are vWF-Activated Platelets Supporting the Activation of the Intrinsic Pathway in CCM?
3.3. Is the Extrinsic Pathway Activated When CCM Lesions Are Established?
3.4. CCM Lesions Have Stable Thrombi with Polyhedrocytes
3.5. Tertiary Hemostasis May Be Disrupted in CCM
3.6. Cavernomas Have Anticoagulant Regions
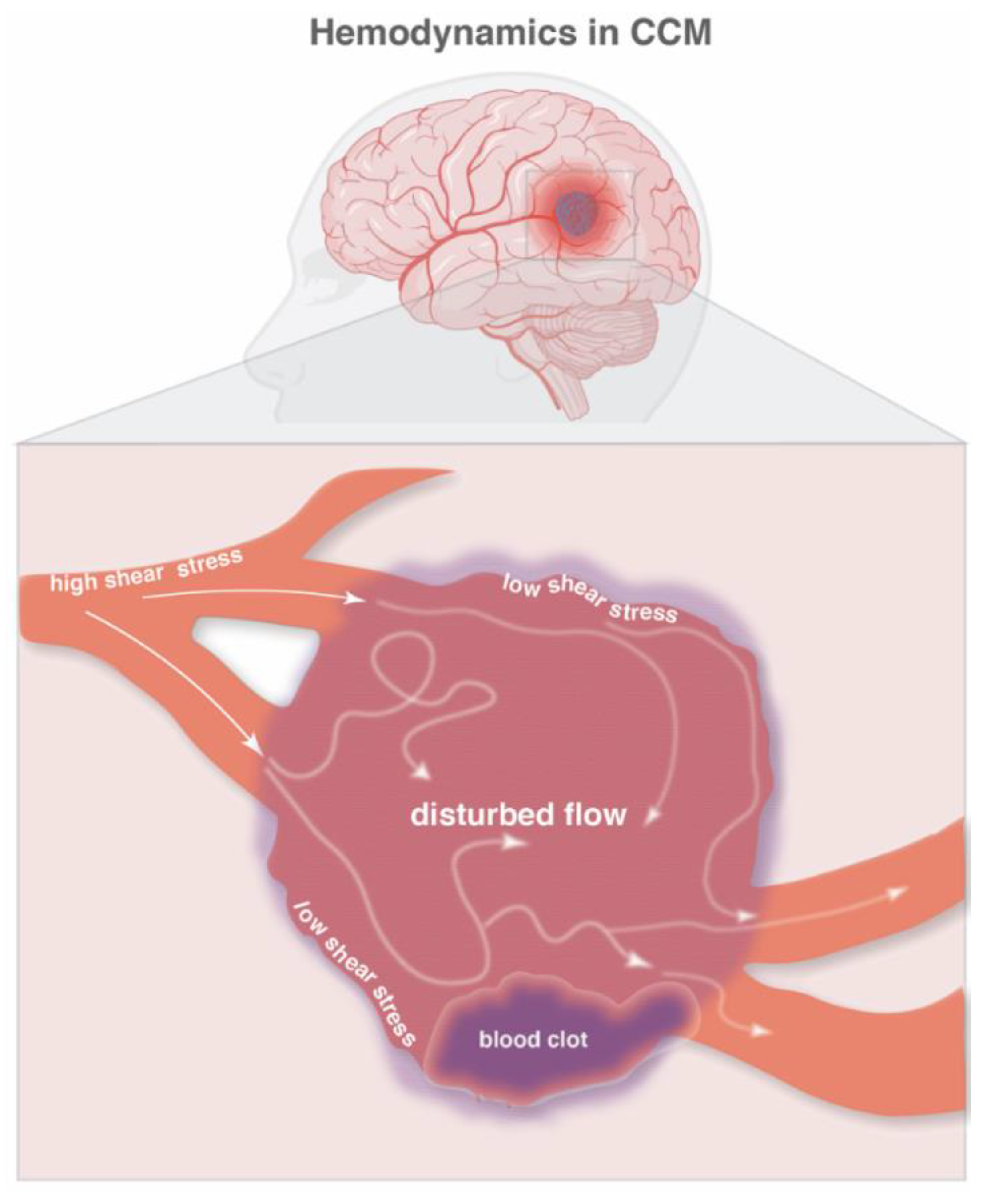
4. Hemodynamics and Hypoxia in CCM
5. Are Platelets Enhancing EndMT in CCM-Deficient Endothelial Cells?
6. Immunothrombosis and Neuroinflammation in CCM
6.1. The Role of PARs in CCM
6.2. Immune Cells and Cytokines in CCM
6.3. The Role of the Microbiome in Innate Immunity and Lesion Severity
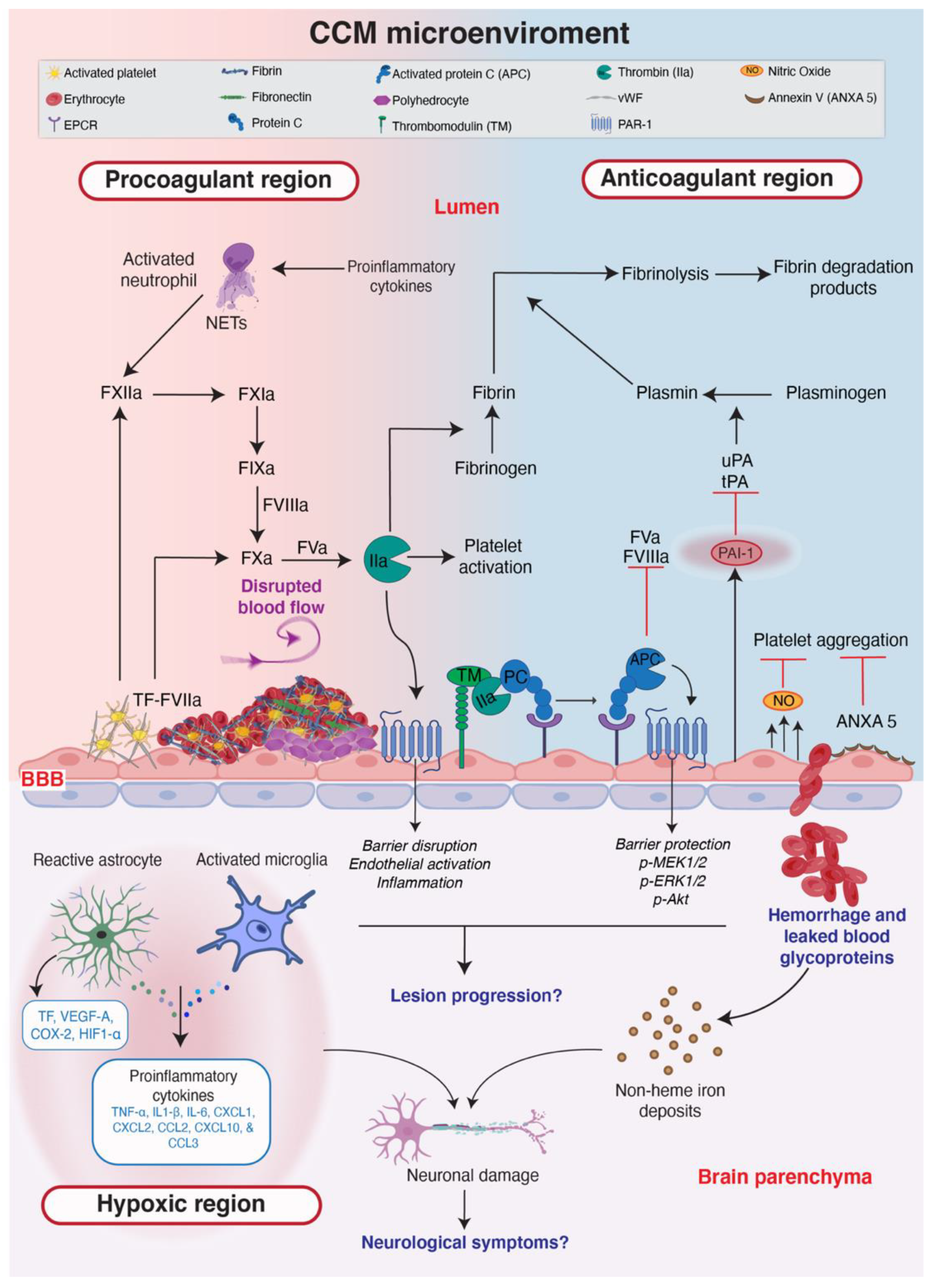
7. The CCM Microenvironment
8. Anticoagulants for Patients with CCM
9. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cerebral Cavernous Malformation Clinical Trials. Secondary Cerebral Cavernous Malformation Clinical Trials. Available online: https://www.clinicaltrials.gov/ct2/results?cond=Cerebral+Cavernous+Malformation&term=&cntry=&state=&city=&dist= (accessed on 17 October 2022).
- Choquet, H.; Pawlikowska, L.; Lawton, M.T.; Kim, H. Genetics of cerebral cavernous malformations: Current status and future prospects. J. Neurosurg. Sci. 2015, 59, 211–220. [Google Scholar] [PubMed]
- Liquori, C.L.; Berg, M.J.; Squitieri, F.; Leedom, T.P.; Ptacek, L.; Johnson, E.W.; Marchuk, D.A. Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am. J. Hum. Genet. 2007, 80, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinverno, M.; Maderna, C.; Abu Taha, A.; Corada, M.; Orsenigo, F.; Valentino, M.; Pisati, F.; Fusco, C.; Graziano, P.; Giannotta, M.; et al. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat. Commun. 2019, 10, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detter, M.R.; Snellings, D.A.; Marchuk, D.A. Cerebral Cavernous Malformations Develop Through Clonal Expansion of Mutant Endothelial Cells. Circ. Res. 2018, 123, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.A.; Snellings, D.A.; Su, Y.R.S.; Hong, C.C.; Castro, M.; Tang, A.T.; Detter, M.R.; Hobson, N.; Girard, R.; Romanos, S.; et al. PIK3CA and CCM mutations fuel cavernomas through a cancer-like mechanism. Nature 2021, 594, 271–276. [Google Scholar] [CrossRef]
- Snellings, D.A.; Hong, C.C.; Ren, A.A.; Lopez-Ramirez, M.A.; Girard, R.; Srinath, A.; Marchuk, D.A.; Ginsberg, M.H.; Awad, I.A.; Kahn, M.L. Cerebral Cavernous Malformation from Mechanism to Therapy. Circ. Res. 2021, 129, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef]
- Cuttano, R.; Rudini, N.; Bravi, L.; Corada, M.; Giampietro, C.; Papa, E.; Morini, M.F.; Maddaluno, L.; Baeyens, N.; Adams, R.H.; et al. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 2016, 8, 6–24. [Google Scholar] [CrossRef]
