Abstract
A new series of sulfonamides, 8a-b, 10, 12, and 14a-b, were synthesized by N-sulfonation reaction with sulfonyl chlorides 6a-b. Five new series of chalcone-sulfonamide hybrids (16-20)a-f were prepared via Claisen–Schmidt condensation of the newly obtained sulfonamides with aromatic aldehydes 15a-f in basic medium. Chalcones substituted with chlorine at position 4 of each series were used as precursors for the generation of their five-membered heterocyclic pyrazoline (22-23)a-d, (24-25)a-b and carbothioamide 27a-f derivatives. The synthesized compounds were evaluated for their anticancer and antituberculosis activities. To determine their anticancer activity, compounds were screened against sixty human cancer cell lines at a single dose (10 μM). Compounds 17a-c were highly active against LOX IMVI (melanoma), with IC50 values of 0.34, 0.73 and 0.54 μM, respectively. Chalcone 18e showed remarkable results against the entire panel of leukemia cell lines with IC50 values between 0.99–2.52 μM. Moreover, compounds 20e and 20f displayed growth inhibition of Mycobacterium tuberculosis H37Rv at concentrations below 10 μM. Although they showed low selectivity in cytotoxicity tests against the Vero cell line, further optimization could advance the potential biological activity of the selected compounds.
1. Introduction
Finding effective treatments for diseases with high mortality rates such as cancer and tuberculosis (TB) remains at the top of the biomedical research agenda worldwide. The number of fatalities associated with cancer have decreased over the last decades as a result of improvements in prevention, diagnosis and treatment [1]. However, the necessity to combine cytotoxic drugs in conventional chemotherapy demonstrates the difficulty in treating this complex and multifactorial disease [2]. TB is the second leading cause of mortality due to infectious agents after COVID [3], despite the availability of affordable and successful treatments [3]. The emergence of multidrug-resistant TB is one of the major threats to the control of the disease, and selecting a new combination of drugs to shorten the treatment period remains as a challenge [4,5,6].
Molecular hybridization, involving the combination of two or more pharmacophoric moieties into a single chemical entity, has become an attractive approach for overcoming limitations in the administration of active chemical entities for treating complex diseases. A successful hybrid compound should access multiple targets and lead to different mechanisms of action [2]. There are multiple known drug-like fragments, and we have selected the sulfonamide functionality in combination with a privileged small nitrogen heterocyclic such as pyrazole, or with its α,β-unsaturated ketone precursors such as chalcones.
On one hand, sulfonamide moiety prevails as the most promising candidate for the design of molecular hybrids [7]. The versatility of the chemical structure, being easily modified; the compatibility with most functional groups; a strong electron withdrawal nature; and the capacity to coordinate metal ions from metalloenzymes forming tetrahedral complexes stabilized by hydrogen bounds are remarkable features [8,9], without mentioning the variety of biological properties that sulfonamides have exhibited including anti-inflammatory [10], anticancer [11,12], antibacterial [13], antiviral [14], antidiabetic [15], and antimalarial [16] activities, among others.
On the other hand, chalcones are compounds that have shown an extensive number of applications in the field of medicinal chemistry due to their wide range of pharmacological properties [17]. A wide variety of synthetic chalcones have been shown in applications such as analgesics [18], antioxidants [19], anti-inflammatories [20], antibacterial [21], antitumor [17], antiviral [22], antihypertensive [23], antidiabetic [24], and antituberculosis [25]. The biological activity of chalcones is attributed to their effect as cell blockers, influenced by the interactions of the α,β-unsaturated carbonyl system with various biomolecules [20,26]. The combination of sulfonamide moiety with chalcones to generate new hybrid compounds with potential therapeutic applications has been described [27,28] (Figure 1), and also used as key precursors for potentially bioactive heterocyclic derivatives [29,30,31,32].
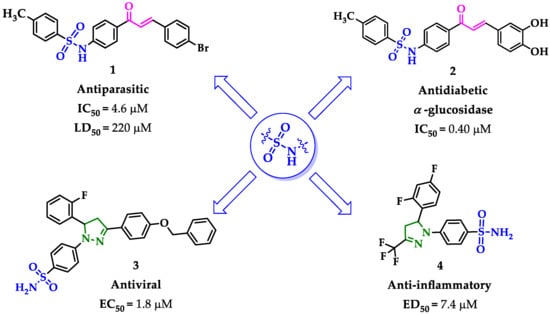
Figure 1.
Sulfonamide moiety as promising candidate for molecular hybridization.
Regarding pyrazoles, their structural and electronic properties allow them to interact with various biological entities involved in the development of several pathologies; moreover, they are responsible for the great variety of biological properties bearing this type of moiety [33]. In addition, it is known that compounds containing sulfur and nitrogen have received significant attention in the field of medicinal chemistry due to the ability of these atoms to generate donor ligands and coordinate complexes with metal cations of zinc, iron, nickel, and copper, which play important roles in different biological processes [34].
Synthetic pyrazoline derivatives have exhibited antitumor [35,36,37], antibacterial [38], immunosuppressive [39], anti-inflammatory [40], anti-diabetic [41], antidepressant [42], antimalarial [43], antiviral [44], and antihypertensive [45] activities. Particularly, the 4,5-dihydro-1H-pyrazole-1-carbothioamides have also shown a wide variety of pharmacological properties such as anticancer [46,47,48,49,50], anticonvulsant [51,52], antituberculosis [53], antimicrobial [54], and anti-inflammatory [55] agents (Figure 1).
In this paper we extend our ongoing research on chalcone derivatives by the synthesis of new nitrogen-containing five-membered heterocyclic hybrids bearing a sulfonamide moiety and performing the study of their anticancer and antituberculosis properties.
2. Results and Discussion
2.1. Chemistry
All hybrids were synthesized from the corresponding benzenesulfonyl chlorides 6a and 6b. In our previous work we described the procedure for the preparation of 6a [56]. Compound 6b was obtained by the chlorosulfonation reaction of acetophenone 5b (Scheme 1), using chlorosulfonic acid and thionyl chloride as chlorinating agents in a 6:2 ratio regarding 5b. The structure of 6b was confirmed by spectroscopic techniques including FTIR, 1H NMR, 13C NMR and mass spectrometry. The IR spectrum shows absorption bands corresponding to =C-H, C=O, C=C and C-O-C bonds at 3110, 1657, 1590, and 1292 cm−1, respectively. An absorption band of the S=O bond was clearly observable at 1167 cm−1. The aliphatic region of the 1H NMR spectrum shows three singlets at 2.56, 4.11, and 4.05 ppm corresponding to COCH3, 4A-OCH3 and, 2A-OCH3 protons, respectively. In the aromatic region only two signals at 8.40 and 6.56 ppm were observed; the downfield signal corresponds to H6A due the inductive effect generated by the sulfonyl group on the system. In the 13C NMR spectrum, ten signals were observed as expected for compound 6b. The molecular ion peak in the mass spectrum revealed the characteristic isotopic profile for one chlorine atom m/z 278/280 (M+/(M+2)+).
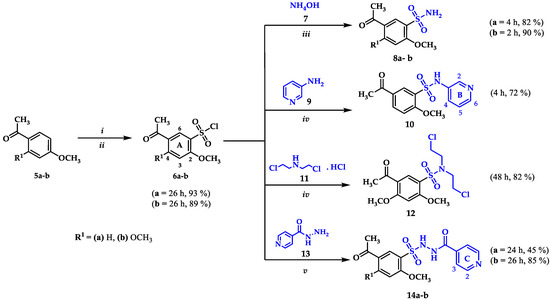
Scheme 1.
General procedure for the synthesis of sulfonamide precursors 8a-b, 10, 12, and 14a-b from benzenesulfonyl chlorides 6a-b: (i) HSO3Cl, SOCl2, 0 °C, 0.5 h, (ii) r.t, 26 h, (iii) EtOH, r.t, (iv) EtOH, TEA, r.t, (v) Water, Na2CO3, r.t.
Sulfonamides 8a and 8b were synthetized through N-sulfonation reactions of 6a-b with ammonia at room temperature, using ethanol to promote product precipitation (Scheme 1). The IR spectra of both sulfonamides presents the two characteristic stretching vibration bands for a primary N-H bond in the region between 3342–3201 cm−1. The 1H NMR spectra confirms the structure of the compounds by the observation of broad singlets at 7.24 and 7.06 ppm corresponding to NH2 protons for 8a and 8b, respectively. The obtention of 8b was also confirmed by single-crystal X-ray data (Figure 2a).
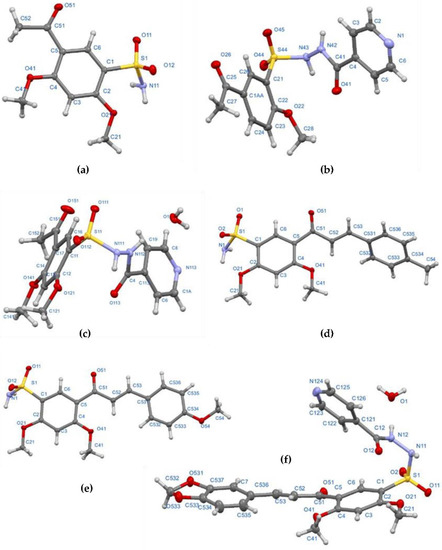
Figure 2.
Molecular structure of compounds 8b (a), 14a (b), 14b (c), 17c (d), 17d (e) and 20f (f) determined from single-crystal X-ray diffraction, showing the atom-labelling scheme. Single crystal diffraction experimental details can be found in Table S1 (see Supplementary Materials).
Compounds 10 and 12 were prepared using amines 9 and 11 and triethylamine to neutralize the reaction in ethanol as a solvent (Scheme 1), conditions reported in our previous work [55]. Compounds 10 and 12 were obtained in 72% and 82% yields. The substitution reaction for the synthesis of the secondary sulfonamide 10 was verified by observation in the 1H NMR spectrum of a singlet corresponding to the sulfonamidic proton at 10.45 ppm and the expected signals for the protons in the pyridinic ring H2B, H6B, and H4B at 8.30 (J = 1.7 Hz), 8.21 (d, J = 4.5 Hz), and 7.51 ppm (d, J = 8.3 Hz), in addition to the overlapped signal of proton H5B with H3A at 7.24 ppm. The sulfonamidic protons in compound 12 were absent and its structure was confirmed by the observed signals of protons N-CH2 and CH2 at 3.53 and 3.68 ppm.
In order to obtain the sulfonamides derived from isoniazid, the reaction between 6a and 13 was carried out under the same reaction conditions used for the synthesis of 10 and 12; however, a complex mixture of products was observed. A number of conditions were tested, including the use of KOH, NaOH and base-free; in all cases, either a complex mixture or traces of the desired product were obtained. To overcome this issue, an alternative route was sought, including a trial using water as solvent, Na2CO3 1 N to keep a basic pH (8–10) level during the reaction and final work-up with HCl 1 N to reach pH 2–3 once the reaction was finished, but no product was isolated [57]. However, an adjustment of the final pH at 7–8 allowed compounds 14a and 14b in moderate yields (Scheme 1). The 1H NMR spectra showed all the expected signals for C-NH, S-NH (9.69–11.13 ppm), and the pyridinic protons of the target compounds. Both structures were elucidated by single-crystal X-ray diffraction (Figure 2b,c).
Sulfonamides 8a-b, 10, 12, and 14a-b reacted with aromatic aldehydes (15a-f) by Claisen–Schmidt condensation in the presence of ethanol and aqueous NaOH (Scheme 2). This procedure resulted in good yields of the corresponding chalcone–sulfonamide hybrids (16-20)a-f (41–88%). Compounds 16a-f and 17a-f were initially obtained in salt form as evidenced in the 1H NMR spectrum of 16d (Figure 3a and Figure 4 red). This behavior is observed due to the acidic character of the sulfonamidic protons by the strong electron withdrawing effect of the sulfonyl group and the resonance stabilization of the conjugate base formed in the basic reaction medium. Further neutralization of each compound with HCl 1% (v/v) allowed the isolation of 16d in its neutral form (Figure 3b and Figure 4 black), and it was verified by the observation of a broad signal at 10.45 ppm corresponding to the sulfonamidic protons. Chalcones 18a-f were directly obtained in their neutral form due to the absence of labile protons in their structures. The 1H NMR spectra for the five new series of chalcones showed the expected signals for Hα and Hβ, confirming the condensation reaction. The coupling constants in the range 15.4–15.8 Hz determined the E configuration for the C=C double bond formed. In addition, the structures of compounds 17c-d and 20f were elucidated by single-crystal X-ray diffraction (Figure 2d–f).
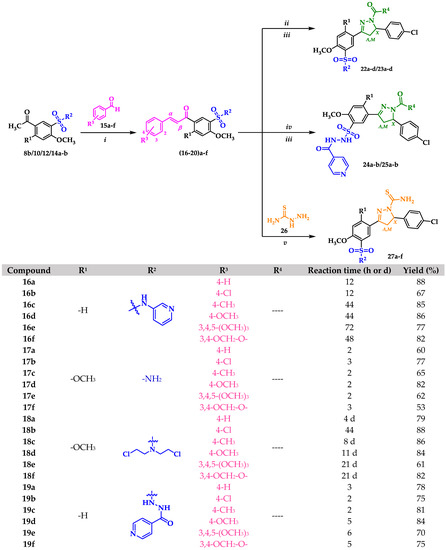
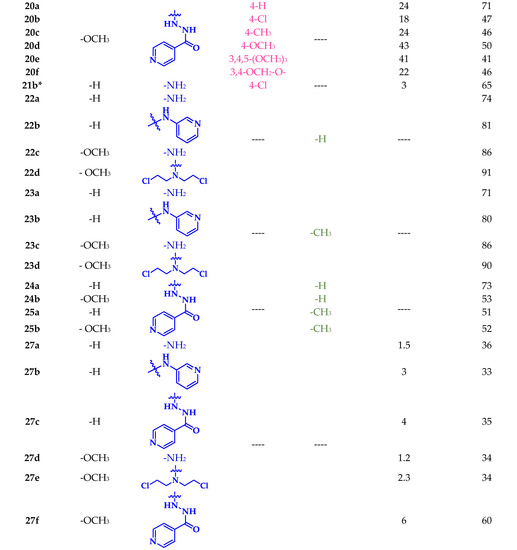
Scheme 2.
General procedure for the synthesis of new chalcone–sulfonamide (16-20)a-f, pyrazoline–sulfonamide (22-23)a-d and (24-25)a-b, and carbothioamide–sulfonamide 27a-f: (i) EtOH, NaOH 50% (w/v), r.t; (ii) NH2-NH2∙H2O, EtOH, reflux; (iii) Ac2O or HCOOH, r.t; (iv) NH2-NH2∙H2O, EtOH, r.t; (v) EtOH, NaOH, 60 °C. * Reported in our previous work [56].
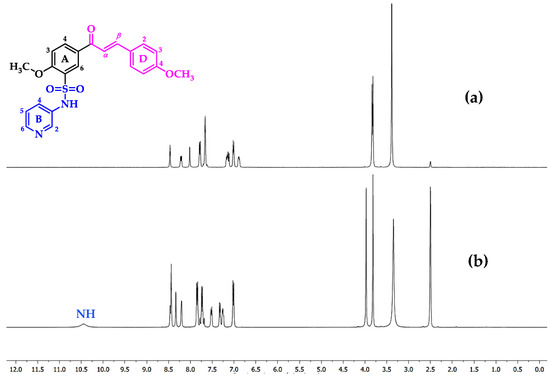
Figure 3.
1H NMR spectrum (400 MHz, DMSO-d6) of compound 16d: (a) salt form (basic media) and (b) neutral.
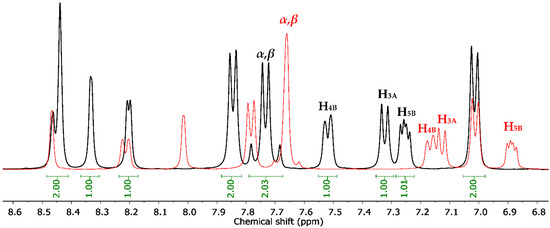
Figure 4.
Chemical shift between 1H NMR signals of compound 16d in salt form (red) and neutral (black).
For the synthesis of the nitrogen-containing five-membered heterocyclic hybrids the p-chloro-substituted chalcones of each series were selected. Compound 21b was initially subjected to a cyclocondensation reaction with hydrazine monohydrate in ethanol under reflux; however, no significant progress in the reaction was observed. Thus, the same procedure was carried out, and after 40 min of reaction, the mixture was brought to room temperature, 1 mL of acetic anhydride was slowly added, and the progress of the reaction was checked. After 3 h a new product had formed, and the total consumption of the precursor was observed. The acid dependence of the reaction suggests that the carbonyl group must first be activated for the cyclization to proceed through a hydrazone-type intermediate [58]. In the 1H NMR spectrum, the absence of signals corresponding to α,β-unsaturation and the presence of one stereogenic and two diastereotopic protons revealed the obtention of the AMX spin system characteristic of pyrazoline derivatives. The spectrum of compound 23a showed three doublet of doublets at 5.53, 3.85, and 3.09 ppm corresponding to HX, HM, and HA, with coupling constants of 2JMA = 18.0 Hz, 3JXM = 11.6 Hz, and 3JXA = 4.3 Hz. Also, a singlet integrating for three protons at 2.28 ppm was assigned to the acetyl group and confirmed the functionalization of the product. The methodology was extended to the preparation of N-formylated (22a-d) and N-acetylated (23a-d) pyrazolines in good to excellent yields (Scheme 2).