- Zhou, Z.; Rawnsley, D.R.; Goddard, L.M.; Pan, W.; Cao, X.J.; Jakus, Z.; Zheng, H.; Yang, J.; Arthur, J.S.; Whitehead, K.J.; et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev. Cell 2015, 32, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef]
- Lopez-Ramirez, M.A.; Pham, A.; Girard, R.; Wyseure, T.; Hale, P.; Yamashita, A.; Koskimaki, J.; Polster, S.; Saadat, L.; Romero, I.A.; et al. Cerebral cavernous malformations form an anticoagulant vascular domain in humans and mice. Blood 2019, 133, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskimaki, J.; Zhang, D.D.; Li, Y.; Saadat, L.; Moore, T.; Lightle, R.; Polster, S.P.; Carrion-Penagos, J.; Lyne, S.B.; Zeineddine, H.A.; et al. Transcriptome clarifies mechanisms of lesion genesis versus progression in models of Ccm3 cerebral cavernous malformations. Acta Neuropathol. Commun. 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Koskimaki, J.; Girard, R.; Li, Y.; Saadat, L.; Zeineddine, H.A.; Lightle, R.; Moore, T.; Lyne, S.; Avner, K.; Shenkar, R.; et al. Comprehensive transcriptome analysis of cerebral cavernous malformation across multiple species and genotypes. JCI Insight 2019, 4, e126167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsenigo, F.; Conze, L.L.; Jauhiainen, S.; Corada, M.; Lazzaroni, F.; Malinverno, M.; Sundell, V.; Cunha, S.I.; Brannstrom, J.; Globisch, M.A.; et al. Mapping endothelial-cell diversity in cerebral cavernous malformations at single-cell resolution. Elife 2020, 9, e61413. [Google Scholar] [CrossRef]
- Yau, A.C.Y.; Globisch, M.A.; Onyeogaziri, F.C.; Conze, L.L.; Smith, R.; Jauhiainen, S.; Corada, M.; Orsenigo, F.; Huang, H.; Herre, M.; et al. Inflammation and neutrophil extracellular traps in cerebral cavernous malformation. Cell. Mol. Life Sci. 2022, 79, 206. [Google Scholar] [CrossRef]
- Globisch, M.A.; Onyeogaziri, F.C.; Jauhiainen, S.; Yau, A.; Orsenigo, F.; Conze, L.L.; Arce, M.; Corada, M.; Smith, R.; Rorsman, C.; et al. Immunothrombosis and vascular heterogeneity in cerebral cavernous malformation. Blood 2022. ahead of print. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- McDonald, D.A.; Shi, C.; Shenkar, R.; Stockton, R.A.; Liu, F.; Ginsberg, M.H.; Marchuk, D.A.; Awad, I.A. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke 2012, 43, 571–574. [Google Scholar] [CrossRef]
- Stassen, J.M.; Arnout, J.; Deckmyn, H. The hemostatic system. Curr. Med. Chem. 2004, 11, 2245–2260. [Google Scholar] [CrossRef] [PubMed]
- Sniecinski, R.M.; Chandler, W.L. Activation of the Hemostatic System During Cardiopulmonary Bypass. Anesth. Analg. 2011, 113, 1319–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Eddleston, M.; de la Torre, J.C.; Oldstone, M.B.; Loskutoff, D.J.; Edgington, T.S.; Mackman, N. Astrocytes are the primary source of tissue factor in the murine central nervous system. A role for astrocytes in cerebral hemostasis. J. Clin. Investig. 1993, 92, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Kozareva, V.; Martin, C.; Osorno, T.; Rudolph, S.; Guo, C.; Vanderburg, C.; Nadaf, N.; Regev, A.; Regehr, W.G.; Macosko, E. A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature 2021, 598, 214–219. [Google Scholar] [CrossRef]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef]