Under the same reaction conditions, chalcones 19b and 20b decomposed according to the 1H NMR spectrum of the major product. In a second approach, the reaction was carried out at room temperature, and after 3 h the precursor was completely consumed, giving rise to pyrazolines 24a-b and 25a-b (Scheme 2).
Sulfonamide–carbothioamide hybrids 27a-f were synthesized by condensation between chalcones (16-21)b and thiosemicarbazide in basic medium (Scheme 2). The reaction did not proceed at room temperature, but an increase to 60 °C was enough to observe the consumption of the precursor. The medium was neutralized with HCl 1% (v/v), the solid form was filtered, and the filtrate was purified by column chromatography on silica gel. This methodology afforded the expected compounds in moderate yields (33–60%). The structure of the new hybrids was consistent with the spectral data. Both 1H and 13C NMR revealed formation of the AMX system. Interestingly, the new carbothioamide protons denoted as C-NH2 were diastereotopic.
2.2. Anticancer Activity
The new molecular hybrids bearing a sulfonamide moiety were submitted to the Developmental Therapeutics Program (DTP) at the National Cancer Institute (NCI) for a preliminary analysis of their structures based on the COMPARE algorithm. All synthesized compounds were selected for single-dose trial at a concentration of 10 μM against 60 human cancer cell lines corresponding to nine human cancer panels: leukemia, non-small-cell lung, melanoma, colon, central nervous system (CNS), ovary, renal, breast, and prostate cancer. The anticancer activity of the compounds was measured as a reduction in growth percentage for each of the cancer cell lines. Compounds 17a-c and 18e consistently reduced the growth of most cancer cells at 10 µM concentration (Figure 5).
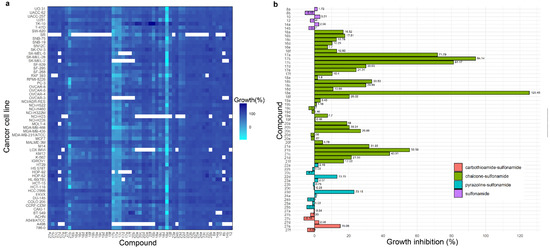
Figure 5.
Anticancer activity of the synthesized hybrids at 10 µM. (a) Heatmap showing the growth of the cancer cells in presence of the compounds. Reduced growth of the cells is represented in cyan and dark blue represents the high growth of the cells. Blank cells indicate lack of data. (b) Mean growth inhibition (GI) percentage for each compound. A GI % value over 100% indicates there is no net growth of tumor cells over the course of the experiment; instead, compound causes the death of the respective cancer cell.
Initial studies carried out at a single dose revealed that in general, sulfonamides (8b, 10, 12, 14a, and 14b), pyrazolines (22a-d, 23a-d, 24a-b and 25a-b), and carbothioamides (27a-f) displayed low antiproliferative activity against the cancer cell lines evaluated, regardless of their substituent in the sulfonamide group and the N-1 of the five-membered rings. However, the chalcone–sulfonamide hybrids showed an increase in antitumoral activity, indicating the α,β-unsaturated carbonyl system could be responsible for imparting antiproliferative properties to the evaluated compounds.
A comparative analysis of the results obtained for the new series of chalcones and compounds 21a-f (reported in our previous work) suggest that the antitumoral properties of these hybrids are influenced by the chlorine substituent in para position of the D ring. It is well known that the inclusion of halogenated atoms can modify the interaction strength and hydrophobicity of the molecules, increasing the specificity and selectivity in the processes of recognition, and binding with biomolecules such as proteins and DNA [59]. We found that the antitumoral activity is highly affected by steric factors in the sulfonamide substituent, being the chalcones containing the -NH2 group the most active and the isoniazid derivatives the least active [60].
Chalcones 17a-c (4-H, 4-Cl, and 4-CH3, respectively) exhibited significant activity against most cancer cell lines evaluated. Compound 17a displayed inhibitory activity against leukemia (GI = 70.81–94.75%), colon cancer (GI = 72.48–96.15%), renal cancer (GI = 66.21–100.00%), and breast cancer (GI = 73.49–100.00%) panels. This compound showed remarkable results against RXF 393 (renal cancer) and MDA-MB-468 (breast cancer) cell lines, being able to inhibit net growth and cause a cytotoxic effect with lethality values of 29.73% and 43.37%, respectively. Chalcone 17b was the most active compound of the series and similar results were observed, leukemia (GI = 92.85–100.00%), colon cancer (GI = 92.23–100.00%), renal cancer (GI = 78.47–100.00%), and breast cancer (GI = 73.94–100.00%) panels were the most sensitive to this compound. Hybrid 17b also displayed important cytotoxic effects against SR (leukemia) with a lethality of 19.50%; NCI-H226 (non-small cell lung cancer) with 25.96%; COLO 205, HCT-15, SW-620 (colon cancer) with values of 30.00%, 41.55%, and 39.92%, respectively; LOX IMVI (melanoma) with 50.40%; and RXF 393 (renal cancer) and MDA-MB-468 (breast cancer) with a 40.37% and 57.82% of lethality, respectively. Chalcone 17c exhibited similar behavior to its analogs; the most sensitive panels were leukemia (GI = 78.87–94.34%), colon cancer (GI = 83.01–100.00%), and breast cancer (GI = 78.66–100.00%); and the cancer cell lines NCI-H460 (non-small cell lung cancer), RXF 393 (renal cancer), and MDA-MB-468 (breast cancer) with lethality values of 3.51%, 28.21%, and 36.18%, correspondingly.
Among the synthesized hybrids, compound 18e was the most active against the totality of cancer cell lines as evidenced by a value of 125.48% in the mean GI%, denoting excellent inhibitory activity and significant cytotoxic effect in most assays. The GI% found for the evaluated cell lines rates between 76.62% and 100.00% except for HS 578T (21.70%). In several cases, compound 18e displayed remarkable lethality and the best results were found for COLO 205, HCC-2998, and HCT-116 cell lines in the colon cancer panel with values of 89.76%, 91.08%, and 87.74%, respectively, but also for LOX IMVI and SK-MEL-2 from the melanoma panel with 84.53% and 94.49%, and for UO-31 (renal cancer) with a 93.86% of lethality.
According to the obtained data for single-dose trial, chalcone–sulfonamide hybrids 17a-c and 18e fulfilled the pre-determined threshold inhibition criteria of NCI and were selected for five-dose screening against 60 cancer cell lines at five different concentrations (100, 10, 1.0, 0.1, and 0.01 μM) to determine IC50 (half inhibitory concentration) and LC50 (half lethal concentration) values. Table 1 summarizes the recorded results for the mentioned compounds.

Table 1.
In vitro cytotoxic activity recorded for hybrids 17a-c and 18e against the panel of 60 cancer cell lines in five-dose screen, expressed as IC50 and LC50 in micromolar (μM) a.
Results obtained from the five-dose screen revealed that compounds 17a, 17b and 17c are potent antiproliferative agents, displaying IC50 values against most cell lines between 0.34−17.6 μM, 0.73−18.6 μM, and 0.54−11.5 μM, respectively. Despite the wide IC50 range showed by chalcone 18e (0.99 - > 100 μM), the compound demonstrated potent activity against several cell lines.
Hybrid 17a showed the best results against K-562 (leukemia) with IC50 = 1.50 μM, HCT-116 (colon cancer) with IC50 = 1.49 μM, LOX IMVI (melanoma) with IC50 = 0.34 μM, and MCF7 and MDA-MB-468 (breast cancer) with IC50 values of 0.97 and 1.20 μM, respectively. Compound 17b was highly active against CCRF-CEM (leukemia) with IC50 = 1.52 μM, HOP-92 (non-small cell lung cancer) with IC50 = 1.46 μM, HCT-116 and HCT-15 (colon cancer) with IC50 values of 0.75 μM and 1.46 μM, and LOX IMVI (melanoma) with IC50 = 0.73 μM. This chalcone showed lower IC50 values in comparison with its analogs, exhibiting the best-marked cytotoxic effects against several cancer cell lines including BT-549 (breast cancer) with 5.97 μM, and COLO 205 and HCT-116 (colon cancer) with LC50 values of 5.88 μM and 5.13 μM, respectively. The highest IC50 values were obtained for chalcone 17c against SNB-75 (CNS Cancer) at 1.84 μM, and LOX IMVI (melanoma) and MDA-MB-468 (breast cancer) with values of 0.54 μM and 1.96 μM. The entire leukemia panel was highly sensitive to compound 18e, displaying IC50 values between 0.99–2.52 μM, the SR cancer cell line being most susceptible. Significant IC50 values were found for NCI-H460 and NCI-H522 (non-small cell lung cancer) with 1.95 μM and 1.66 μM, correspondingly. In addition, chalcone 18e showed important cytotoxic activity against several cell lines including LOX IMVI (melanoma) and UO-31 (renal cancer) with respective LC50 values of 5.79 μM and 5.51 μM.
The results shown above suggest that compounds with structures analogous to 17b constitute important templates for the future development of potential antitumor agents.
2.3. Antituberculosis Activity, Cytotoxicity and Synergism
All new sulfonamide hybrids were subjected to in vitro growth inhibition screening against Mycobacterium bovis BCG (ATCC 35734) and Mycobacterium tuberculosis H37Rv (ATCC 27294) strains (ATCC-USA, Virginia, USA). Antituberculosis activity was carried out using the agar dilution spot culture growth inhibition assay [61]. Initially, the compounds were evaluated at a concentration of 10 mg/L, and those showing inhibition were further evaluated at lower concentrations (5, 1, 0.5, 0.1 and 0.01 mg/L). Isoniazid was used as positive control. The results for each compound were expressed as minimum inhibitory concentration values (MICs), as shown in Table 2. The synthesized sulfonamides, pyrazolines and carbothioamides were inactive and only chalcones showed inhibitory effects with MIC values between 9.0–29 μM, showing the importance of an α,β-unsaturated carbonyl system as a pharmacophore unit. Isoniazid derivatives exhibited the most potent antitubercular activity. The substituents 4-H, 4-OCH3, 3,4,5-(OCH3)3, and 3,4-OCH2O- enhanced the inhibitory activity. The most active chalcones were 20a, 20e, and 20f obtained from chloride 6b (R1 = OCH3), displaying MIC values of 11 μM, 9.0 μM, and 9.8 μM against Mycobacterium tuberculosis H37Rv and 20a and 20e against Mycobacterium bovis BCG with MIC values of 11 μM and 9.0 μM, respectively.
Table 2.
Anti-TB and cytotoxic activity for compounds 17a, 17f, 19a, 19d, 20a, 20d-f.
The growth of M. tuberculosis H37Rv in liquid medium in presence of the compounds 20e and 20f at 1×MIC and 10×MIC concentrations was followed by optical density measurements (Figure 6). Both compounds showed similar potency in liquid medium with moderate growth observed after 21 days of incubation at 1×MIC, while at 10×MIC a modest growth was observed for 20e, while for 20f no significant growth was observed in comparison with day 0. The cultivation of solid media without compounds was undertaken for each sample for evaluating cidal or static effects of the compounds. Both 20e and 20f were found to be bacteriostatic even at 10×MIC concentrations, because growth resumed in all experiments.
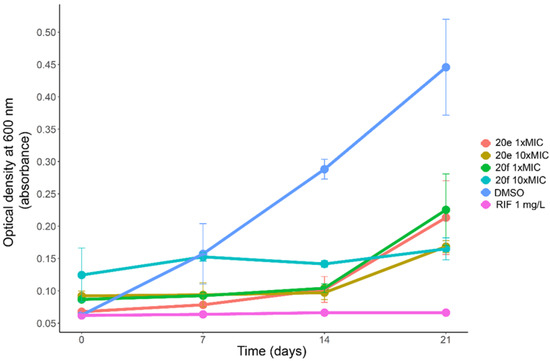
Figure 6.
Growth curves of M. tuberculosis H37Rv in liquid media containing 1×MIC and 10×MIC concentrations of the compounds 20e and 20f.
Additionally, cytotoxicity studies were conducted on the Vero cell line for the mentioned hybrids as shown in Table 2. The results revealed low IC50 values (<10 μM) and therefore high cytotoxicity levels against Vero cells. Selectivity index (SI) was calculated for the screened compounds and none of the chalcones showed selectivity against Mycobacterium tuberculosis H37Rv strains compared to the Vero cell (SI < 1).
Additionally, the compounds with most potent activity were tested against a panel of M. tuberculosis strains from six different lineages that were resistant to isoniazid and rifampicin (Table 3), including an orphan strain with no match in spolDB4 [62]: A Beijing strain known to be an emerging pathogen in several areas [63,64]; a LAM 9 strain, which is the predominant genotypic family from Latin American and Mediterranean (LAM) lineage [65]; a Haarlem strain, which is ubiquitous worldwide [66]; the ATCC 35838 RR, RR (rifampicin-resistant strain); ATCC 35822 RI (isoniazid- resistant strain); and ATCC 27294 (Table 3).
Table 3.
Antituberculosis activity of resistant strains of Mycobacterium tuberculosis, expressed in Minimum Inhibitory Concentration (MIC) in micromolar (µM) units.
All the evaluated compounds were inactive against M. tuberculosis Haarlem and M. tuberculosis ATCC 35838 RR (MIC >10 mg/L). On the other hand, compounds 20e and 20f were highly active against M. tuberculosis LAM 9 (SIT 42), showing MIC values of 8.97 µM and 9.77 µM, respectively. Against the M. tuberculosis Beijing strain, the compounds with lower MIC values were 20a and 20e. The compounds 19a, 20a, 20e, and 20f displayed MIC values <12 µM against the M. tuberculosis ATCC 35838 RR strain.
Moreover, the possibility of synergism between compounds 20a and 20f in combination with antibiotics rifampicin, isoniazid, levofloxacin, and amikacin was evaluated and the results are shown in Table 4. Compound 20f in combination with rifampicin displayed a fractional inhibitory concentration index (FICI) of 0.37 (≤0.5), suggesting synergism. The other combinations of compounds and TB drugs did not show interaction.
Table 4.
MIC values of the compounds before and after combinations with antibiotics and the FICI index in M. tuberculosis H37Rv.
3. Materials and Methods
3.1. General
All chemicals and solvents were purchased from Sigma-Aldrich and Merck unless stated otherwise. Melting points were measured using a Stuart SMP10 melting point device (Bibby Scientific Ltd., Staffordshire, UK) and are uncorrected. ATR-FTIR spectra were recorded on a Shimadzu IRAffinity–1 (Shimadzu Corp., Columbia, USA). The 1H and 13C NMR spectra were run on a BRUKER DPX 400 spectrometer (Bruker, Billerica, USA) operating at 400 and 100 MHz, respectively, using DMSO–d6 and CDCl3 as solvents and TMS as internal standard. Elemental analyses were performed on a Thermo Finnigan Flash EA1112 CHN elemental analyzer (Thermo Fischer Scientific Inc., Madison, USA) and the values are within ±0.4% of the theoretical values. The mass spectra were recorded on a SHIMADZU–GCMS–QP 2010 spectrometer (Shimadzu Corp., Kyoto, Japan) with an electronic impact source operating at 70 eV. Thin layer chromatography (TLC) was performed on 0.2 mm pre-coated aluminum plates of silica gel 60 F254 (Merck, Dramstand, Germany) and spots visualized with ultraviolet irradiation. The single-crystal X-ray data were collected in a Diffractometer Bruker D8 Venture (Bruker Daltonics GmbH & Co. KG, Bremen, Germany) at “Centro de Instrumentación Científico y Técnico”, (CICT) in “Universidad de Jaén” (UJA).
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of Acetophenone 5b
Three hundred and forty-two milligrams of crushed KOH (6.10 mmol) and 9 mL of DMSO were stirred for 30 min at room temperature. To this mixture, a solution of 422 mg of 2’,4’-dihydroxyacetophenone (2.77 mmol) in 6 mL of DMSO was added and stirred for 15 min, then cooled in an ice-water bath. A quantity of 0.40 mL of methyl iodide (6.42 mmol) was slowly added and left under constant stirring at room temperature for 24 h.
The reaction mixture was poured into ice-water and extracted with chloroform; the organic phase was evaporated under reduced pressure and the product purification was carried out by CC, using silica gel as stationary phase and a mixture of CHCl3: petroleum ether (20:1) as mobile phase.
1-(2,4-Dimethoxyphenyl)ethan-1-one (5b)
Beige solid; 79% yield; m.p. 37–38 °C. FTIR (ATR) (cm−1): 3011 (C-H Ar), 1650 (C=O), 1599 (C=C), 1255 (C-O-C (a)), 1018 (C-O-C (s)). 1H NMR (400 MHz, CDCl3) δ ppm 7.82 (d, J = 8.8 Hz, 1H, H6A), 6.51 (dd, J = 8.8, 2.3 Hz, 1H, H1A), 6.45 (d, J = 2.3 Hz, 1H, H3A), 3.88 (s, 3H, 4A-OCH3), 3.84 (s, 3H, 2A-OCH3), 2.56 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ ppm 197.9 (C), 164.7 (C), 161.2 (C), 132.8 (CH), 121.3 (C), 105.2 (CH), 98.4 (CH), 55.64 (CH3), 55.56 (CH3), 31.9 (CH3). MS (EI, m/z (%)): 180 (M+, 57), 165 (100), 122 (44), 107 (35), 77 (45), 43 (38). Anal. calcd. for C10H12O3: C, 66.65; H, 6.71. Found: C, 66.50; H, 6.89.