- Weibel, E.R.; Palade, G.E. New Cytoplasmic Components in Arterial Endothelia. J. Cell Biol. 1964, 23, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Vanhooreibeke, K.; De Meyer, S.F. von Willebrand Factor and Platelet Glycoprotein Ib: A Thromboinflammatory Axis in Stroke. Front. Immunol. 2019, 10, 2884. [Google Scholar] [CrossRef] [PubMed]
- Much, C.D.; Sendtner, B.S.; Schwefel, K.; Freund, E.; Bekeschus, S.; Otto, O.; Pagenstecher, A.; Felbor, U.; Rath, M.; Spiegler, S. Inactivation of Cerebral Cavernous Malformation Genes Results in Accumulation of von Willebrand Factor and Redistribution of Weibel-Palade Bodies in Endothelial Cells. Front. Mol. Biosci. 2021, 8, 622547. [Google Scholar] [CrossRef]
- Fredenburgh, J.C.; Gross, P.L.; Weitz, J.I. Emerging anticoagulant strategies. Blood 2017, 129, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renne, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faxalv, L.; Boknas, N.; Strom, J.O.; Tengvall, P.; Theodorsson, E.; Ramstrom, S.; Lindahl, T.L. Putting polyphosphates to the test: Evidence against platelet-induced activation of factor XII. Blood 2013, 122, 3818–3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boknas, N.; Faxalv, L.; Strom, J.O.; Tengvall, P.; Theodorsson, E.; Ramstrom, S.; Lindahl, T.L. Response: Platelets do not generate activated factor XII--how inappropriate experimental models have led to misleading conclusions. Blood 2014, 124, 1692–1694. [Google Scholar] [CrossRef] [Green Version]
- Rusu, L.; Andreeva, A.; Visintine, D.J.; Kim, K.; Vogel, S.M.; Stojanovic-Terpo, A.; Chernaya, O.; Liu, G.Q.; Bakhshi, F.R.; Haberichter, S.L.; et al. G protein-dependent basal and evoked endothelial cell vWF secretion. Blood 2014, 123, 442–450. [Google Scholar] [CrossRef] [Green Version]
- Noshiro, S.; Mikami, T.; Kataoka-Sasaki, Y.; Sasaki, M.; Ohnishi, H.; Ohtaki, S.; Wanibuchi, M.; Mikuni, N.; Kocsis, J.D.; Honmou, O. Co-expression of tissue factor and IL-6 in immature endothelial cells of cerebral cavernous malformations. J. Clin. Neurosci. 2017, 37, 83–90. [Google Scholar] [CrossRef]
- Cox, E.M.; Bambakidis, N.C.; Cohen, M.L. Pathology of cavernous malformations. Handb. Clin. Neurol. 2017, 143, 267–277. [Google Scholar] [CrossRef]
- Abe, M.; Fukudome, K.; Sugita, Y.; Oishi, T.; Tabuchi, K.; Kawano, T. Thrombus and encapsulated hematoma in cerebral cavernous malformations. Acta Neuropathol. 2005, 109, 503–509. [Google Scholar] [CrossRef]
- Tanriover, G.; Sozen, B.; Seker, A.; Kilic, T.; Gunel, M.; Demir, N. Ultrastructural analysis of vascular features in cerebral cavernous malformations. Clin. Neurol. Neurosurg. 2013, 115, 438–444. [Google Scholar] [CrossRef]
- Cho, J.; Mosher, D.F. Enhancement of thrombogenesis by plasma fibronectin cross-linked to fibrin and assembled in platelet thrombi. Blood 2006, 107, 3555–3563. [Google Scholar] [CrossRef]
- Cho, J.; Mosher, D.F. Role of fibronectin assembly in platelet thrombus formation. J. Thromb. Haemost. 2006, 4, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Reininger, A.J. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia 2008, 14, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Lebedeva, T.; Nagaswami, C.; Hayes, V.; Massefski, W.; Litvinov, R.I.; Rauova, L.; Lowery, T.J.; Weisel, J.W. Clot contraction: Compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood 2014, 123, 1596–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernysh, I.N.; Nagaswami, C.; Kosolapova, S.; Peshkova, A.D.; Cuker, A.; Cines, D.B.; Cambor, C.L.; Litvinov, R.I.; Weisel, J.W. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep.-Uk 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Wolberg, A.S.; Aleman, M.M.; Leiderman, K.; Machlus, K.R. Procoagulant Activity in Hemostasis and Thrombosis: Virchow’s Triad Revisited. Anesth. Analg. 2012, 114, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Tutwiler, V.; Mukhitov, A.R.; Peshkova, A.D.; Le Minh, G.; Khismatullin, R.R.; Vicksman, J.; Nagaswami, C.; Litvinov, R.I.; Weisel, J.W. Shape changes of erythrocytes during blood clot contraction and the structure of polyhedrocytes. Sci. Rep. 2018, 8, 17907. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Shenkar, R.; Du, H.; Duckworth, E.; Raja, H.; Batjer, H.H.; Awad, I.A. Immune response in human cerebral cavernous malformations. Stroke 2009, 40, 1659–1665. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Shenkar, R.; Zeineddine, H.A.; Girard, R.; Fam, M.D.; Austin, C.; Moore, T.; Lightle, R.; Zhang, L.; Wu, M.; et al. B-Cell Depletion Reduces the Maturation of Cerebral Cavernous Malformations in Murine Models. J. Neuroimmune Pharmacol. 2016, 11, 369–377. [Google Scholar] [CrossRef]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef]
- Loskutoff, D.J.; Vanmourik, J.A.; Erickson, L.A.; Lawrence, D. Detection of an Unusually Stable Fibrinolytic Inhibitor Produced by Bovine Endothelial-Cells. Proc. Natl. Acad. Sci. USA 1983, 80, 2956–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunitada, S.; Fitzgerald, G.A.; Fitzgerald, D.J. Inhibition of Clot Lysis and Decreased Binding of Tissue-Type Plasminogen-Activator as a Consequence of Clot Retraction. Blood 1992, 79, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Orsenigo, F. Endothelial adherens junctions at a glance. J. Cell Sci. 2013, 126, 2545–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt, C. P07204 TRBM_HUMAN. Available online: https://www.uniprot.org/uniprotkb/P07204/entry (accessed on 17 October 2022).