3.2.2. General Procedure for the Synthesis of Benzenesulfonyl Chloride 6b
A mixture of chlorosulfonic acid (60 mmol) and thionyl chloride (20 mmol) was stirred in an ice bath for 30 min. Subsequently, corresponding acetophenone 1a-b (10 mmol) was added portion-wise and the reaction mixture was stirred at room temperature for 26 h. Next, it was quenched in ice-water and the precipitate obtained was filtered and washed with water. Compound 6b was purified by CC using silica gel as stationary phase and a mixture of CHCl3: EtOAc (10:1) as mobile phase. Product 6a did not require further purification. NMR spectra of all synthesized compounds from here in, can be found in File S1 (see Supplementary Materials).
5-Acetyl-2,4-dimethoxybenzenesulfonyl Chloride (6b)
White solid; 89% yield; m.p. 152–155 °C. FTIR (ATR) (cm−1): 3110 (C-H Ar), 1657 (C=O), 1590 (C=C), 1292 (C-O-C), 1167 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.40 (s, 1H, H6A), 6.56 (s, 1H, H3A), 4.11 (s, 3H, 4A-OCH3), 4.05 (s, 3H, 2A-OCH3), 2.56 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ ppm 195.5 (C), 165.9 (C), 161.8 (C), 133.9 (CH), 124.7 (C), 120.4 (C), 96.2 (CH), 57.2 (CH3), 56.6 (CH3), 31.8 (CH3). MS (EI, m/z (%)): 278/280 (M+/M+2+, 31/12), 263 (100), 149 (35), 135 (49), 43 (54). Anal. calcd. for C10H11ClO5S: C, 43.09; H, 3.98; S, 11.50. Found: C, 43.12; H, 3.88; S, 11.54.
3.2.3. General Procedure for the Synthesis of Sulfonamides 8a-b
A mixture of the appropriate chloride 6a-b (0.31 mmol) and ammonia 3 (1 mL) in ethanol 96% (1 mL) was stirred at room temperature and the progress was monitored by TLC. After completion, the solid formed was filtered and washed with ethanol and water. Compounds 8a-b did not require further purification.
5-Acetyl-2-methoxybenzenesulfonamide (8a)
Pale pink crystals; 82% yield; m.p. 195–198 °C. FTIR (ATR) ν(cm−1): 3329 and 3208 (N-H), 3098 (C-H Ar), 1657 (C=O), 1159 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.29 (d, J = 2.3 Hz, 1H, H6A), 8.19 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 7.32 (d, J = 8.7 Hz, 1H, H3A), 7.24 (s, 2H, NH2), 4.00 (s, 3H, OCH3), 2.56 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 195.7 (C), 159.6 (C), 134.4 (CH), 131.4 (C), 128.8 (C), 127.7 (CH), 112.6 (CH), 56.7 (CH3), 26.4 (CH3). MS (EI, m/z (%)): 229 (M+, 22), 214 (100), 119 (25), 76 (57), 43 (87). Anal. calcd. for C9H11NO4S: C, 47.15; H, 4.84; N, 6.11; S, 13.98. Found: C, 47.09; H, 4.93; N, 6.15; S, 14.01.
5-Acetyl-2,4-dimethoxybenzenesulfonamide (8b)
Colorless crystals; 90% yield; m.p. 226–228 °C. FTIR (ATR) (cm−1): 3342 and 3201 (N-H), 3121 (C-H Ar), 1650 (C=O), 1155 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.09 (s, 1H, H6A), 7.06 (s, 2H, NH2), 6.83 (s, 1H, H3A), 4.02 (s, 6H, 2A-OCH3 and 4A-OCH3), 2.50 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 195.6 (C), 163.5 (C), 160.8 (C), 130.4 (CH), 123.9 (C), 118.5 (C), 97.1 (CH), 56.7 (CH3), 56.6 (CH3), 31.6 (CH3). MS (EI, m/z (%)): 259 (M+, 95), 244 (100), 134 (91), 76 (79), 43 (100). Anal. calcd. for C10H13NO5S: C, 46.32; H, 5.05; N, 5.40; S, 12.37. Found: C, 46.43; H, 5.05; N, 5.42; S, 12.30. Crystals suitable for single-crystal X-ray diffraction were obtained from ethanolic solution, and the crystal data for 8b were deposited at CCDC with reference CCDC2191609.
3.2.4. General Procedure for the Synthesis of Sulfonamide 10
To a mixture of chloride 6a (0.33 mmol) and amine 9 (0.36 mmol) in ethanol 96% (1.5 mL), TEA (0.15 mL) was added and left under stirring at room temperature. Once the reaction was complete, the medium was neutralized with TEA and the solid obtained was filtered and washed with ethanol to afford compound 10. No further purification was required.
5-Acetyl-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (10)
Pink solid; 72% yield; m.p. 217–218 °C. FTIR (ATR) (cm−1): 3073 (C-H Ar), 1682 (C=O), 1595 (C=C), 1267 (C-N), 1152 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.45 (s, 1H, NH), 8.30 (d, J = 1.7 Hz, 1H, H2B), 8.27 (d, J = 2.0 Hz, 1H, H6A), 8.21 (d, J = 4.5 Hz, 1H, H6B), 8.18 (dd, J = 8.8, 2.0 Hz, 1H, H4A), 7.51 (d, J = 8.3 Hz, 1H, H4B), 7.30–7.24 (m, 2H, H3A and H5B), 3.94 (s, 3H, OCH3), 2.52 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 195.9 (C), 159.8 (C), 145.3 (CH), 141.5 (CH), 136.2 (CH), 134.5 (C), 130.2 (CH), 129.2 (C), 127.4 (CH), 126.4 (C), 124.3 (CH), 113.2 (CH), 57.0 (CH3), 26.6 (CH3). MS (EI, m/z (%)): 306 (M+, 40), 213 (46), 119 (97), 91 (63), 43 (100), 39 (51). Anal. calcd. for C14H14N2O4S: C, 54.89; H, 4.61; N, 9.14; S, 10.47. Found: C, 54.90; H, 4.68; N, 9.13; S, 10.42.
3.2.5. General Procedure for the Synthesis of Sulfonamide 12
To a mixture of chloride 6b (0.33 mmol) and amine 11 (0.36 mmol) in ethanol (1.5 mL), TEA (0.15 mL) was added and left under stirring at room temperature. Once the reaction was complete, the medium was neutralized with TEA and the solid obtained was filtered and washed with ethanol. Compound 12 did not require further purification.
5-Acetyl-N,N-bis(2-chloroethyl)-2,4-dimethoxybenzenesulfonamide (12)
White solid; 82% yield; m.p. 190–192 °C. FTIR (ATR) (cm−1): 3111 (C-H Ar), 2982 (C-H), 1661 (C=O), 1591 (C=C), 1143 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.12 (s, 1H, H6A), 6.87 (s, 1H, H3A), 4.06–4.01 (m, 6H, 2A-OCH3 and 4A-OCH3), 3.68 (t, J = 6.8 Hz, 4H, Cl-CH2), 3.53 (t, J = 6.8 Hz, 4H, N-CH2), 2.51 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 195.4 (C), 164.4 (C), 161.1 (C), 133.3 (CH), 119.1 (C), 119.0 (C), 97.6 (CH), 57.0 (CH3), 56.8 (CH3), 49.6 (CH2), 42.1 (CH2), 31.7 (CH3). MS (EI, m/z (%)): 384 (M+, 1), 334 (83), 243 (100), 195 (50), 149 (76). Anal. calcd. for C14H19Cl2NO5S: C, 43.76; H, 4.98; N, 3.65; S, 8.34. Found: C, 43.92; H, 5.06; N, 3.60; S, 8.17.
3.2.6. General Procedure for the Synthesis of Sulfonamides 14a-b
To a mixture of isoniazid 13 (0.29 mmol) and chloride 6a for 14a or 6b for 14b (0.31 mmol) in water (2 mL), 1 N sodium carbonate was added to reach pH 8–10 and was stirred at room temperature maintaining this pH. Once the reaction was finished, the pH was adjusted to 7–8 by adding HCl 10% v/v if necessary, the precipitate was filtered and washed with water. No further purification was required.
5-Acetyl-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (14a)
Beige crystals; 45% yield; m.p. 191–194 °C; FTIR (ATR) (cm−1): 3303 (N-H), 3234 (N-H), 3091 (C-H Ar), 1666 (C=O), 1595 (C=C), 1162 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.13 (s, 1H, S-NH), 10.08 (s, 1H, C-NH), 8.87 (s, 2H, H2C), 8.46–8.33 (m, 2H, H6A and H4A), 7.76 (s, 2H, H3C), 7.49 (d, J = 8.7 Hz, 1H, H3A), 4.16 (s, 3H, OCH3), 2.68 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 195.6 (C), 164.1 (C), 161.0 (C), 150.0 (CH), 139.3 (C), 135.7 (CH), 129.7 (CH), 128.5 (C), 127.4 (C), 121.4 (CH), 113.0 (CH), 57.0 (CH3), 26.4 (CH3). MS (EI, m/z (%)): 349 (M+, 1), 106 (100), 78 (66), 51 (50), 43 (52). Anal. calcd. for C15H15N3O5S: C, 51.57; H, 4.33; N, 12.03; S, 9.18. Found: C, 51.60; H, 4.41; N, 12.03; S, 9.16. Crystals suitable for single-crystal X-ray diffraction were obtained from ethanolic solution, and the crystal data for 14a were deposited at CCDC with reference CCDC 2191607.
5-Acetyl-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (14b)
Pale yellow crystals; 85% yield; m.p. 188–189 °C. FTIR (ATR) (cm−1): 3163 (C-H Ar), 1676 (C=O), 1659 (C=O), 1592 (C=C), 1166 (S=O), 1288 (C-N). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.91 (d, J = 3.6 Hz, 1H, S-NH), 9.69 (d, J = 3.6 Hz, 1H, C-NH), 8.69 (d, J = 5.0 Hz, 2H, H2C), 8.06 (s, 1H, H6A), 7.57 (d, J = 5.0 Hz, 2H, H3C), 6.82 (s, 1H, H3A), 4.03–4.00 (m, 6H, 2A-OCH3 and 4A-OCH3), 2.46 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 195.3 (C), 164.5 (C), 164.1 (C), 162.4 (C), 150.3 (CH), 139.1 (C), 132.8 (CH), 121.2 (CH), 119.2 (C), 118.5 (C), 97.3 (CH), 57.1 (CH3), 56.7 (CH3), 31.6 (CH3). MS (EI, m/z (%)): 379 (M+, 6), 243 (40), 149 (32), 106 (88), 78 (82), 43 (100). Anal. calcd. for C16H17N3O6S: C, 50.65; H, 4.52; N, 11.08; S, 8.45. Found: C, 50.50; H, 4.52; N, 11.13; S, 8.40. Crystals suitable for single-crystal X-ray diffraction were obtained from ethanolic solution, and the crystal data for 14b were deposited at CCDC with reference CCDC 2191610.
3.2.7. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 16a-f and 17a-f
To a mixture of sulfonamide 8b or 10 (0.20 mmol) and the corresponding aldehyde 15a-f (0.24 mmol) in ethanol (1.5 mL), 2 drops of 50% (w/v) NaOH solution were added and the reaction mixture was stirred at room temperature. The sodium salt formed was filtered and washed with small portions of ethanol. Next, it was poured into water and neutralized by adding 10% (w/v) HCl aqueous solution. The precipitate was filtered and washed with water to afford the corresponding compounds 16a-f and 17a-f. No further purification was required.
5-Cinnamoyl-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (16a)
White solid; 88% yield; m.p. 229–230 °C. FTIR (ATR) (cm−1): 3079 (C-H Ar), 1659 (C=O), 1601 (C=C), 1340 (C-N), 1158 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.48 (d, J = 1.4 Hz, 1H, H6A), 8.23 (dd, J = 8.4, 1.4 Hz, 1H, H4A), 8.01 (d, J = 1.8 Hz, 1H, H2B), 7.86–7.75 (m, 3H, Hα and H2D), 7.72–7.61 (m, 2H, Hβ and H6B), 7.50–7.41 (m, 3H, H3D and H4D), 7.20–7.08 (m, 2H, H3A and H4B), 6.88 (dd, J = 8.1, 4.6 Hz, 1H, H5B), 3.84 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 187.4 (C), 160.4 (C), 146.8 (C), 143.6 (CH), 143.4 (CH), 137.0 (CH), 134.8 (C), 134.7 (C), 132.5 (CH), 130.6 (CH), 129.9 (CH), 129.0 (CH), 128.9 (C), 128.8 (CH), 125.5 (CH), 122.9 (CH), 121.9 (CH), 112.1 (CH), 56.2 (CH3). MS (EI, m/z (%)): 394 (M+, 8), 368 (36), 236 (26), 97 (56), 57 (100), 43 (77). Anal. calcd. for C21H18N2O4S: C, 63.94; H, 4.60; N, 7.10; S, 8.13. Found: C, 63.90; H, 4.69; N, 7.24; S, 8.10.
(E)-5-(3-(4-Chlorophenyl)acryloyl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (16b)
White solid; 67% yield; m.p. 256–258 °C. FTIR (ATR) (cm−1): 3068 (C-H Ar), 1661 (C=O), 1604 (C=C), 1312 (C-N), 1152 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.47 (d, J = 2.3 Hz, 1H, H6A), 8.26 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 8.04 (d, J = 2.6 Hz, 1H, H2B), 7.86 (d, J = 8.6 Hz, 2H, H2D), 7.82 (d, J = 15.7 Hz, 1H, Hα), 7.71 (dd, J = 4.5, 1.5 Hz, 1H, H6B), 7.67 (d, J = 15.7 Hz, 1H, Hβ), 7.51 (d, J = 8.6 Hz, 2H, H3D), 7.23–7.18 (m, 1H, H4B), 7.15 (d, J = 8.7 Hz, 1H, H3A), 6.92 (dd, J = 8.3, 4.5 Hz, 1H, H5B), 3.84 (s, 3H, OCH3). 13C RMN (101 MHz, DMSO-d6) δ ppm 187.3 (C), 160.5 (C), 146.4 (C), 143.5 (CH), 142.0 (CH), 137.3 (CH), 135.0 (C), 134.6 (C), 133.7 (C), 132.7 (CH), 130.5 (CH), 129.9 (CH), 129.0 (CH), 128.9 (C), 125.6 (CH), 122.9 (CH), 122.7 (CH), 112.1 (CH), 56.2 (CH3). MS (EI, m/z (%)): 428/430 (M+/M+2+, 9/3), 313 (29), 236 (37), 97 (60), 57 (100). Anal. calcd. for C21H17ClN2O4S: C, 58.81; H, 4.00; N, 6.53; S, 7.48. Found: C, 58.79; H, 3.93; N, 6.50; S, 7.48.
(E)-2-Methoxy-N-(pyridin-3-yl)-5-(3-(p-tolyl)acryloyl)benzenesulfonamide (16c)
Beige solid; 85% yield; m.p. 231–234 °C. FTIR (ATR) (cm−1): 2947 (C-H), 1659 (C=O), 1598 (C=C), 1339 (C-N), 1156 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.47 (d, J = 2.4 Hz, 1H, H6A), 8.22 (dd, J = 8.6, 2.4 Hz, 1H, H4A), 8.01 (d, J = 2.6 Hz, 1H, H2B), 7.75–7.64 (m, 5H, Hα, Hβ, H2D and H6B), 7.27 (d, J = 7.7 Hz, 2H, H3D), 7.19–7.11 (m, 2H, H3A and H4B), 6.89 (dd, J = 8.3, 4.6 Hz, 1H, H5B), 3.84 (s, 3H, OCH3), 2.35 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 187.3 (C), 160.3 (C), 146.8 (C), 143.6 (CH), 143.5 (CH), 140.6 (C), 137.0 (CH), 134.7 (C), 132.4 (CH), 132.0 (C), 129.8 (CH), 129.6 (CH), 129.0 (C), 128.8 (CH), 125.4 (CH), 122.8 (CH), 120.8 (CH), 112.0 (CH), 56.2 (CH3), 21.1 (CH3). MS (EI, m/z (%)): 408 (M+, 38), 315 (24), 178 (36), 145 (68), 115 (78), 39 (100). Anal. calcd. for C22H20N2O4S: C, 64.69; H, 4.94; N, 6.86; S, 7.85. Found: C, 64.70; H, 5.01; N, 6.87; S, 7.79.
(E)-2-Methoxy-5-(3-(4-methoxyphenyl)acryloyl)-N-(pyridin-3-yl)benzenesulfonamide (16d)
Pale yellow solid; 86% yield; m.p. 228–230 °C. FTIR (ATR) (cm−1): 3072 (C-H Ar), 1653 (C=O), 1593 (C=C), 1220 (C-O-C), 1159 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.47 (d, J = 2.4 Hz, 1H, H6A), 8.21 (dd, J = 8.6, 2.4 Hz, 1H, H4A), 8.01 (d, J = 2.6 Hz, 1H, H2B), 7.78 (d, J = 8.3 Hz, 2H, H2D), 7.71–7.61 (m, 3H, Hα, Hβ and H6B), 7.18–7.10 (m, 2H, H4B and H3A), 7.01 (d, J = 8.3 Hz, 2H, H3D), 6.89 (dd, J = 8.3, 4.6 Hz, 1H, H5B), 3.84 (s, 3H, 2A-OCH3), 3.82 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 187.2 (C), 161.3 (C), 160.2 (C), 146.8 (C), 143.6 (CH), 143.4 (CH), 137.0 (CH), 134.6 (C), 132.3 (CH), 130.6 (CH), 129.8 (CH), 129.2 (C), 127.3 (C), 125.4 (CH), 122.8 (CH), 119.3 (CH), 114.5 (CH), 112.0 (CH), 56.2 (CH3), 55.4 (CH3). MS (EI, m/z (%)): 424 (M+, 98), 237 (58), 161 (85), 133 (81), 95 (100), 39 (77). Anal. calcd. for C22H20N2O5S: C, 62.25; H, 4.75; N, 6.60; S, 7.55. Found: C, 62.30; H, 4.82; N, 6.61; S, 7.47.