- UniProt, C. P15306 TRBM_MOUSE. Available online: https://www.uniprot.org/uniprotkb/P15306/entry (accessed on 17 October 2022).
- Esmon, C.T. Inflammation and the activated protein C anticoagulant pathway. Semin. Thromb. Hemost. 2006, 32, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Esmon, N.L.; Owen, W.G.; Esmon, C.T. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J. Biol. Chem. 1982, 257, 859–864. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A.; Balasubramanian, V.; Garcia-Cardena, G.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ. Res. 2005, 96, E48–E57. [Google Scholar] [CrossRef] [Green Version]
- Hamik, A.; Lin, Z.Y.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerzsten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem. 2007, 282, 13769–13779. [Google Scholar] [CrossRef] [Green Version]
- Gkaliagkousi, E.; Ritter, J.; Ferro, A. Platelet-derived nitric oxide signaling and regulation. Circ. Res. 2007, 101, 654–662. [Google Scholar] [CrossRef] [Green Version]
- Galan, A.M.; van Heerde, W.L.; Escolar, G.; Ordinas, A.; Sixma, J.; de Groot, P.G. Antithrombotic action of annexin V proved as efficient as direct inhibition of tissue factor or thrombin. Eur. J. Clin. Investig. 2006, 36, 633–639. [Google Scholar] [CrossRef]
- Reutelingsperger, C.P.M.; Hornstra, G.; Hemker, H.C. Isolation and Partial-Purification of a Novel Anticoagulant from Arteries of Human Umbilical-Cord. Eur. J. Biochem. 1985, 151, 625–629. [Google Scholar] [CrossRef]
- Daly, C.; Pasnikowski, E.; Burova, E.; Wong, V.; Aldrich, T.H.; Griffiths, J.; Ioffe, E.; Daly, T.J.; Fandl, J.P.; Papadopoulos, N.; et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15491–15496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultstrom, M.; Fromell, K.; Larsson, A.; Persson, B.; Nilsson, B.; Quaggin, S.E.; Betsholtz, C.; Frithiof, R.; Lipcsey, M.; Jeansson, M. Angiopoietin-2 Inhibition of Thrombomodulin-Mediated Anticoagulation-A Novel Mechanism That May Contribute to Hypercoagulation in Critically Ill COVID-19 Patients. Biomedicines 2022, 10, 1333. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Q.; Li, J.J.; Hu, L.; Lee, M.; Karpatkin, S. Thrombin induces increased expression and secretion of angiopoietin-2 from human umbilical vein endothelial cells. Blood 2002, 99, 1646–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenny Zhou, H.; Qin, L.; Zhang, H.; Tang, W.; Ji, W.; He, Y.; Liang, X.; Wang, Z.; Yuan, Q.; Vortmeyer, A.; et al. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat. Med. 2016, 22, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Sforza, D.M.; Putman, C.M.; Cebral, J.R. Hemodynamics of Cerebral Aneurysms. Annu. Rev. Fluid Mech. 2009, 41, 91–107. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wang, Q.; Xu, F.; Zhang, X.; Zeng, Z.; Yan, Y.; Lu, Z.; Xue, G.; Zuo, Q.; Luo, Y.; et al. Underlying mechanism of hemodynamics and intracranial aneurysm. Chin. Neurosurg. J. 2021, 7, 44. [Google Scholar] [CrossRef]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef]
- Choquet, H.; Nelson, J.; Pawlikowska, L.; McCulloch, C.E.; Akers, A.; Baca, B.; Khan, Y.; Hart, B.; Morrison, L.; Kim, H. Association of cardiovascular risk factors with disease severity in cerebral cavernous malformation type 1 subjects with the common Hispanic mutation. Cerebrovasc. Dis. 2014, 37, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Rodel, C.J.; Otten, C.; Donat, S.; Lourenco, M.; Fischer, D.; Kuropka, B.; Paolini, A.; Freund, C.; Abdelilah-Seyfried, S. Blood Flow Suppresses Vascular Anomalies in a Zebrafish Model of Cerebral Cavernous Malformations. Circ. Res. 2019, 125, e43–e54. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Coleman, P.; Chen, J.; Ting, K.K.; Choi, J.P.; Zheng, X.; Vadas, M.A.; Gamble, J.R. Low fluid shear stress conditions contribute to activation of cerebral cavernous malformation signalling pathways. Biochim. Biophys. Acta, Mol. Basis Dis. 2019, 1865, 165519. [Google Scholar] [CrossRef]
- Dejana, E.; Lampugnani, M.G. Endothelial cell transitions. Science 2018, 362, 746–747. [Google Scholar] [CrossRef] [PubMed]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFbeta. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Zigler, M.; Kamiya, T.; Brantley, E.C.; Villares, G.J.; Bar-Eli, M. PAR-1 and Thrombin: The Ties That Bind the Microenvironment to Melanoma Metastasis. Cancer Res. 2011, 71, 6561–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosnier, L.O.; Sinha, R.K.; Burnier, L.; Bouwens, E.A.; Griffin, J.H. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012, 120, 5237–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senden, N.H.M.; Jeunhomme, T.M.A.A.; Heemskerk, J.W.M.; Wagenvoord, R.; van’t Veer, C.; Hemker, H.C.; Buurman, W.A. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J. Immunol. 1998, 161, 4318–4324. [Google Scholar]
- Braach, N.; Frommhold, D.; Buschmann, K.; Pflaum, J.; Koch, L.; Hudalla, H.; Staudacher, K.; Wang, H.J.; Isermann, B.; Nawroth, P.; et al. RAGE Controls Activation and Anti-Inflammatory Signalling of Protein C. PLoS ONE 2014, 9, e89422. [Google Scholar] [CrossRef]
- Bouwens, E.A.; Stavenuiter, F.; Mosnier, L.O. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J. Thromb. Haemost. 2013, 11 (Suppl. S1), 242–253. [Google Scholar] [CrossRef] [Green Version]
- Soh, U.J.; Trejo, J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through beta-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar] [CrossRef] [Green Version]
- Molinar-Inglis, O.; Birch, C.A.; Nicholas, D.; Orduna-Castillo, L.; Cisneros-Aguirre, M.; Patwardhan, A.; Chen, B.; Grimsey, N.J.; Coronel, L.J.; Lin, H.; et al. aPC/PAR1 confers endothelial anti-apoptotic activity via a discrete, beta-arrestin-2-mediated SphK1-S1PR1-Akt signaling axis. Proc. Natl. Acad. Sci. USA 2021, 118, e2106623118. [Google Scholar] [CrossRef]
- Uchiba, M.; Okajima, K.; Oike, Y.; Ito, Y.; Fukudome, K.; Isobe, H.; Suda, T. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ. Res. 2004, 95, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Feistritzer, C.; Lenta, R.; Riewald, M. Protease-activated receptors-1 and-2 can mediate endothelial barrier protection: Role in factor Xa signaling. J. Thromb. Haemost. 2005, 3, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Yang, L.; Rezaie, A.R. Factor X/Xa elicits protective signaling responses in endothelial cells directly via PAR-2 and indirectly via endothelial protein C receptor-dependent recruitment of PAR-1. J. Biol. Chem. 2010, 285, 34803–34812. [Google Scholar] [CrossRef] [Green Version]
- Benelhaj, N.E.; Maraveyas, A.; Featherby, S.; Collier, M.E.W.; Johnson, M.J.; Ettelaie, C. Alteration in endothelial permeability occurs in response to the activation of PAR2 by factor Xa but not directly by the TF-factor VIIa complex. Thromb. Res. 2019, 175, 13–20. [Google Scholar] [CrossRef]
- Mirza, H.; Yatsula, V.; Bahou, W.F. The proteinase activated receptor-2 (PAR-2) mediates mitogenic responses in human vascular endothelial cells. J. Clin. Investig. 1996, 97, 1705–1714. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Sennlaub, F.; Beauchamp, M.H.; Fan, L.; Joyal, J.S.; Checchin, D.; Nim, S.; Lachapelle, P.; Sirinyan, M.; Hou, X.; et al. Proangiogenic effects of protease-activated receptor 2 are tumor necrosis factor-alpha and consecutively Tie2 dependent. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Jindal, R.; King, K.R.; Tilles, A.W.; Yarmush, M.L. The Inflammatory Response to Double Stranded DNA in Endothelial Cells Is Mediated by NF kappa B and TNF alpha. PLoS ONE 2011, 6, e19910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, R.; Zeineddine, H.A.; Koskimaki, J.; Fam, M.D.; Cao, Y.; Shi, C.; Moore, T.; Lightle, R.; Stadnik, A.; Chaudagar, K.; et al. Plasma Biomarkers of Inflammation and Angiogenesis Predict Cerebral Cavernous Malformation Symptomatic Hemorrhage or Lesional Growth. Circ. Res. 2018, 122, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Lyne, S.B.; Girard, R.; Koskimaki, J.; Zeineddine, H.A.; Zhang, D.D.; Cao, Y.; Li, Y.; Stadnik, A.; Moore, T.; Lightle, R.; et al. Biomarkers of cavernous angioma with symptomatic hemorrhage. JCI Insight 2019, 4, e128577. [Google Scholar] [CrossRef] [PubMed]
- Girard, R.; Zeineddine, H.A.; Fam, M.D.; Mayampurath, A.; Cao, Y.; Shi, C.; Shenkar, R.; Polster, S.P.; Jesselson, M.; Duggan, R.; et al. Plasma Biomarkers of Inflammation Reflect Seizures and Hemorrhagic Activity of Cerebral Cavernous Malformations. Transl. Stroke Res. 2018, 9, 34–43. [Google Scholar] [CrossRef]
- Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef]
- Sugama, Y.; Tiruppathi, C.; Offakidevi, K.; Andersen, T.T.; Fenton, J.W., 2nd; Malik, A.B. Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: A mechanism for stabilizing neutrophil adhesion. J. Cell Biol. 1992, 119, 935–944. [Google Scholar] [CrossRef]