(E)-2-Methoxy-N-(pyridin-3-yl)-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonamide (16e)
Yellow solid; 77% yield; m.p. 239–241 °C. FTIR (ATR) (cm−1): 3077 (C-H Ar), 1660 (C=O), 1595 (C=C), 1272 (C-O-C), 1126 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.49 (d, J = 2.3 Hz, 1H, H6A), 8.27 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 8.02 (d, J = 2.6 Hz, 1H, H2B), 7.78 (d, J = 15.5 Hz, 1H, Hα), 7.69–7.64 (m, 2H, Hβ and H6B), 7.20 (s, 2H, H2D), 7.19–7.13 (m, 2H, H3A and H4B), 6.89 (dd, J = 8.3, 4.6 Hz, 1H, H5B), 3.88 (s, 6H, 3D-OCH3), 3.85 (s, 3H, OCH3), 3.73 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 187.5 (C), 160.3 (C), 153.1 (C), 146.8 (C), 143.9 (CH), 143.5 (CH), 139.7 (C), 137.0 (CH), 134.9 (C), 132.5 (CH), 130.3 (C), 129.8 (CH), 129.1 (C), 125.5 (CH), 122.8 (CH), 121.2 (CH), 111.9 (CH), 106.4 (CH), 60.2 (CH3), 56.2 (CH3), 56.2 (CH3). MS (EI, m/z (%)): 484 (M+, 13), 264 (7), 109 (35), 95 (50), 57 (47), 43 (100). Anal. calcd. for C24H24N2O7S: C, 59.49; H, 4.99; N, 5.78; S, 6.62. Found: C, 59.51; H, 4.80; N, 5.79; S, 6.58.
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (16f)
Yellow solid; 82% yield; m.p. 220–223 °C. FTIR (ATR) (cm−1): 3076 (C-H Ar), 1659 (C=O), 1596 (C=C), 1236 (C-O-C), 1159 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.46 (d, J = 2.4 Hz, 1H, H6A), 8.24 (dd, J = 8.6, 2.4 Hz, 1H, H4A), 8.00 (d, J = 2.7 Hz, 1H, H2B), 7.72–7.60 (m, 3H, Hα, Hβ and H6B), 7.57 (s, 1H, H2D), 7.29 (d, J = 8.1 Hz, 1H, H6D), 7.18–7.09 (m, 2H, H3A and H4B), 6.98 (d, J = 8.1 Hz, 1H, H5D), 6.88 (dd, J = 8.3, 4.6 Hz, 1H, H5B), 6.10 (s, 2H, CH2), 3.83 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 187.2 (C), 160.2 (C), 149.5 (C), 148.1 (C), 146.8 (C), 143.6 (CH), 143.4 (CH), 136.9 (CH), 134.8 (C), 132.4 (CH), 129.7 (CH), 129.2 (C), 129.1 (C), 125.6 (CH), 125.4 (CH), 122.8 (CH), 119.9 (CH), 111.9 (CH), 108.5 (CH), 107.0 (CH), 101.6 (CH2), 56.2 (CH3). MS (EI, m/z (%)): 438 (M+, 55), 251 (42), 165 (56), 89 (86), 66 (57), 39 (100). Anal. calcd. for C22H18N2O6S: C, 60.27; H, 4.14; N, 6.39; S, 7.31. Found: C, 60.34; H, 4.10; N, 6.39; S, 7.50.
5-Cinnamoyl-2,4-dimethoxybenzenesulfonamide (17a)
Beige solid; 60% yield; m.p. 230–231 °C. FTIR (ATR) (cm−1): 3403 and 3375 (N-H), 3097 (C-H Ar), 1638 (C=O), 1596 (C=C), 1153 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.04 (s, 1H, H6A), 7.77–7.71 (m, 2H, H2D), 7.63–7.52 (m, 2H, Hα and Hβ), 7.48–7.42 (m, 3H, H3D and H4D), 7.10 (s, 2H, NH2), 6.89 (s, 1H, H3A), 4.04 (s, 6H, 2A-OCH3 and 4A-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.6 (C), 162.9 (C), 160.5 (C), 142.1 (CH), 134.7 (C), 130.6 (CH), 130.4 (CH), 129.0 (CH), 128.5 (CH), 126.6 (CH), 124.1 (C), 119.5 (C), 97.2 (CH), 56.8 (CH3). MS (EI, m/z (%)): 347 (M+, 36), 319 (34), 256 (55), 131 (62), 103 (100), 77 (69). Anal. calcd. for C17H17NO5S: C, 58.78; H, 4.93; N, 4.03; S, 9.23. Found: C, 58.59; H, 4.90; N, 4.15; S, 9.22.
(E)-5-(3-(4-Chlorophenyl)acryloyl)-2,4-dimethoxybenzenesulfonamide (17b)
Pale yellow solid; 77% yield; m.p. 260–262 °C. FTIR (ATR) (cm−1): 3397 and 3238 (N-H), 3106 (C-H Ar), 1642 (C=O), 1597 (C=C), 1154 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.04 (s, 1H, H6A), 7.77 (d, J = 8.6 Hz, 2H, H2D), 7.54–7.59 (m, 2H, Hα and Hβ), 7.50 (d, J = 8.6 Hz, 2H, H3D), 7.10 (s, 2H, NH2), 6.88 (s, 1H, H3A), 4.06–4.02 (m, 6H, 2A-OCH3 and 4A-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.5 (C), 163.0 (C), 160.7 (C), 140.6 (CH), 134.9 (C), 133.7 (C), 130.7 (CH), 130.2 (CH), 129.1 (CH), 127.3 (CH), 124.1 (C), 119.4 (C), 97.2 (CH), 56.85 (CH3), 56.83 (CH3). MS (EI, m/z (%)): 381/383 (M+/M+2+, 22/8), 353 (36), 256 (100), 137 (55), 102 (70), 75 (43). Anal. calcd. for C17H16ClNO5S: C, 53.47; H, 4.22; N, 3.67; S, 8.40. Found: C, 53.50; H, 4.30; N, 3.63; S, 8.32.
(E)-2,4-Dimethoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonamide (17c)
Pale yellow crystals; 65% yield; m.p. 206–208 °C. FTIR (ATR) (cm−1): 3369 and 3256 (N-H), 2987 (C-H), 1654 (C=O), 1600 (C=C), 1152 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.04 (s, 1H, H6A), 7.65 (d, J = 7.7 Hz, 2H, H2D), 7.59 (d, J = 15.8 Hz, 1H, Hβ), 7.50 (d, J = 15.8 Hz, 1H, Hα), 7.28 (d, J = 7.7 Hz, 2H, H3D), 7.11 (s, 2H, NH2), 6.90 (s, 1H, H3A), 4.08–4.03 (m, 6H, 2A-OCH3 and 4A-OCH3), 2.36 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.7 (C), 162.9 (C), 160.5 (C), 142.3 (CH), 140.6 (C), 132.0 (C), 130.6 (CH), 129.7 (CH), 128.6 (CH), 125.6 (CH), 124.0 (C), 119.6 (C), 97.2 (CH), 56.8 (CH3), 21.1 (CH3). MS (EI, m/z (%)): 361 (M+, 21), 346 (53), 281 (25), 115 (70), 105 (100), 91 (47). Anal. calcd. for C18H19NO5S: C, 59.82; H, 5.30; N, 3.88; S, 8.87. Found: C, 59.77; H, 5.30; N, 3.95; S, 8.79. Crystals suitable for single-crystal X-ray diffraction were obtained from ethanolic solution, and the crystal data for 17c were deposited at CCDC with reference CCDC 2191611.
(E)-2,4-Dimethoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonamide (17d)
Yellow crystals; 82% yield; m.p. 215–218 °C. FTIR (ATR) (cm−1): 3398 and 3199 (N-H), 1638 (C=O), 1592 (C=C), 1251 (C-O-C), 1152 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.01 (s, 1H, H6A), 7.70 (d, J = 8.4 Hz, 2H, H2D), 7.56 (d, J = 15.8 Hz, 1H, Hβ), 7.40 (d, J = 15.8 Hz, 1H, Hα), 7.09 (s, 2H, NH2), 7.00 (d, J = 8.4 Hz, 2H, H3D), 6.88 (s, 1H, H3A), 4.05–4.01 (m, 6H, 2A-OCH3 and 4A-OCH3), 3.81 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.6 (C), 162.7 (C), 161.2 (C), 160.3 (C), 142.3 (CH), 130.4 (CH), 130.3 (CH), 127.3 (C), 124.2 (CH), 124.0 (C), 119.8 (C), 114.5 (CH), 97.2 (CH), 56.71 (CH3), 56.70 (CH3), 55.3 (CH3). MS (EI, m/z (%)): 377 (M+, 48), 297 (44), 244 (27), 161 (52), 133 (47), 121 (100). Anal. calcd. for C18H19NO6S: C, 57.28; H, 5.07; N, 3.71; S, 8.50. Found: C, 57.09; H, 5.14; N, 3.65; S, 8.52. Crystals suitable for single-crystal X-ray diffraction were obtained from ethanolic solution, and the crystal data for 17d were deposited at CCDC with reference CCDC 2191612.
(E)-2,4-Dimethoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonamide (17e)
Beige solid; 62% yield; m.p. 253–256 °C. FTIR (ATR) (cm−1): 3381 and 3253 (N-H), 2972 (C-H Ar), 1653 (C=O), 1607 (C=C), 1270 (C-O-C), 1149 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.98 (s, 1H, H6A), 7.53–7.43 (m, 2H, Hβ and Hα), 7.09 (s, 2H, NH2), 7.07 (s, 2H, H2D), 6.89 (s, 1H, H3A), 4.05–4.01 (m, 6H, 2A-OCH3 and 4A-OCH3), 3.84 (s, 6H, 3D-OCH3), 3.71 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 189.2 (C), 162.7 (C), 160.3 (C), 153.1 (C), 142.8 (CH), 139.6 (C), 130.2(CH, C), 126.2 (CH), 124.0 (C), 119.8 (C), 106.0 (CH), 97.2 (CH), 60.1 (CH3), 56.74 (CH3), 56.69 (CH3), 56.0 (CH3). MS (EI, m/z (%)): 437 (M+, 100), 406 (40), 355 (24), 181 (43), 127 (35), 43 (29). Anal. calcd. for C20H23NO8S: C, 54.91; H, 5.30; N, 3.20; S, 7.33. Found: C, 55.00; H, 5.28; N, 3.35; S, 7.38.
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-2,4-dimethoxybenzenesulfonamide (17f)
Pale yellow solid; 53% yield; m.p. 257–258 °C. FTIR (ATR) (cm−1): 3389 and 3300 (N-H), 3109 (C-H Ar), 1646 (C=O), 1595 (C=C), 1244 (C-O-C), 1154 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.99 (s, 1H, H6A), 7.52 (d, J = 15.7 Hz, 1H, Hβ), 7.42–7.35 (m, 2H, Hα and H2D), 7.24 (d, J = 8.0 Hz, 1H, H6D), 7.08 (s, 2H, NH2), 6.98 (d, J = 8.0 Hz, 1H, H5D), 6.87 (s, 1H, H3A), 6.09 (s, 2H, CH2), 4.06–4.00 (m, 6H, 2A-OCH3 and 4A-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.7 (C), 162.7 (C), 160.3 (C), 149.4 (C), 148.1 (C), 142.3 (CH), 130.4 (CH), 129.1 (C), 125.2 (CH), 124.7 (CH), 124.0 (C), 119.8 (C), 108.6 (CH), 106.8 (CH), 101.6 (CH2), 97.2 (CH), 56.7 (CH3). MS (EI, m/z (%)): 391 (M+, 100), 377 (22), 69 (27), 55 (31), 43 (67). Anal. calcd. for C18H17NO7S: C, 55.24; H, 4.38; N, 3.58; S, 8.19. Found: C, 55.09; H, 4.40; N, 3.63; S, 8.13.
3.2.8. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 18a-f
To a mixture of sulfonamide 12 (0.20 mmol) and the corresponding aldehyde 15a-f (0.23 mmol) in ethanol (1.5 mL), 2 drops of 50% (w/v) NaOH solution were added and the reaction mixture was stirred at room temperature. The resulting precipitate was collected by filtration under vacuum, washed with ethanol and purified by column chromatography on silica gel, using a mixture of CHCl3:EtOAc (10:1) as eluent.
N,N-bis(2-chloroethyl)-5-cinnamoyl-2,4-dimethoxybenzenesulfonamide (18a)
Beige solid; 79% yield; m.p. 184–186 °C. FTIR (ATR) (cm−1): 3065 (C-H Ar), 2949 (C-H), 1645 (C=O), 1599 (C=C), 1145 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.29 (s, 1H, H6A), 7.66 (d, J = 15.8 Hz, 1H, Hβ), 7.59 (dd, J = 6.6, 3.0 Hz, 2H, H2D), 7.42–7.34 (m, 4H, H3D, H4D and Hα), 6.54 (s, 1H, H3A), 4.04 (s, 3H, 4A-OCH3), 4.00 (s, 3H, 2A-OCH3), 3.64 (s, 8H, N-CH2 and Cl-CH2). 13C NMR (101 MHz, CDCl3) δ ppm 189.5 (C), 163.8 (C), 160.8 (C), 143.9 (CH), 135.1 (C), 134.9 (CH), 130.6 (CH), 129.1 (CH), 128.6 (CH), 126.4 (CH), 121.6 (C), 120.1 (C), 95.9 (CH), 56.6 (CH3), 56.5 (CH3), 51.3 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 472 (M+, 4), 331 (100), 283 (38), 131 (52), 103 (41). Anal. calcd. for C21H23Cl2NO5S: C, 53.40; H, 4.91; N, 2.97; S, 6.79. Found: C, 53.25; H, 4.90; N, 3.06; S, 6.68.
(E)-N,N-bis(2-Chloroethyl)-5-(3-(4-chlorophenyl)acryloyl)-2,4-dimethoxybenzenesulfonamide (18b)
White solid; 88% yield; m.p. 186–187 °C. FTIR (ATR) (cm−1): 3084 (C-H Ar), 2914 (C-H), 1645 (C=O), 1601 (C=C), 1149 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.30 (s, 1H, H6A), 7.60 (d, J = 15.8 Hz, 1H, Hβ), 7.51 (d, J = 8.4 Hz, 2H, H2D), 7.39–7.31 (m, 3H, H3D and Hα), 6.53 (s, 1H, H3A), 4.04 (s, 3H, 4A-OCH3), 4.00 (s, 3H, 2A-OCH3), 3.63 (s, 8H, N-CH2 and Cl-CH2). 13C NMR (101 MHz, CDCl3) δ ppm 189.0 (C), 163.9 (C), 161.0 (C), 142.2 (CH), 136.4 (C), 135.0 (CH), 133.6 (C), 129.7 (CH), 129.3 (CH), 126.8 (CH), 121.4 (C), 120.2 (C), 95.9 (CH), 56.6 (CH3), 56.5 (CH3), 51.3 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 507 (M+, 4), 365 (100), 317 (26), 165 (30), 135 (24). Anal. calcd. for C21H22Cl3NO5S: C, 49.77; H, 4.38; N, 2.76; S, 6.33. Found: C, 49.68; H, 4.45; N, 2.79; S, 6.09.
(E)-N,N-bis(2-Chloroethyl)-2,4-dimethoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonamide (18c)
Pale yellow solid; 86% yield; m.p. 186–189 °C. FTIR (ATR) (cm−1): 2923 (C-H), 1644 (C=O), 1597 (C=C), 1276 (C-O-C), 1150 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.27 (s, 1H, H6A), 7.62 (d, J = 15.8 Hz, 1H, Hβ), 7.48 (d, J = 7.9 Hz, 2H, H2D), 7.30 (d, J = 15.8 Hz, 1H, Hα), 7.20 (d, J = 7.9 Hz, 2H, H3D), 6.53 (s, 1H, H3A), 4.03 (s, 3H, 4A-OCH3), 3.99 (s, 3H, 2A-OCH3), 3.63 (s, 8H, N-CH2 and Cl-CH2), 2.38 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ ppm 189.6 (C), 163.7 (C), 160.7 (C), 144.1 (CH), 141.1 (C), 134.8 (CH), 132.3 (C), 129.8 (CH), 128.6 (CH), 125.4 (CH), 121.7 (C), 119.9 (C), 95.9 (CH), 56.6 (CH3), 56.5 (CH3), 51.3 (CH2), 42.4 (CH2), 21.6 (CH3). MS (EI, m/z (%)): 486 (M+, 4), 345 (100), 297 (28), 145 (39), 105 (56). Anal. calcd. for C22H25Cl2NO5S: C, 54.33; H, 5.18; N, 2.88; S, 6.59. Found: C, 54.52; H, 5.22; N, 2.57; S, 6.68.