- Kaplanski, G.; Marin, V.; Fabrigoule, M.; Boulay, V.; Benoliel, A.M.; Bongrand, P.; Kaplanski, S.; Farnarier, C. Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood 1998, 92, 1259–1267. [Google Scholar] [CrossRef]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.T.; Sullivan, K.R.; Hong, C.C.; Goddard, L.M.; Mahadevan, A.; Ren, A.; Pardo, H.; Peiper, A.; Griffin, E.; Tanes, C.; et al. Distinct cellular roles for PDCD10 define a gut-brain axis in cerebral cavernous malformation. Sci. Transl. Med. 2019, 11, eaaw3521. [Google Scholar] [CrossRef]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Malinverno, M.; Globisch, M.A.; Maderna, C.; Corada, M.; Orsenigo, F.; Conze, L.L.; Rorsman, C.; Sundell, V.; Arce, M.; et al. Propranolol Reduces the Development of Lesions and Rescues Barrier Function in Cerebral Cavernous Malformations A Preclinical Study. Stroke 2021, 52, 1418–1427. [Google Scholar] [CrossRef]
- Guzeloglu-Kayisli, O.; Amankulor, N.M.; Voorhees, J.; Luleci, G.; Lifton, R.P.; Gunel, M. KRIT1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes and pyramidal cells of the adult human cerebral cortex. Neurosurgery 2004, 54, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Seker, A.; Pricola, K.L.; Guclu, B.; Ozturk, A.K.; Louvi, A.; Gunel, M. CCM2 expression parallels that of CCM1. Stroke 2006, 37, 518–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanriover, G.; Boylan, A.J.; DiLuna, M.L.; Pricola, K.L.; Louvi, A.; Gunel, M. PDCD10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery 2008, 62, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Chen, L.L.; Two, A.M.; Zhang, H.F.; Min, W.; Gunel, M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc. Natl. Acad. Sci. USA 2011, 108, 3737–3742. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, H.; He, Y.; Jiang, Q.; Tanaka, Y.; Park, I.H.; Pober, J.S.; Min, W.; Zhou, H.J. Mural Cell-Specific Deletion of Cerebral Cavernous Malformation 3 in the Brain Induces Cerebral Cavernous Malformations. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2171–2186. [Google Scholar] [CrossRef]
- Bouchard, B.A.; Shatos, M.A.; Tracy, P.B. Human brain pericytes differentially regulate expression of procoagulant enzyme complexes comprising the extrinsic pathway of blood coagulation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1–9. [Google Scholar] [CrossRef]
- Lopez-Ramirez, M.A.; Lai, C.C.; Soliman, S.I.; Hale, P.; Pham, A.; Estrada, E.J.; McCurdy, S.; Girard, R.; Verma, R.; Moore, T.; et al. Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. J. Clin. Investig. 2021, 131, e139570. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.T.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, O.; Hirano, K.; Nishimura, J.; Kubo, C.; Kanaide, H. Mechanism of endothelium-dependent relaxation induced by thrombin in the pig coronary artery. Eur. J. Pharmacol. 1998, 351, 67–77. [Google Scholar] [CrossRef]
- Motley, E.D.; Eguchi, K.; Patterson, M.M.; Palmer, P.D.; Suzuki, H.; Eguchi, S. Mechanism of endothelial nitric oxide synthase phosphorylation and activation by thrombin. Hypertension 2007, 49, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, C.; Virtuoso, A.; Maggio, N.; Papa, M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Timis, T.L.; Florian, I.A.; Susman, S.; Florian, I.S. Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations-A Short Overview. Int. J. Mol. Sci. 2021, 22, 6141. [Google Scholar] [CrossRef]
- Flemming, K.D.; Link, M.J.; Christianson, T.J.; Brown, R.D., Jr. Use of antithrombotic agents in patients with intracerebral cavernous malformations. J. Neurosurg. 2013, 118, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Gruschwitz, J.; Dinh, B.B.K.; Wanke, I.; Kockro, R.A.; Eisenring, C.V.; Gasciauskaite, G. Antithrombotic therapy of Cerebral cavernous malformations. Interdiscip. Neurosur. 2020, 22, 100851. [Google Scholar] [CrossRef]
- Zuurbier, S.M.; Hickman, C.R.; Tolias, C.S.; Rinkel, L.A.; Leyrer, R.; Flemming, K.D.; Bervini, D.; Lanzino, G.; Wityk, R.J.; Schneble, H.-M.; et al. Long-term antithrombotic therapy and risk of intracranial haemorrhage from cerebral cavernous malformations: A population-based cohort study, systematic review, and meta-analysis. Lancet Neurol. 2019, 18, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Paz, S.; Salem, M.M.; Maragkos, G.A.; Ascanio, L.C.; Enriquez-Marulanda, A.; Lee, M.; Kicielinski, K.P.; Moore, J.M.; Thomas, A.J.; Ogilvy, C.S. Role of aspirin and statin therapy in patients with cerebral cavernous malformations. J. Clin. Neurosci. 2020, 78, 246–251. [Google Scholar] [CrossRef]