(E)-N,N-bis(2-Chloroethyl)-2,4-dimethoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonamide (18d)
Yellow solid; 84% yield; m.p. 168–171 °C. FTIR (ATR) (cm−1): 3016 (C-H Ar), 2847 (C-H), 1642 (C=O), 1593 (C=C), 1143 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.26 (s, 1H, H6A), 7.61 (d, J = 15.8 Hz, 1H, Hβ), 7.54 (d, J = 8.8 Hz, 2H, H2D), 7.22 (d, J = 15.8 Hz, 1H, Hα), 6.91 (d, J = 8.7 Hz, 2H, H3D), 6.53 (s, 1H, H3A), 4.03 (s, 3H, 4A-OCH3), 3.99 (s, 3H, 2A-OCH3), 3.84 (s, 3H, 4D-OCH3), 3.63 (s, 8H, N-CH2 and Cl-CH2). 13C NMR (101 MHz, CDCl3) δ ppm 189.6 (C), 163.7 (C), 161.8 (C), 160.6 (C), 144.0 (CH), 134.7 (CH), 130.4 (CH), 127.7 (C), 124.2 (CH), 121.9 (C), 119.9 (C), 114.5 (CH), 95.9 (CH), 56.6 (CH3), 56.5 (CH3), 55.5 (CH3), 51.3 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 502 (M+, 4), 361 (46), 267 (28), 161 (39), 121 (100). Anal. calcd. for C22H25Cl2NO6S: C, 52.60; H, 5.02; N, 2.79; S, 6.38. Found: C, 52.43; H, 5.03; N, 2.54; S, 6.35.
(E)-N,N-bis(2-Chloroethyl)-2,4-dimethoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonamide (18e)
Yellow solid; 61% yield; m.p. 186–188 °C. FTIR (ATR) (cm−1): 2957 (C-H), 1647 (C=O), 1600 (C=C), 1273 (C-O-C), 1145 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.24 (s, 1H, H6A), 7.52 (d, J = 15.8 Hz, 1H, Hβ), 7.19 (d, J = 15.8 Hz, 1H, Hα), 6.80 (s, 2H, H2D), 6.53 (s, 1H, H3A), 4.03 (s, 3H, 4A-OCH3), 3.98 (s, 3H, 2A-OCH3), 3.91–3.87 (m, 9H, 3D-OCH3 and 4D-OCH3), 3.63 (s, 8H, N-CH2 and Cl-CH2). 13C NMR (101 MHz, CDCl3) δ 189.7 (C), 163.7 (C), 160.7 (C), 153.6 (C), 144.3 (CH), 140.6 (C), 134.5 (CH), 130.5 (C), 125.8 (CH), 121.7 (C), 119.9 (C), 105.9 (CH), 96.0 (CH), 61.1 (CH3), 56.6 (CH3), 56.5 (CH3), 56.4 (CH3), 51.3 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 562 (M+, 2), 334 (45), 243 (100), 83 (79), 43 (51). Anal. calcd. for C24H29Cl2NO8S: C, 51.25; H, 5.20; N, 2.49; S, 5.70. Found: C, 51.05; H, 5.43; N, 2.52; S, 5.91.
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-N,N-bis(2-chloroethyl)-2,4-dimethoxybenzenesulfonamide (18f)
Yellow solid; 82% yield; m.p. 193–196 °C. FTIR (ATR) (cm−1): 2885 (C-H), 1643 (C=O), 1595 (C=C), 1250 (C-O-C), 1150 (S=O). 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, H6A), 7.57 (d, J = 15.7 Hz, 1H, Hβ), 7.18 (d, J = 15.7 Hz, 1H, Hα), 7.13–7.02 (m, 2H H2D and H6D), 6.82 (d, J = 7.9 Hz, 1H, H5D), 6.53 (s, 1H, H3A), 6.02 (s, 2H, O-CH2), 4.06–3.97 (m, 6H, 2A-OCH3 and 4A-OCH3), 3.63 (s, 8H, N-CH2 and Cl-CH2). 13C NMR (101 MHz, CDCl3) δ ppm 189.3 (C), 163.7 (C), 160.7 (C), 150.0 (C), 148.5 (C), 143.9 (CH), 134.8 (CH), 129.5 (C), 125.4 (CH), 124.5 (CH), 121.8 (C), 120.0 (C), 108.8 (CH), 106.8 (CH), 101.7 (CH2), 95.9 (CH), 56.6 (CH3), 56.5 (CH3), 51.3 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 516 (M+, 7), 375 (49), 311 (47), 281 (36), 135 (100). Anal. calcd. for C22H23Cl2NO7S: C, 51.17; H, 4.49; N, 2.71; S, 6.21. Found: C, 51.10; H, 4.58; N, 2.82; S, 6.22.
3.2.9. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 19a-f
To a mixture of sulfonamide 14a (0.20 mmol) and the corresponding aldehyde 15a-f (0.24 mmol) in ethanol (1.5 mL), 2 drops of a 50% w/v NaOH solution were added and stirred at room temperature. After completion of the reaction, the medium was neutralized using an aqueous 10% v/v HCl solution, the precipitate was filtered and washed with water. The products 19a-f were purified by CC on silica gel, using a CHCl3:EtOH (15:1) mixture as eluent.
5-Cinnamoyl-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (19a)
Beige solid; 78% yield; m.p. 227–229 °C. FTIR (ATR) (cm−1): 3212 (N-H), 3013 (C-H Ar), 1682 (C=O), 1654 (C=O), 1597 (C=C), 1155 (S=O). 1H NMR (400 MHz, DMSO-d6) δ 10.97 (s, 1H, S-NH), 9.95 (s, 1H, C-NH), 8.67 (d, J = 6.0 Hz, 2H, H2C), 8.50 (dd, J = 8.8, 2.3 Hz, 1H, H4A), 8.43 (d, J = 2.3 Hz, 1H, H6A), 7.90 (d, J = 15.6 Hz, 1H, Hα), 7.88–7.83 (m, 2H, H2D), 7.72 (d, J = 15.6 Hz, 1H, Hβ), 7.58 (d, J = 6.0 Hz, 2H, H3C), 7.48–7.43 (m, 3H, H3D and H4D), 7.38 (d, J = 8.8 Hz, 1H, H3A), 4.04 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 186.8 (C), 164.2 (C), 161.1 (C), 150.4 (CH), 144.1 (CH), 138.9 (C), 135.9 (CH), 134.6 (C), 130.7 (CH), 130.2 (CH), 129.1 (C), 128.9 (CH), 128.9 (CH), 127.7 (C), 121.5 (CH), 121.2 (CH), 113.0 (CH), 57.1 (CH3). MS (EI, m/z (%)): 437 (M+, 1), 238 (61), 135 (88), 106 (100), 78 (77), 51 (63). Anal. calcd. for C22H19N3O5S: C, 60.40; H, 4.38; N, 9.61; S, 7.33. Found: C, 60.36; H, 4.30; N, 9.55; S, 7.45.
(E)-5-(3-(4-Chlorophenyl)acryloyl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (19b)
White solid; 75%; m.p. 242–244 °C. FTIR (ATR) (cm−1): 3235 (N-H), 3030 (C-H Ar), 1681 (C=O), 1655 (C=O), 1600 (C=C), 1155 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.99 (s, 1H, S-NH), 9.99 (s, 1H, C-NH), 8.67 (d, J = 5.3 Hz, 2H, H2C), 8.52 (d, J = 8.2 Hz, 1H, H4A), 8.42 (s, 1H, H6A), 7.99–7.89 (m, 3H, H2D and Hα), 7.71 (d, J = 15.5 Hz, 1H, Hβ), 7.57 (d, J = 5.3 Hz, 2H, H3C), 7.52 (d, J = 8.1 Hz, 2H, H3D), 7.38 (d, J = 8.2 Hz, 1H, H3A), 4.03 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 186.6 (C), 164.2 (C), 161.2 (C), 150.4 (CH), 142.7 (CH), 138.9 (C), 136.0 (CH), 135.2 (C), 133.6 (C), 130.7 (CH), 130.3 (CH), 129.02 (C), 128.98 (CH), 127.8 (C), 122.2 (CH), 121.2 (CH), 113.1 (CH), 57.1 (CH3). MS (EI, m/z (%)): 471/473 (M+/M+2+, 5/2), 272 (42), 106 (100), 78 (98), 51 (88). Anal. calcd. for C22H18ClN3O5S: C, 55.99; H, 3.84; N, 8.90; S, 6.79. Found: C, 56.08; H, 3.70; N, 8.91; S, 6.75.
(E)-N’-Isonicotinoyl-2-methoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonohydrazide (19c)
White solid; 81% yield; m.p. 250-252 °C. FTIR (ATR) (cm-1): 3508 (N-H), 3033 (C-H Ar), 1691 (C=O), 1658 (C=O), 1599 (C=C), 1162 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.97 (s, 1H, S-NH), 9.94 (s, 1H, C-NH), 8.67 (d, J = 5.0 Hz, 2H, H2C), 8.49 (d, J = 8.7 Hz, 1H, H4A), 8.42 (s, 1H, H6A), 7.84 (d, J = 15.5 Hz, 1H, Hα), 7.75 (d, J = 7.8 Hz, 2H, H2D), 7.69 (d, J = 15.5 Hz, 1H, Hβ), 7.58 (d, J = 5.0 Hz, 2H, H3C), 7.37 (d, J = 8.7 Hz, 1H, H3A), 7.26 (d, J = 7.8 Hz, 2H, H3D), 4.03 (s, 3H, OCH3), 2.34 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 186.7 (C), 164.2 (C), 161.0 (C), 150.4 (CH), 144.2 (CH), 140.8 (C), 138.9 (C), 135.8 (CH), 131.9 (C), 130.2 (CH), 129.5 (CH), 129.2 (C), 129.0 (CH), 127.7 (C), 121.2 (CH), 120.4 (CH), 113.0 (CH), 57.1 (CH3), 21.1(CH3). MS (EI, m/z (%)): 451 (M+, 3), 252 (61), 135 (69), 106 (100), 78 (77), 51 (58). Anal. calcd. for C23H21N3O5S: C, 61.18; H, 4.69; N, 9.31; S, 7.10. Found: C, 61.01; H, 4.76; N, 9.47; S, 7.11.
(E)-N’-Isonicotinoyl-2-methoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonohydrazide (19d)
Pale yellow solid; 84% yield; m.p. 216–217 °C. FTIR (ATR) (cm−1): 3233 (N-H), 3042 (C-H Ar), 1676 (C=O), 1655 (C=O), 1597 (C=C), 1256 (C-O-C), 1154 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.97 (s, 1H, S-NH), 9.93 (s, 1H, C-NH), 8.67 (d, J = 5.9 Hz, 2H, H2C), 8.48 (dd, J = 8.8, 2.3 Hz, 1H, H4A), 8.41 (d, J = 2.3 Hz, 1H, H6A), 7.83 (d, J = 8.6 Hz, 2H, H2D), 7.76 (d, J = 15.5 Hz, 1H, Hα), 7.69 (d, J = 15.5 Hz, 1H, Hβ), 7.58 (d, J = 5.9 Hz, 2H, H3C), 7.36 (d, J = 8.8 Hz, 1H, H3A), 7.01 (d, J = 8.6 Hz, 2H, H3D), 4.03 (s, 3H, OCH3), 3.82 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 186.6 (C), 164.2 (C), 161.4 (C), 160.9 (C), 150.3 (CH), 144.2 (CH), 138.9 (C), 135.7 (CH), 130.8 (CH), 130.1 (CH), 129.4 (C), 127.7 (C), 127.2 (C), 121.2 (CH), 118.9 (CH), 114.4 (CH), 113.0 (CH), 57.0 (CH3), 55.4 (CH3). MS (EI, m/z (%)): 467 (M+, 3), 300 (32), 268 (82), 78 (74), 51 (100). Anal. calcd. for C23H21N3O6S: C, 59.09; H, 4.53; N, 8.99; S, 6.86. Found: C, 59.20; H, 4.53; N, 9.06; S, 6.72.
(E)-N’-Isonicotinoyl-2-methoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonohydrazide (19e)
Yellow solid; 70% yield; m.p. 202–204 °C. FTIR (ATR) (cm−1): 3260 (N-H), 3049 (C-H Ar), 1678 (C=O), 1636 (C=O), 1595 (C=C), 1279 (C-O-C), 1123 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.98 (d, J = 2.8 Hz, 1H, S-NH), 9.95 (d, J = 2.8 Hz, 1H, C-NH), 8.68 (d, J = 6.1 Hz, 2H, H2C), 8.51 (dd, J = 8.8, 2.3 Hz, 1H, H4A), 8.42 (d, J = 2.3 Hz, 1H, H6A), 7.86 (d, J = 15.5 Hz, 1H, Hα), 7.68 (d, J = 15.5 Hz, 1H, Hβ), 7.58 (d, J = 6.1 Hz, 2H, H3C), 7.38 (d, J = 8.8 Hz, 1H. H3A), 7.22 (s, 2H, H2D), 4.04 (s, 3H, OCH3), 3.85 (s, 6H, 3D-OCH3), 3.72 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 186.7 (C), 164.2 (C), 161.0 (C), 153.1 (C), 150.4 (CH), 144.6 (CH), 139.9 (C), 138.9 (C), 135.9 (CH), 130.19 (C), 130.16 (CH), 129.3 (C), 127.8 (C), 121.2 (CH), 120.8 (CH), 112.9 (CH), 106.7 (CH), 60.2 (CH3), 57.1 (CH3), 56.2 (CH3). MS (EI, m/z (%)): 527 (M+, 0.07), 328 (25), 297 (14), 135 (100), 106 (50), 78 (72), 51 (96). Anal. calcd. for C25H25N3O8S: C, 56.92; H, 4.78; N, 7.97; S, 6.08. Found: C, 57.00; H, 4.71; N, 7.80; S, 6.09.
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (19f)
Yellow solid; 75% yielf; m.p. 245–248 °C. FTIR (ATR) (cm−1): 3230 (N-H), 3041 (C-H Ar), 1689 (C=O), 1648 (C=O), 1596 (C=C), 1220 (C-O-C), 1153 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.97 (s, 1H, S-NH), 9.93 (s, 1H, C-NH), 8.67 (d, J = 6.2 Hz, 2H,H2C), 8.49 (dd, J = 8.8, 2.3 Hz, 1H, H4A), 8.42 (d, J = 2.3 Hz, 1H, H6A), 7.79 (d, J = 15.4 Hz, 1H, Hα), 7.69–7.61 (m, 2H, Hβ and H2D), 7.58 (d, J = 6.2 Hz, 2H, H3C), 7.36 (d, J = 8.8 Hz, 1H, H3A), 7.30 (dd, J = 8.2, 1.7 Hz, 1H, H6D), 6.98 (d, J = 8.2 Hz, 1H, H5D), 6.10 (s, 2H, CH2), 4.03 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 186.5 (C), 164.2 (C), 161.0 (C), 150.4 (CH), 149.6 (C), 148.1 (C), 144.2 (CH), 138.9, 135.8 (CH), 130.1 (CH), 129.3 (C), 129.2 (C), 127.7 (C), 126.1 (CH), 121.2 (CH), 119.4 (CH), 113.0 (CH), 108.5 (CH), 107.0 (CH), 101.7 (CH2), 57.1 (CH3). MS (EI, m/z (%)): 481 (0.09), 282 (99.8), 135 (99.9), 106 (99.9), 78 (99.9), 51 (100). Anal. calcd. for C23H19N3O7S: C, 57.37; H, 3.98; N, 8.73; S, 6.66. Found: C, 57.32; H, 4.01; N, 8.59; S, 6.79.
3.2.10. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 20a-f
To a mixture of sulfonamide 14b (0.20 mmol) and the respective aldehyde 15a-f (0.24 mmol) in ethanol (1.5 mL), 2 drops of a 50% w/v NaOH solution were added and stirred at room temperature. The reaction media was neutralized with an aqueous 10% v/v HCl, the precipitate was filtered and washed with water. The products 20a-f were purified by CC on silica gel and a DCM:MeOH (20:1) mixture as mobile phase.
5-Cinnamoyl-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (20a)
Beige crystals; 71% yield; m.p. 143–145 °C. FTIR (ATR) (cm−1): 3135 (N-H), 2988 (C-H Ar), 1673 (C=O), 1650 (C=O), 1597 (C=C), 1157 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (d, J = 3.0 Hz, 1H, S-NH), 9.73 (d, J = 3.0 Hz, 1H, C-NH), 8.67 (d, J = 5.5 Hz, 2H, H2C), 7.99 (s, 1H, H6A), 7.70–7.65 (m, 2H, H3D), 7.58 (d, J = 5.5 Hz, 2H, H3C), 7.53 (d, J = 15.9 Hz, 1H, Hβ), 7.48 (d, J = 15.9 Hz, 1H, Hα), 7.45–7.40 (m, 3H, H3D and H4D), 6.88 (s, 1H, H3A), 4.06–4.02 (m, 6H, 2A-OCH3 and 4A-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.5 (C), 164.1 (C), 163.9 (C), 162.1 (C), 150.3 (CH), 142.3 (CH), 139.0 (C), 134.6 (C), 132.8 (CH), 130.5 (CH), 129.0 (CH), 128.4 (CH), 126.5 (CH), 121.2 (CH), 119.5 (C), 119.2 (C), 97.4 (CH), 57.1 (CH3), 56.8 (CH3). MS (EI, m/z (%)): 467 (M+, 0.3), 300 (18), 165 (52), 103 (79), 77 (94), 51 (100). Anal. calcd. for C23H21N3O6S: C, 59.09; H, 4.53; N, 8.99; S, 6.86. Found: C, 59.01; H, 4.55; N, 9.07; S, 6.70.
(E)-5-(3-(4-Chlorophenyl)acryloyl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (20b)
Pale yellow solid; 47% yield; m.p. 196–198 °C. FTIR (ATR) (cm−1): 3183 (N-H), 2987 (C-H Ar), 1678 (C=O), 1650 (C=O), 1601 (C=C), 1157 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (s, 1H, S-NH), 9.73 (s, 1H, C-NH), 8.67 (d, J = 5.8 Hz, 2H, H2C), 7.99 (s, 1H, H6A), 7.72 (d, J = 8.4 Hz, 2H, H2D), 7.58 (d, J = 5.8 Hz, 2H, H3C), 7.52–7.46 (m, 4H, Hα, Hβ and H3D), 6.87 (s, 1H, H3A), 4.06–4.01 (m, 6H, 2A-OCH3 and 4A-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.3 (C), 164.1 (C), 163.9 (C), 162.2 (C), 150.3 (CH), 140.7 (CH), 139.0 (C), 134.9 (C), 133.6 (C), 132.9 (CH), 130.1 (CH), 129.0 (CH), 127.2 (CH), 121.2 (CH), 119.4 (C), 119.3 (C), 97.3 (CH), 57.1 (CH3), 56.8 (CH3). MS (EI, m/z (%)): 501/504 (M+/M+2+, 0.16/0.10), 274 (29), 165 (95), 106 (100), 78 (99.7), 51 (99.1). Anal. calcd. for
C23H20ClN3O6S: C, 55.04; H, 4.02; N, 8.37; S, 6.39. Found: C, 54.99; H, 4.07; N, 8.39; S, 6.52.
(E)-N’-Isonicotinoyl-2,4-dimethoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonohydrazide (20c)
White solid; 46% yield; m.p. 195–197 °C. FTIR (ATR) (cm−1): 3161 (N-H), 2987 (C-H Ar), 1677 (C=O), 1650 (C=O), 1598 (C=C), 1157 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (s, 1H, S-NH), 9.72 (s, 1H, C-NH), 8.67 (d, J = 6.1 Hz, 2H, H2C), 7.97 (s, 1H, H6A), 7.60–7.54 (m, 4H, H3C and H2D), 7.49 (d, J = 15.8 Hz, 1H, Hβ), 7.41 (d, J = 15.8 Hz, 1H, Hα), 7.23 (d, J = 7.7 Hz, 2H, H3D), 6.87 (s, 1H, H3A), 4.05–4.01 (m, 6H, 2A-OCH3 and 4A-OCH3), 2.33 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.6 (C), 164.1 (C), 163.8 (C), 162.0 (C), 150.3 (CH), 142.5 (CH), 140.5 (C), 139.0 (C), 132.8 (CH), 131.9 (C), 129.6 (CH), 128.5 (CH), 125.6 (CH), 121.2 (CH), 119.6 (C), 119.2 (C), 97.3 (CH), 57.1 (CH3), 56.8 (CH3), 21.0 (CH3). MS (EI, m/z (%)): 481 (M+, 0.6), 282 (25), 123 (66), 106 (100), 78 (97), 51 (86). Anal. calcd. for C24H23N3O6S: C, 59.86; H, 4.81; N, 8.73; S, 6.66. Found: C, 59.81; H, 4.72; N, 8.79; S, 6.74.
(E)-N’-Isonicotinoyl-2,4-dimethoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonohydrazide (20d)
Pale yellow solid; 50% yield; m.p. 214–217 °C. FTIR (ATR) (cm−1): 3327 (N-H), 3231 (N-H), 2986 (C-H Ar), 1699 (C=O), 1651 (C=O), 1593 (C=C), 1223 (C-O-C), 1150 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (s, 1H, S-NH), 9.71 (s, 1H, C-NH), 8.68 (d, J = 5.5 Hz, 2H, H2C), 7.96 (s, 1H, H6A), 7.63 (d, J = 8.3 Hz, 2H, H2D), 7.58 (d, J = 5.5 Hz, 2H, H3C), 7.48 (d, J = 15.8 Hz, 1H, Hβ), 7.33 (d, J = 15.8 Hz, 1H, Hα), 6.98 (d, J = 8.3 Hz, 2H, H3D), 6.87 (s, 1H, H3A), 4.05–4.00 (m, 6H, 2A-OCH3 and 4A-OCH3), 3.80 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.5 (C), 164.1 (C), 163.7 (C), 161.9 (C), 161.2 (C), 150.3 (CH), 142.5 (CH), 139.0 (C), 132.7 (CH), 130.3 (CH), 127.2 (C), 124.2 (CH), 121.2 (CH), 119.8 (C), 119.1 (C), 114.5 (CH), 97.3 (CH), 57.0 (CH3), 56.7 (CH3), 55.3 (CH3). MS (EI, m/z (%)): 497 (M+, 3), 282 (96), 139 (100), 111 (50), 75 (13). Anal. calcd. for C24H23N3O7S: C, 57.94; H, 4.66; N, 8.45; S, 6.45. Found: C, 57.85; H, 4.60; N, 8.55; S, 6.32.
(E)-N’-Isonicotinoyl-2,4-dimethoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonohydrazide (20e)
Yellow solid; 42% yield; m.p. 162–165 °C. FTIR (ATR) (cm−1): 3222 (N-H), 1700 (C=O), 3002 (C-H Ar) 1678 (C=O), 1597 (C=C), 1276 (C-O-C), 1150 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.94 (s, 1H, S-NH), 9.72 (s, 1H, C-NH), 8.67 (d, J = 4.2 Hz, 2H, H2C), 7.96 (s, 1H, H6A), 7.58 (d, J = 5.0 Hz, 2H, H3C), 7.52–7.38 (m, 2H, Hα and Hβ), 7.04 (s, 2H, H2D), 6.88 (s, 1H, H3A), 4.02 (s, 6H, 2A-OCH3 and 4A-OCH3), 3.82 (s, 6H, 3D-OCH3), 3.70 (s, 3H, 4D-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 189.2 (C), 164.6 (C), 164.3 (C), 162.5(C), 153.6 (C), 150.8 (CH), 143.2 (CH), 140.1 (C), 139.5 (C), 133.1 (CH), 130.7 (C), 126.6 (CH), 121.7 (CH), 120.3 (C), 119.7 (C), 106.5 (CH), 97.8 (CH), 60.6 (CH3), 57.6 (CH3), 57.2 (CH3), 56.5 (CH3). MS (EI, m/z (%)): 557 (M+, 0.4), 358 (78), 165 (62), 106 (100), 78 (82), 51 (75). Anal. calcd. for C26H27N3O9S: C, 56.01; H, 4.88; N, 7.54; S, 5.75. Found: C, 55.97; H, 4.82; N, 7.64; S, 5.73.
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (20f)
Yellow crystals; 46% yield; m.p. 150–152 °C. FTIR (ATR) (cm−1): 3227 (N-H), 2988 (C-H Ar), 1649 (C=O), 1593 (C=C), 1242 (C-O-C), 1154 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.92 (d, J = 3.4 Hz, 1H, S-NH), 9.68 (d, J = 3.4 Hz, 1H, C-NH), 8.67 (d, J = 5.3 Hz, 2H, H2C), 7.95 (s, 1H, H6A), 7.58 (d, J = 5.3 Hz, 2H, H3C), 7.44 (d, J = 16.5 Hz, 1H, Hβ), 7.36–7.29 (m, 2H, Hα and H2D), 7.15 (d, J = 8.0 Hz, 1H, H6D), 6.94 (d, J = 8.0 Hz, 1H, H5D), 6.86 (s, 1H, H3A), 6.09 (s, 2H, CH2), 4.06–3.99 (m, 6H, 2A-OCH3 and 4A-OCH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 188.5 (C), 164.1 (C), 163.7 (C), 161.9 (C), 150.3 (CH),149.4 (C), 148.0 (C), 142.5 (CH), 139.0 (C), 132.6 (CH), 129.0 (C), 125.1 (CH), 124.7 (CH), 121.2 (CH), 119.8 (C), 119.1 (C), 108.5 (CH), 106.7 (CH), 101.6 (CH2), 97.3 (CH), 57.0 (CH3), 56.7 (CH3). MS (EI, m/z (%)): 511 (M+, 0.2), 312 (43), 135 (100), 106 (82), 78 (70), 51 (70). Anal. calcd. for C24H21N3O8S: C, 56.35; H, 4.14; N, 8.22; S, 6.27. Found: C, 56.43; H, 4.19; N, 8.20; S, 6.14. Crystals suitable for single-crystal X-ray diffraction were obtained from ethanolic solution, and the crystal data for 20f were deposited at CCDC with reference CCDC 2191613.
3.2.11. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 22a-d
A mixture of the respective chalcone (16-18)b or 21b (0.27 mmol) and hydrazine monohydrate (5.3 mmol) in ethanol (1.0 mL) was heated at reflux for 0.67 h. The reaction mixture was cooled, then 1.0 mL of formic acid was slowly added and stirred overnight. The solid formed was filtered and washed with small amounts of water and ethanol to afford compounds 22a-d without further purification.
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxybenzenesulfonamide (22a)
White solid; 74% yield; m.p. 230–231 °C. FTIR (ATR) (cm−1): 3332 and 3230 (N-H), 3109 (C-H Ar), 2849 (-(C=O)-H), 1653 (C=O), 1600 (C=N), 1148 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 (s, 1H, CHO), 8.19 (d, J = 2.3 Hz, 1H, H6A), 7.87 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 7.39 (d, J = 8.3 Hz, 2H, H2D), 7.32–7.17 (m, 5H, NH2, H3A and H3D), 5.53 (dd, J = 11.6, 4.8 Hz, 1H, HX), 3.98–3.87 (m, 4H, HM and OCH3), 3.17 (dd, J = 18.1, 4.8 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 160.0 (CHO), 157.8 (C), 155.4 (C), 140.3 (C), 132.8 (CH), 132.3 (C), 131.9 (C), 128.9 (CH), 128.0 (CH), 125.9 (CH), 122.7 (C), 113.3 (CH), 58.3 (CH), 56.7 (CH3), 42.3 (CH2). MS (EI, m/z (%)): 393/395 (M+/M+2+, 94/38), 254 (48), 228 (66), 213 (75), 57 (100), 43 (87). Anal. calcd. for C17H16ClN3O4S: C, 51.84; H, 4.09; N, 10.67; S, 8.14. Found: C, 51.99; H, 4.00; N, 10.75; S, 8.12.
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (22b)
White solid; 81% yield; m.p. 241–243 °C. FTIR (ATR) (cm−1): 3192 (N-H), 3076 (C-H Ar), 1668 (C=O), 1604 (C=N), 1595 (C=C), 1159 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.39 (s, 1H, NH), 8.90 (s, 1H, CHO), 8.32 (d, J = 2.7 Hz, 1H, H2B), 8.25–8.18 (m, 2H, H6A and H6B), 7.87 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 7.51 (dt, J = 8.5, 2.7 Hz, 1H, H4B), 7.39 (d, J = 8.2 Hz, 2H, H3D), 7.30–7.24 (m, 4H, H3A, H5B and H2D), 5.52 (dd, J = 11.9, 5.2 Hz, 1H, HX), 3.92 (s, 3H, OCH3), 3.87 (dd, J = 18.1, 11.9 Hz, 1H, HM), 3.21 (dd, J = 18.1, 5.2 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 159.8 (CHO), 157.6 (C), 154.7 (C), 145.2 (CH), 141.6 (CH), 140.1 (C), 134.3 (C), 133.9 (CH), 132.0 (C), 128.6 (CH), 127.84 (CH), 127.80 (CH), 127.2 (CH), 126.8 (C), 123.9 (CH), 122.9 (C), 113.3 (CH), 58.1 (CH), 56.6 (CH3), 41.9 (CH2). MS (EI, m/z (%)): 470/472 (M+/M+2+, 15/6), 359 (25), 331 (51), 153 (24), 94 (100), 39 (72). Anal. calcd. for C22H19ClN4O4S: C, 56.11; H, 4.07; N, 11.90; S, 6.81. Found: C, 56.01; H, 4.22; N, 11.98; S, 6.73.
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2,4-dimethoxybenzenesulfonamide (22c)
White solid; 86% yield; m.p. 269–271 °C. FTIR (ATR) (cm−1): 3319 and 3219 (N-H), 3122 (C-H Ar), 2847 (-(C=O)-H), 1651 (C=O), 1602 (C=N), 1590 (C=C), 1146 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.89 (s, 1H, CHO), 8.25 (s, 1H, H6A), 7.40 (d, J = 8.5 Hz, 2H, H3D), 7.24 (d, J = 8.5 Hz, 2H, H2D), 7.09 (s, 2H, NH2), 6.82 (s, 1H, H3A), 5.48 (dd, J = 11.7, 4.8 Hz, 1H, HX), 3.99 (s, 3H, 4A-CH3), 3.97–3.88 (m, 4H, 2A-CH3 and HM), 3.15 (dd, J = 18.7, 4.8 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 162.2 (C), 159.7 (CHO), 159.2 (C), 154.3 (C), 140.4 (C), 132.0 (C), 128.8 (CH), 128.2 (CH), 127.6 (CH), 124.2 (C), 110.8 (C), 97.3 (CH), 57.7 (CH), 56.6 (CH3), 56.5 (CH3), 45.3 (CH2). MS (EI, m/z (%)): 423/425 (M+/M+2+, 100/50), 284 (69), 243 (83), 162 (56), 89 (50), 77 (43). Anal. calcd. for C18H18ClN3O5S: C, 51.01; H, 4.28; N, 9.91; S, 7.56. Found: C, 50.99; H, 4.35; N, 9.97; S, 7.39.
N,N-bis(2-Chloroethyl)-5-(5-(4-chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2,4-dimethoxybenzenesulfonamide (22d)
White solid; 91% yield; m.p. 222–224 °C. FTIR (ATR) (cm−1): 2971 (C-H Ar), 2844 (-(C=O)-H), 1665 (C=O), 1605 (C=N), 1558 (C=C), 1152 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.93 (s, 1H, CHO), 8.47 (s, 1H, H6A), 7.31 (d, J = 8.4 Hz, 2H, H3D), 7.19 (d, J = 8.4 Hz, 2H, H2D), 6.49 (s, 1H, H3A), 5.43 (dd, J = 11.8, 4.9 Hz, 1H, HX), 4.00 (s, 3H, 4A-CH3), 3.93–3.83 (m, 4H, 2A-CH3 and HM), 3.64 (s, 8H, N-CH2 and Cl-CH2), 3.26 (dd, J = 18.6, 4.9 Hz, 1H, HA). 13C NMR (101 MHz, CDCl3) δ ppm 163.2 (C), 160.3 (CHO), 159.8 (C), 153.7 (C), 139.4 (C), 133.8 (C), 132.8 (CH), 129.3 (CH), 127.3 (CH), 120.6 (C), 112.7 (C), 95.9 (CH), 58.5 (CH), 56.5 (CH3), 56.2 (CH3), 51.2 (CH2), 45.6 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 549 (M+, 100), 343 (37), 315 (37), 178 (32), 42 (36). Anal. calcd. for C22H24Cl3N3O5S: C, 48.14; H, 4.41; N, 7.66; S, 5.84. Found: C, 48.02; H, 4.52; N, 7.70; S, 5.71.
3.2.12. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 23a-d
A mixture of the corresponding chalcone (16-18)b or 21b (0.27 mmol) and hydrazine monohydrate (5.3 mmol) in ethanol (1.0 mL) was heated at reflux for 0.67 h. The reaction mixture was cooled, then 1.0 mL of acetic anhydride was carefully added and stirred overnight. The product was filtered and washed with small portions of water and ethanol. Compounds 23a-d did not require further purification.
5-(1-Acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxybenzenesulfonamide (23a)
White solid; 71% yield; m.p. 229–232 °C. FTIR (ATR) (cm−1): 3384 and 3281 (N-H), 3049 (C-H Ar), 1675 (C=O), 1605 (C=N), 1591 (C=C), 1153 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.17 (d, J = 2.4 Hz, 1H, H6A), 7.87 (dd, J = 8.7, 2.4 Hz, 1H, H4A), 7.37 (d, J = 8.2 Hz, 2H, H2D), 7.27 (d, J = 8.7 Hz, 1H, H3A), 7.24–7.16 (m, 4H, NH2 and H3D), 5.53 (dd, J = 11.6, 4.3 Hz, 1H, HX), 3.94 (s, 3H, OCH3), 3.85 (dd, J = 18.0, 11.6 Hz, 1H, HM), 3.09 (dd, J = 18.0, 4.3 Hz, 1H, HA), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 167.8 (C), 157.7 (C), 153.5 (C), 141.4 (C), 132.6 (CH), 132.0 (C), 131.9 (C), 128.8 (CH), 127.7 (CH), 125.8 (CH), 123.0 (C), 113.2 (CH), 59.2 (CH), 56.7 (CH3), 42.1 (CH2), 21.9 (CH3). MS (EI, m/z (%)): 407/409 (M+/M+2+, 34/13), 365 (100), 254 (23), 213 (23), 57 (25), 43 (79). Anal. calcd. for C18H18ClN3O4S: C, 53.01; H, 4.45; N, 10.30; S, 7.86. Found: C, 52.98; H, 4.33; N, 10.51; S, 7.92.