- Anfossi, G.; Trovati, M.; Mularoni, E.; Massucco, P.; Calcamuggi, G.; Emanuelli, G. Influence of Propranolol on Platelet-Aggregation and Thromboxane-B2 Production from Platelet-Rich Plasma and Whole-Blood. Prostaglandins Leukot. Essent. Fat. Acids 1989, 36, 1–7. [Google Scholar] [CrossRef]
- Weksler, B.B.; Gillick, M.; Pink, J. Effect of propranolol on platelet function. Blood 1977, 49, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Lanfranconi, S.; Scola, E.; Bertani, G.A.; Zarino, B.; Pallini, R.; d’Alessandris, G.; Mazzon, E.; Marino, S.; Carriero, M.R.; Scelzo, E.; et al. Propranolol for familial cerebral cavernous malformation (Treat_CCM): Study protocol for a randomized controlled pilot trial. Trials 2020, 21, 401. [Google Scholar] [CrossRef] [PubMed]
- Detter, M.R.; Shenkar, R.; Benavides, C.R.; Neilson, C.A.; Moore, T.; Lightle, R.; Hobson, N.; Shen, L.; Cao, Y.; Girard, R.; et al. Novel Murine Models of Cerebral Cavernous Malformations. Angiogenesis 2020, 23, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Maderna, C.; Pisati, F.; Tripodo, C.; Dejana, E.; Malinverno, M. A murine model of cerebral cavernous malformations with acute hemorrhage. Iscience 2022, 25, 103943. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.; Arnould, M.; De Luca, C.; Otten, C.; Abdelilah-Seyfried, S.; Heredia, A.; Leutenegger, A.L.; Schwaninger, M.; Tournier-Lasserve, E.; Boulday, G. Novel Chronic Mouse Model of Cerebral Cavernous Malformations. Stroke 2020, 51, 1272–1278. [Google Scholar] [CrossRef]
- Zhou, H.J.; Qin, L.F.; Jiang, Q.; Murray, K.N.; Zhang, H.F.; Li, B.; Lin, Q.; Graham, M.; Liu, X.R.; Grutzendler, J.; et al. Caveolae-mediated Tie2 signaling contributes to CCM pathogenesis in a brain endothelial cell-specific Pdcd10-deficient mouse model. Nat. Commun. 2021, 12, 504. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| CCM Article | Murine Blood Glycoproteins | Shown in Humans? |
|---|---|---|
| Lopez-Ramirez et al., 2019 [12] | APC | N |
| Yau et al., 2022 [16] | Fibrinogen/fibrin | N |
| Globisch et al., 2022 [17] | vWF | Y |
| Fibrinogen/fibrin | Y | |
| Fibronectin | N |
| CCM Article | Murine Blood Cells | Shown in Humans |
|---|---|---|
| Shi et al., 2009 [49] | N/A | Macrophages T-cells B-cells |
| Shi et al., 2016 [50] | B-cells | N/A |
| Yau et al., 2022 [16] | Macrophages | N |
| Neutrophils T-cells B-cells Platelets | Y N N N | |
| Globisch et al., 2022 [17] | Activated platelets | N |
| Polyhedrocytes | Y |
| CCM Article | In Situ Brain Cytokines/Chemokines | Circulating Cytokines/Chemokines | Associated Symptoms |
|---|---|---|---|
| Noshiro et al., 2012 [37] (Clinical; mRNA) | ↑ IL-6 | N/A | Increased number of hemorrhage events Increased occurrence of bleeding in brain stem |
| Girard et al., 2018 [98] (Clinical) | N/A | ↑ MMP2 ↑ ICAM-1 ↓ MMP9 | Increased seizures |
| ↓ VEGF ↓ Endoglin | Recent bleeding | ||
| IL-2 Interferon gamma TNFα IL-1β | Increased incidence of events (bleeds/lesional growth) | ||
| ↓ IL-10 ↓ CCL2/MCP1 ↓ ROBO4 | Iron deposition from bleeding lesions | ||
| Girard et al., 2018 [96] (Clinical) | N/A | ↓ CD14 ↓ IL-6 ↓ VEGF ↑ IL-1β ↑ sROBO4 | Symptomatic hemorrhagic expansion |
| Lyne et al., 2019 [97] (Clinical) | N/A | sCD14 VEGF IL-10 CRP sROBO4 | Bleeding |
| Yau et al., 2022 [16] (Preclinical) | IL-1β TNF CXCL1/KC/GRO CXCL2/MIP-2 CCL2/MCP-1 CXCL10/IP-10 IL-6 CCL3/MIP-1α | N/A | Lesion severity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Globisch, M.A.; Onyeogaziri, F.C.; Smith, R.O.; Arce, M.; Magnusson, P.U. Dysregulated Hemostasis and Immunothrombosis in Cerebral Cavernous Malformations. Int. J. Mol. Sci. 2022, 23, 12575. https://doi.org/10.3390/ijms232012575
Globisch MA, Onyeogaziri FC, Smith RO, Arce M, Magnusson PU. Dysregulated Hemostasis and Immunothrombosis in Cerebral Cavernous Malformations. International Journal of Molecular Sciences. 2022; 23(20):12575. https://doi.org/10.3390/ijms232012575
Chicago/Turabian StyleGlobisch, Maria Ascencion, Favour Chinyere Onyeogaziri, Ross Osborne Smith, Maximiliano Arce, and Peetra Ulrica Magnusson. 2022. "Dysregulated Hemostasis and Immunothrombosis in Cerebral Cavernous Malformations" International Journal of Molecular Sciences 23, no. 20: 12575. https://doi.org/10.3390/ijms232012575
APA StyleGlobisch, M. A., Onyeogaziri, F. C., Smith, R. O., Arce, M., & Magnusson, P. U. (2022). Dysregulated Hemostasis and Immunothrombosis in Cerebral Cavernous Malformations. International Journal of Molecular Sciences, 23(20), 12575. https://doi.org/10.3390/ijms232012575