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (23b)
White solid; 80% yield; m.p. 265–268 °C. FTIR (ATR) (cm−1): 3196 (N-H), 3027 (C-H Ar), 1646 (C=O), 1606 (C=N), 1596 (C=C), 1156 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.42 (s, 1H, NH), 8.32 (s, 1H, H2B), 8.24–8.16 (m, 2H, H6A and H6B), 7.87 (d, J = 8.7 Hz, 1H, H4A), 7.51 (d, J = 8.3 Hz, 1H, H4B), 7.36 (d, J = 8.0 Hz, 2H, H3D), 7.30–7.24 (m, 2H, H3A and H5B), 7.20 (d, J = 8.0 Hz, 2H, H2D), 5.51 (dd, J = 12.0, 4.8 Hz, 1H, HX), 3.90 (s, 3H, OCH3), 3.82 (dd, J = 18.1, 12.0 Hz, 1H, HM), 3.12 (dd, J = 18.1, 4.8 Hz, 1H, HA), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 167.5 (C), 157.5 (C), 152.9 (C), 145.2 (CH), 141.6 (CH), 141.2 (C), 134.4 (C), 133.8 (CH), 131.7 (C), 128.6 (CH), 127.7 (CH), 127.6 (CH), 127.3 (CH), 126.9 (C), 124.0 (CH), 123.3 (C), 113.4 (CH), 59.1 (CH), 56.6 (CH3), 41.9 (CH2), 21.7 (CH3). MS (EI, m/z (%)): 484/485 (M+/M+2+, 10/4), 373 (17), 331 (36), 153 (18), 94 (56), 43 (100). Anal. calcd. for C23H21ClN4O4S: C, 56.96; H, 4.36; N, 11.55; S, 6.61. Found: C, 57.06; H, 4.25; N, 11.30; S, 6.69.
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-2,4-dimethoxybenzenesulfonamide (23c)
White solid; 86% yield; m.p. 159–161 °C. FTIR (ATR) (cm−1): 3441 and 3352 (N-H), 3021 (C-H Ar), 1654 (C=O), 1602 (C=N), 1582 (C=C), 1157 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (s, 1H, H6A), 7.38 (d, J = 8.5 Hz, 2H, H3D), 7.19 (d, J = 8.5 Hz, 2H, H2D), 7.08 (s, 2H, NH2), 6.82 (s, 1H, H3D), 5.48 (dd, J = 11.8, 4.4 Hz, 1H, HX), 3.98 (s, 3H, 4A-OCH3), 3.93–3.85 (m, 4H, 2A-OCH3 and HM), 3.08 (dd, J = 18.6, 4.4 Hz, 1H, HA), 2.26 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 167.3 (C), 162.1 (C), 159.0 (C), 152.6 (C), 141.5 (C), 131.7 (C), 128.6 (CH), 128.2 (CH), 127.4 (CH), 124.1 (C), 111.2 (C), 97.3 (CH), 58.7 (CH), 56.6 (CH3), 56.5 (CH3), 45.1 (CH2), 21.7 (CH3). MS (EI, m/z (%)): 437/439 (M+/M+2+, 27/10), 395 (45), 284 (13), 243 (14), 57 (27), 43 (100). Anal. calcd. for C19H20ClN3O5S: C, 52.12; H, 4.60; N, 9.60; S, 7.32. Found: C, 52.25; H, 4.51; N, 9.72; S, 7.19.
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-N,N-bis(2-chloroethyl)-2,4-dimethoxybenzenesulfonamide (23d)
White solid; 90% yield; m.p. 196–198 °C. FTIR (ATR) (cm−1): 2976 (C-H Ar), 1643 (C=O), 1600 (C=N), 1576 (C=C), 1140 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.45 (s, 1H, H6A), 7.28 (d, J = 8.1 Hz, 2H H3D), 7.16 (d, J = 8.1 Hz, 2H, H2D), 6.48 (s, 1H, H3A), 5.49 (dd, J = 11.9, 4.7 Hz, 1H, HX), 4.00 (s, 3H, 4A-OCH3), 3.91 (s, 3H, 2A-OCH3), 3.82 (dd, J = 18.5, 11.9 Hz, 1H, HM), 3.65 (s, 8H, N-CH2 and Cl-CH2), 3.20 (dd, J = 18.5, 4.7 Hz, 1H, HA), 2.40 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 169.1 (C), 163.1 (C), 159.6 (C), 151.9 (C), 140.7 (C), 133.4 (C), 132.7 (CH), 129.1 (CH), 127.2 (CH), 120.5 (C), 113.3 (C), 95.9 (CH), 59.5 (CH), 56.5 (CH3), 56.2 (CH3), 51.3 (CH2), 45.4 (CH2), 42.4 (CH2), 22.1 (CH3). MS (EI, m/z (%)): 563 (7), 519 (12), 243 (8), 71 (17), 43 (100). Anal. calcd. for C23H26Cl3N3O5S: C, 49.08; H, 4.66; N, 7.47; S, 5.70. Found: C, 49.15; H, 4.39; N, 7.60; S, 5.48.
3.2.13. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 24a-b
A mixture of the respective chalcone 14a-b (0.21 mmol) and hydrazine monohydrate (4.2 mmol) in ethanol (1.0 mL) was stirred at room temperature for 3 h, CCD analysis revealed consumption of the precursor and formation of several products. Subsequently, 1.0 mL of formic acid was slowly added and stirred at room temperature for an additional 4 h; water was added to the mixture and the precipitate formed was filtered and washed with water. Both products were purified by CC using silica gel as stationary phase, and CHCl3:MeOH (15:1) mixture was employed as mobile phase for compound 24a and a DCM:MeOH (20:1) mixture for compound 24b.
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (24a)
Pale yellow solid; 73% yield; m.p. 156–158 °C. FTIR (ATR) (cm−1): 3214 (N-H), 3038 (C-H Ar), 2841 (-(C=O)-H), 1655 (C=O), 1649 (C=O), 1603 (C=N), 1163 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.95 (s, 1H, S-NH), 9.88 (s, 1H, C-NH), 8.87 (s, 1H, CHO), 8.69 (d, J = 6.1 Hz, 2H, H2C), 8.19 (d, J = 2.3 Hz, 1H, H6A), 7.89 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 7.58 (d, J = 6.1 Hz, 2H, H3C), 7.38 (d, J = 8.5 Hz, 2H, H3D), 7.30 (d, J = 8.7 Hz, 1H, H3A), 7.23 (d, J = 8.5 Hz, 2H, H2D), 5.51 (dd, J = 11.8, 5.0 Hz, 1H, HX), 3.96 (s, 3H, OCH3), 3.88 (dd, J = 18.1, 11.8 Hz, 1H, HM), 3.18 (dd, J = 18.1, 5.0 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 164.1 (C), 159.7 (CHO), 159.0 (C), 154.8 (C), 150.4 (CH), 140.1 (C), 138.9 (C), 133.7 (CH), 132.0 (C), 128.6 (CH), 127.8 (CH), 127.7 (C), 127.5 (CH), 122.2 (C), 121.2 (CH), 113.4 (CH), 58.0 (CH), 56.8 (CH3), 42.0 (CH2). MS (EI, m/z (%)): 513/515 (M+/M+2+, 14/6), 314 (43), 123 (43), 106 (95), 85 (100), 47 (78). Anal. calcd. for C23H20ClN5O5S: C, 53.75; H, 3.92; N, 13.63; S, 6.24. Found: C, 53.96; H, 3.85; N, 13.41; S, 6.20.
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (24b)
White solid; 56% yield; m.p. 166–168 °C. FTIR (ATR) (cm−1): 3166 (N-H), 2945 (C-H Ar), 2829 (-(C=O)-H), 1654 (C=O), 1647 (C=O), 1601 (C=N), 1167 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.95 (s, 1H, S-NH), 9.74 (s, 1H, C-NH), 8.86 (s, 1H, CHO), 8.70 (d, J = 5.4 Hz, 2H, H2C), 8.22 (s, 1H, H6A), 7.59 (d, J = 5.4 Hz, 2H, H3C), 7.37 (d, J = 8.3 Hz, 2H, H3D), 7.20 (d, J = 8.3 Hz, 2H, H2D), 6.81 (s, 1H, H3A), 5.45 (dd, J = 11.6, 4.5 Hz, 1H, HX), 3.98 (s, 3H, 4A-OCH3), 3.93–3.85 (m, 4H, 2A-OCH3 and HM), 3.10 (dd, J = 18.7, 4.5 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 164.1 (C), 163.2 (C), 160.8 (C), 159.7 (CHO), 154.0 (CH), 150.4 (C), 140.4 (C), 139.0 (C), 132.0 (C), 130.3 (CH), 128.7 (CH), 127.6 (CH), 121.3 (CH), 119.5 (C), 110.8 (C), 97.5 (CH), 57.7 (CH), 57.0 (CH3), 56.6 (CH3), 45.2 (CH2). MS (EI, m/z (%)): 544 (M+, 0.1), 531 (48), 460 (53), 341 (51), 43 (100). Anal. calcd. for C24H22ClN5O6S: C, 52.99; H, 4.08; N, 12.87; S, 5.89. Found: C, 53.05; H, 4.13; N, 12.66; S, 5.71.
3.2.14. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 25a-b
A mixture of the respective chalcone 14a-b (0.21 mmol) and hydrazine monohydrate (4.2 mmol) in ethanol (1.0 mL) was stirred at room temperature for 3 h. Next, 1.0 mL of acetic anhydride was carefully added and stirred at room temperature for additional 4 h; water was added to the reaction mixture and the precipitate was filtered and washed with water. The products were purified by CC on silica gel, using a CHCl3:MeOH (15:1) mixture as eluent for compound 25a and a DCM:MeOH (20:1) mixture for compound 25b.
5-(1-Acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (25a)
Beige solid; 51% yield; m.p. 146–148 °C. FTIR (ATR) (cm−1): 3207 (N-H), 3014 (C-H Ar), 1655 (C=O), 1638 (C=O), 1603 (C=N), 1164 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.95 (s, 1H, S-NH), 9.87 (s, 1H, C-NH), 8.69 (d, J = 5.7 Hz, 2H, H2C), 8.16 (d, J = 2.3 Hz, 1H, H6A), 7.89 (dd, J = 8.7, 2.3 Hz, 1H, H4A), 7.59 (d, J = 5.7 Hz, 2H, H3C), 7.35 (d, J = 8.1 Hz, 2H, H3D), 7.29 (d, J = 8.7 Hz, 1H, H3A), 7.18 (d, J = 8.1 Hz, 2H, H2D), 5.51 (dd, J = 11.9, 4.7 Hz, 1H, HX), 3.96 (s, 3H, OCH3), 3.83 (dd, J = 18.1, 11.9 Hz, 1H, HM), 3.10 (dd, J = 18.1, 4.7 Hz, 1H, HA), 2.27 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 167.4 (C), 164.1 (C), 158.8 (C), 153.0 (C), 150.3 (CH), 141.2 (C), 138.9 (C), 133.5 (CH), 131.7 (C), 128.5 (CH), 127.8 (C), 127.5 (CH), 127.4 (CH), 122.6 (C), 121.2 (CH), 113.4 (CH), 58.9 (CH), 56.8 (CH), 41.9 (CH2), 21.7 (CH3). MS (EI, m/z (%)): 527/529 (M+/M+2+, 0.5/0.2), 328 (39), 286 (64), 106 (64), 78 (65), 43 (100). Anal. calcd. for C24H22ClN5O5S: C, 54.60; H, 4.20; N, 13.26; S, 6.07. Found: C, 54.79; H, 4.07; N, 13.35; S, 6.14.
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (25b)
White solid; 52% yield; m.p. 159–160 °C. FTIR (ATR) (cm−1): 3235 (N-H), 2927 (C-H Ar), 1675 (C=O), 1626 (C=O), 1600 (C=N), 1162 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.97 (s, 1H, S-NH), 9.77 (s, 1H, C-NH), 8.73–8.69 (m, 2H, H2C), 8.22 (s, 1H, H6A), 7.63–7.59 (m, 2H, H3C), 7.37 (d, J = 8.3 Hz, 2H, H3D), 7.17 (d, J = 8.3 Hz, 2H, H2D), 6.82 (s, 1H, H3A), 5.47 (dd, J = 11.9, 4.5 Hz, 1H, HX), 3.99 (s, 3H, 4A-OCH3), 3.93–3.82 (m, 4H, 2A-OCH3 and HM), 3.05 (dd, J = 18.7, 4.5 Hz, 1H HA), 2.26 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ ppm 167.3 (C), 164.1 (C), 163.1 (C), 160.7 (C), 152.3 (C), 150.4 (CH), 141.5 (C), 139.1 (C), 131.7 (C), 130.3 (CH), 128.6 (CH), 127.4 (CH), 121.3 (CH), 119.4 (C), 111.2 (C), 97.4 (CH), 58.7 (CH), 56.9 (CH3), 56.6 (CH3), 45.1 (CH2), 21.7 (CH3). MS (EI, m/z (%)): 558 (M+, 0.1), 358 (60), 316 (55), 106 (53), 43 (100). Anal. calcd. for C25H24ClN5O6S: C, 53.81; H, 4.34; N, 12.55; S, 5.75. Found: C, 53.69; H, 4.30; N, 12.32; S, 5.93.
3.2.15. General Procedure for the Synthesis of Carbothioamide–Sulfonamide Hybrids 27a-f
To a mixture of the corresponding chalcone (16-21)b (0.24 mmol) and thiosemicarbazide 26 (0.26 mmol) in ethanol (1.0 mL), 3 drops of 50% w/v NaOH were added and stirred at 60 °C. After completion of the reaction, water was added, the medium was neutralized with aqueous 10% v/v HCl, and the solid formed was filtered and washed with water. Purification of the products was carried out by CC on silica gel, using a CHCl3:MeOH (15:1) mixture as the mobile phase.
5-(4-Chlorophenyl)-3-(4-methoxy-3-sulfamoylphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27a)
Beige solid; 36% yield; m.p. 206–208 °C. FTIR (ATR) (cm−1): 3503 and 3384 (N-H), 3326 (N-H), 3080 (C-H Ar), 3256 (N-H), 1604 (C=N), 1567 (C=C), 1162 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.20 (d, J = 2.3 Hz, 1H, H6A), 8.07–8.03 (m, 2H, C-NH and H4A), 7.94 (s, 1H, C-NH), 7.39 (d, J = 8.1 Hz, 2H, H3D), 7.31 (d, J = 8.7 Hz, 1H, H3A), 7.21–7.14 (m, 4H, H2D and NH2), 5.95 (dd, J = 11.4, 3.5 Hz, 1H, HX), 4.01–3.88 (m, 4H, HM and OCH3), 3.14 (dd, J = 17.9, 3.5 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ 176.0 (C), 157.7 (C), 154.0 (C), 141.9 (C), 132.6 (CH), 131.7 (C), 131.4 (C), 128.5 (CH), 127.3 (CH), 126.3 (CH), 122.6 (C), 113.1 (CH), 62.4 (CH), 56.5 (CH3), 42.2 (CH2). MS (EI, m/z (%)): 424/426 (M+/M+2+, 5/2), 365 (16), 211 (22), 71 (54), 57 (100), 43 (85). Anal. calcd. for C17H17ClN4O3S2: C, 48.05; H, 4.03; N, 13.19; S, 15.09. Found: C, 47.96; H, 4.12; N, 13.27; S, 15.11.
5-(4-Chlorophenyl)-3-(4-methoxy-3-(N-(pyridin-3-yl)sulfamoyl)phenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27b)
Pale yellow solid; 33% yield; m.p. 236–238 °C. FTIR (ATR) (cm−1): 3433 and 3352 (N-H), 3069 (C-H Ar), 3269 (N-H), 1601 (C=N), 1584 (C=C), 1162 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.36 (s, 1H, S-NH), 8.32 (s, 1H, H2B), 8.26 (s, 1H, H6A), 8.20 (d, J = 4.8 Hz, 1H, H6B), 8.05–7.97 (m, 3H, H4A and C-NH2), 7.50 (d, J = 8.3 Hz, 1H, H4B), 7.36 (d, J = 8.1 Hz, 2H, H3D), 7.28–7.23 (m, 2H, H3A and H5B), 7.14 (d, J = 8.1 Hz, 2H, H2D), 5.89 (dd, J = 11.2, 3.7 Hz, 1H, HX), 3.94–3.84 (m, 4H, HM and OCH3), 3.13 (dd, J = 18.0, 3.7 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 176.1 (C), 157.7 (C), 153.5 (C), 145.0 (CH), 141.9 (C), 141.4 (CH), 134.4 (C), 134.1 (CH), 131.4 (C), 128.5 (CH), 128.4 (CH), 127.4 (CH), 126.9 (CH), 126.8 (C), 123.9 (CH), 123.1 (C), 113.3 (CH), 62.4 (CH), 56.6 (CH3), 42.1 (CH2). MS (EI, m/z (%)): 502/504 (M+/M+2+, 2/1), 468 (13), 441 (74), 413 (25), 331 (100), 290 (26). Anal. calcd. for C22H20ClN5O3S2: C, 52.64; H, 4.02; N, 13.95; S, 12.77. Found: C, 52.37; H, 4.15; N, 14.09; S, 12.48.
5-(4-Chlorophenyl)-3-(3-((2-isonicotinoylhydrazineyl)sulfonyl)-4-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27c)
Pale yellow solid; 35% yield; m.p. 216–218 °C. FTIR (ATR) (cm−1): 3459 and 3339 (N-H), 3237 (N-H), 3082 (C-H Ar), 1689 (C=O), 1604 (C=N), 1584 (C=C), 1166 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (s, 1H, S-NH), 9.85 (s, 1H, C-NH), 8.67 (d, J = 5.2 Hz, 2H, H2C), 8.21 (d, J = 2.2 Hz, 1H, H6A), 8.07–7.92 (m, 3H, H4A and NH2), 7.58 (d, J = 5.2 Hz, 2H, H3C), 7.34 (d, J = 8.1 Hz, 2H, H3D), 7.29 (d, J = 8.8 Hz, 1H, H3A), 7.11 (d, J = 8.1 Hz, 2H, H2D), 5.89 (dd, J = 11.6, 3.5 Hz, 1H, HX), 3.97 (s, 3H, OCH3), 3.88 (dd, J = 18.1, 11.6 Hz, 1H, HM), 3.10 (dd, J = 18.1, 3.5 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 176.0 (C), 164.2 (C), 159.0 (C), 153.8 (C), 150.3 (CH), 141.9 (C), 138.9 (C), 133.9 (CH), 131.4 (C), 128.4 (CH), 127.95 (CH), 127.87 (C), 127.3 (CH), 122.4 (C), 121.3 (CH), 113.4 (CH), 62.4 (CH), 56.8 (CH3), 42.2 (CH2). MS (EI, m/z (%)): 545 (M+, 0.1), 211 (22), 106 (49), 78 (60), 60 (82), 43 (100). Anal. calcd. for C23H21ClN6O4S2: C, 50.69; H, 3.88; N, 15.42; S, 11.76. Found: C, 50.80; H, 3.95; N, 15.36; S, 11.61.
5-(4-Chlorophenyl)-3-(2,4-dimethoxy-5-sulfamoylphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27d)
Pale yellow solid; 34% yield; m.p. 223–225 °C. FTIR (ATR) (cm−1): 3431 and 3255 (N-H), 3067 (C-H Ar), 1601 (C=N), 1576 (C=C), 1151 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.30 (s, 1H, H6A), 8.00 (s, 1H, C-NH), 7.65 (s, 1H, C-NH), 7.37 (d, J = 8.2 Hz, 2H, H3D), 7.14 (d, J = 8.2 Hz, 2H, H2D), 7.05 (s, 2H, S-NH2), 6.80 (s, 1H, H3A), 5.87 (dd, J = 11.4, 3.2 Hz, 1H, HX), 4.02–3.93 (m, 4H, 4A-CH3 and HM), 3.91 (s, 3H, 2A-CH3), 3.09 (dd, J = 18.7, 3.2 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ ppm 175.8 (C), 162.3 (C), 159.2 (C), 153.7 (C), 142.0 (C), 131.4 (C), 128.53 (CH), 128.47 (CH), 127.2 (CH), 124.3 (C), 110.8 (C), 97.2 (CH), 62.1 (CH), 56.6 (CH3), 56.5 (CH3), 45.5 (CH2). MS (EI, m/z (%)): 454/456 (M+/M+2+, 6/3), 395 (17), 127 16), 85 (22), 60 (51), 43 (100). Anal. calcd. for C18H19ClN4O4S2: C, 47.52; H, 4.21; N, 12.32; S, 14.09. Found: C, 47.36; H, 4.11; N, 12.49; S, 14.07.
3-(5-(N,N-bis(2-Chloroethyl)sulfamoyl)-2,4-dimethoxyphenyl)-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27e)
Beige solid; 34% yield; m.p. 196–198 °C. FTIR (ATR) (cm−1): 3405 and 3254 (N-H), 3152 (C-H Ar), 1595 (C=N), 1579 (C=C), 1148 (S=O). 1H NMR (400 MHz, CDCl3) δ ppm 8.46 (s, 1H, H6A), 7.30 (d, J = 8.4 Hz, 2H, H3D), 7.20–7.05 (m, 3H, H2D and C-NH), 6.47 (s, 1H, H3A), 6.08 (s, 1H, C-NH), 5.94 (dd, J = 11.5, 3.5 Hz, 1H, HX), 4.01 (s, 3H, 4A-OCH3), 3.96–3.87 (m, 4H, 2A-OCH3 and HM), 3.65 (s, 8H, N-CH2 and Cl-CH2), 3.25 (dd, J = 18.5, 3.5 Hz, 1H, HA). 13C NMR (101 MHz, CDCl3) δ ppm 176.7 (C), 163.5 (C), 160.0 (C), 154.0 (C), 140.6 (C), 133.4 (C), 132.7 (CH), 129.2 (CH), 127.1 (CH), 120.7 (C), 112.4 (C), 95.9 (CH), 63.0 (CH), 56.6 (CH3), 56.3 (CH3), 51.2 (CH2), 46.1 (CH2), 42.4 (CH2). MS (EI, m/z (%)): 580 (M+, 3), 211 (32), 102 (59), 50 (100), 42 (94). Anal. calcd. for C22H25Cl3N4O4S2: C, 45.56; H, 4.35; N, 9.66; S, 11.06. Found: C, 45.72; H, 4.21; N, 9.45; S, 11.24.
5-(4-Chlorophenyl)-3-(5-((2-isonicotinoylhydrazineyl)sulfonyl)-2,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27f)
Yellow solid; 60% yield; m.p. 180–182 °C. FTIR (ATR) (cm−1): FTIR (ATR) (cm−1): 3554 and 3449 (N-H), 3317 (N-H), 3154 (C-H Ar), 1677 (C=N), 1582 (C=C), 1159 (S=O). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.91 (s, 1H, S-NH), 9.69 (s, 1H, C-NH), 8.68 (d, J = 5.7 Hz, 2H, H2C), 8.32 (s, 1H, H6A), 8.00 (s, 1H, S=C-NH), 7.74 (s, 1H, S=C-NH), 7.59 (d, J = 5.7 Hz, 2H, H3C), 7.33 (d, J = 8.2 Hz, 2H, H3D), 7.10 (d, J = 8.2 Hz, 2H, H2D), 6.79 (s, 1H, H3A), 5.83 (dd, J = 11.4, 3.3 Hz, 1H, HX), 3.99 (s, 3H, 4A-OCH3), 3.94–3.86 (m, 4H, 2A-OCH3 and HM), 3.05 (dd, J = 18.6, 3.3 Hz, 1H, HA). 13C NMR (101 MHz, DMSO-d6) δ 175.8 (C), 164.2 (C), 163.2 (C), 160.8 (C), 153.3 (C), 150.3 (CH), 142.0 (C), 139.0 (C), 131.3 (C), 130.5 (CH), 128.4 (CH), 127.2 (CH), 121.3 (CH), 119.8 (C), 110.9 (C), 97.4 (CH), 62.0 (CH), 56.9 (CH3), 56.5 (CH3), 45.5 (CH2). MS (EI, m/z (%)): 575 (M+, 0.6), 473 (10), 315 (30), 106 (82), 43 (100). Anal. calcd. for C24H23ClN6O5S2: C, 50.13; H, 4.03; N, 14.61; S, 11.15. Found: C, 50.06; H, 4.24; N, 14.60; S, 11.39.
3.3. Anticancer Activity
All compounds selected by the US National Cancer Institute (NCI) were initially tested at a single dose (10−5 M) on the full panel of 60 cancer cell lines represented by leukemia, melanoma, lung, colon, central nervous system (CNS), ovarian, kidney, prostate and breast cancer. Only compounds that were able to satisfy the predetermined inhibition criteria against a minimum number of cell lines proceeded to five-dose assays (100, 10, 1.0, 1.0, 0.1 and 0.01 μM).
In these studies, human cancer cell lines were cultured in a RPMI-1640 medium containing 5% fetal bovine serum and 2 mM of L-glutamine. Following the protocol for a typical assay, cells were inoculated into 96-well plates and incubated at 37 °C, 5% CO2, 95% air, and 100% relative humidity for 24 h prior to the addition of the compounds to be tested. After 24 h, two plates of each cell line were fixed in situ with trichloroacetic acid (TCA) to represent a measure of the cell population for each cell line at the time of sample addition (Tz). Compounds were dissolved in DMSO to 400-fold the maximum desired concentration for analysis and subsequently diluted to twice the maximum desired concentration with medium containing 50 μg/mL gentamicin. An additional series of four dilutions at 10-fold concentration was performed to provide a total of five concentrations of the compound plus the control. Aliquots of 100 μL of these dilutions were taken and added to 96-well plates already containing 100 μL of the medium, resulting in the final required concentrations of the sample. The plates were incubated for 48 h at the same conditions. For adherent cells, the assay was terminated with the addition of cold TCA. Cells were fixed in situ by the slow addition of 50 μL of 50% (w/v) cold ATC (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant was discarded, and the plates were washed five times with tap water and air dried. A quantity of 100 μL of 0.4% (w/v) sulforhodamine B (SRB) solution in 1% acetic acid was added to each well, and each plate was incubated for 10 min at room temperature. Upon completion of staining, unbound pigment was removed by five washes with 1% acetic acid and the plates were air-dried. The stains were solubilized with 10 mM Trizma base and absorbance was recorded on an automated plate reader at a wavelength of 515 nm. Using the absorbance measurements [time zero (Tz), growth control in the absence of the sample and growth test in the presence of the sample at the five concentration levels (Ti)], the percent growth was calculated for each sample at the concentration levels. The percent growth was calculated as: [(Ti − Tz)/(C − Tz)] × 100 for concentrations for which Ti > Tz, and [(Ti − Tz)/Tz] × 100 for concentrations for which Ti < Tz. Two dose-response parameters were calculated for each compound. The 50% growth inhibition (GI50) was calculated from [(Ti − Tz)/(C − Tz)] × 100 = 50, which is the sample concentration at which the net increase in protein is 50% lower in treated cells (as measured by SRB staining) compared to the net increase in protein seen in control cells and the LC50 (sample concentration at which a 50% reduction in protein measured at the end of treatment compared to the beginning), indicating a net loss of cells; calculated from [(Ti − Tz)/Tz] × 100 = −50. Values were calculated for each of these two parameters if the activity level was reached; however, if the effect was not reached or was exceeded, the value for that parameter was expressed as greater or less than the maximum or minimum of the concentration tested [67].
3.4. Antituberculosis Activity
The agar dilution spot culture growth inhibition assay (SPOTi) [61] was performed to evaluate the minimum inhibitory concentration (MIC) values of the synthesized compounds against Mycobacterium bovis BCG and Mycobacterium tuberculosis H37Rv (ATCC 27294). A stock solution of 100 mg/mL in DMSO of each compound was prepared and then dilutions were made in DMSO until reaching concentrations of 10, 5, 1, 0.5, 0.1 and 0.01 mg/mL. Initially, each compound was evaluated at a concentration of 10 mg/L. The compounds that showed antitubercular activity at this concentration were evaluated at lower concentrations (5, 1, 0.5, 0.1 and 0.01 mg/L). Antituberculosis activity was assessed in a 24-well plate, and 2 μL of the DMSO dilution from each compound was dispensed into each well. Two mL of Middlebrook 7H10 culture medium (HiMedia, Mumbai, India) supplemented with glycerol 0.5% and ADNaCl (Albumin, Dextrose and Sodium Chloride) for M. bovis BCG, or with glycerol 0.5% with OADC (10% oleic acid, albumin, dextrose and catalase) for M. tuberculosis were added to each well to reach desired concentration of the compounds in solid medium. Dilutions of the bacterial cultures were prepared at a cell density of 106 CFU/mL in Middlebrook 7H9 from strains of M. bovis BCG and M. tuberculosis H37Rv grown for 4 weeks in supplemented Middlebrook 7H9 medium at a temperature of 37 °C. A volume of 2 μL of the inoculum was carefully dispensed into the middle of each well and the plates were incubated at 37 °C for 2–3 weeks. Isoniazid was included as a positive control at concentrations of 10, 1, 0.1, 0.1, 0.05 and 0.01 mg/L. The plates were carefully observed, and MIC values were recorded as the lowest concentration of compound that completely inhibited bacterial growth upon visual inspection [68]. The experiment was performed in triplicate.
Growth curves were performed for M. tuberculosis H37Rv in liquid medium Middlebrook 7H9 supplemented with glycerol 0.5% with OADC (10% oleic acid, albumin, dextrose and catalase). The compounds 20e and 20f were added from the start of the experiment at the desired concentration from the prepared dilutions in DMSO. The day of the start of the experiment and every 7 days until day 21, an aliquot of 1 mL was removed from the medium and optical density was measured at 600 nm using an equipment MultiskanTM FC Microplate Photometer. In addition, every day of measurement of optical density, an aliquot of the liquid culture was removed, ten-fold diluted to 10−4 with supplemented Middlebrook 7H9 medium, and each ten-fold dilution plated in Petri dishes by triplicate in supplemented Middlebrook 7H10 agar media without compounds or antibiotics. After 3 weeks of incubation the number of colonies were counted in each Petri dish.
Compounds displaying MIC values < 10 mg/L against M. tuberculosis H37Rv were also evaluated against six resistant strains of M. tuberculosis using the same agar dilution method: An orphan strain, a rifampicin-resistant strain (ATCC 35838), an isoniazid-resistant strain (ATCC 35822), and three multi-resistant strains (Haarlem, LAM9 SIT 42, Beijing). Levofloxacin was used as a positive control and DMSO as a negative control.
3.5. Cytotoxicity on Vero Cell Line
The VERO African green monkey kidney cell line (ATCC CCL-81) was cultured in Gibco RPMI Media 1640 culture medium, supplemented with 10% Fetal Bovine Serum (FBS), and 0.1% antibiotics (penicillin, streptomycin) 50 μg/mL. The cells were kept at 37 °C, 5% CO2 and 100% relative humidity, until obtaining a 70% confluence.
To evaluate the cytotoxic effect of the compounds, a cell viability test was performed, using the MTT reductase assay [69], which consisted of sowing in a plate of 96 wells, a cell density of 104 in each well with supplemented medium. The cells were then treated (in triplicate) with compounds that had an MIC less than or equal to 10μg/mL. The cells were exposed for 24 h to each compound in different concentrations (0.1, 0.5, 1, 5, 10, 20, 50, and 100 μM). After 24 h the compound was removed and the cells were washed with PBS 0.01M, then 150 μL of freshly prepared MTT (0.25 mg/mL) was added to each well and incubated for 3–4 h, until the presence of formazan was noticed in the negative control, then the MTT was removed and 100 μL of DMSO was added to dissolve the formazan. For this to dissolve, it was left at room temperature for 10 min. Next, a reading at 560 nm in a FLUOROstar® Omega multiplied reader (BMG Labtech, Ortenberg, Germany) was performed. IC50 values were determined by interpolation from mean absorbance data of 100% viability (negative control) [70].
3.6. Synergism
Synergism assays were performed to compare the MICs of antituberculosis drugs alone and in combination with the compounds that presented lower MICs against M. tuberculosis H37Rv (20a and 20f). The experimental protocol known as a “checkerboard” assay described by Ying et al. in 2021 [71] was employed for this purpose. The synergism assays were carried out in 96-well plates with Middlebrook 7H9 medium supplemented with OADC. The drugs and compounds were evaluated at different ranges of concentrations according to their potency: rifampin 0.048 mg/L to 0.78 mg/L, isoniazid 0.006 mg/L to 1 mg/L, levofloxacin 0.063 mg/L to 1 mg/L, amikacin 0.188 mg/L to 3 mg/L, and compounds 20e and 20f from 0.625 mg/L to 20 mg/L. Plate readings were performed in a MultiskanTM FC multiplate photometer (Thermo ScientificTM, Singapore) at 540 nm. The fractional inhibitory concentration index (FICI) was calculated using the formula: FICI = (MICAB/MICA) + (MICBA/MICB), where MICAB is the MIC of A in the presence of B, and MICBA is the MIC of B in the presence of A, and MICA is the MIC of A, and MICB is the MIC of B. Synergism is suggested by a FICI index ≤ 0.5, whereas a FICI index ≥ 4.0 suggests antagonism. A FICI index between 0.5 and 4.0, suggests no interaction between the active ingredients [72].
4. Conclusions
Five new series of chalcone–sulfonamide hybrids (16-20) a-f were synthesized via Claisen–Schmidt condensation of sulfonamides 8a-b, 10, 12, and 14a-b with aromatic aldehydes 15a-f. Twelve pyrazolines (22-23)a-d and (24-25)a-b and six carbothioamides 27a-f were obtained from 4-Cl-substituted chalcones of each series. All hybrids were preliminarily subjected to in vitro anticancer screening against 60 NCI human cancer cell lines at a single concentration of 10 μM. Chalcones 17a-c and 18e were the most active with mean values of GI % of 71.79%, 94.14%, 81.17%, and 100.00%, respectively, and a 25.48% of lethality for 18e. A subsequent screening of these compounds at five concentration levels revealed remarkable growth inhibition values against LOX IMVI (Melanoma) with GI50 of 0.34 μM, 0.73 μM, and 0.54 μM for 17a-c. The entire leukemia panel was highly sensitive to compound 18e displaying GI50 values between 0.99–2.52 μM. Anticancer trial results suggest that compounds with similar structures to 17a-c and 18e could be important templates for the future design and development of potential antiproliferative agents. Antituberculosis activity was measured against M. tuberculosis H37Rv and chalcones 17a, 17f, 19a, 19d, 20a, and 20d-f displaying MIC values between 8.97–28.79 μM. From additional studies performed against the Vero cell line, the tested compounds showed high cytotoxic effects indicating that structures should be optimized in order to improve selectivity.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232012589/s1.
Author Contributions
Conceptualization: L.F.C., A.I. and B.I; Funding acquisition: D.I., J.Q., R.A., J.C. and B.I.; Investigation: L.F.C., D.I., O.M.V., R.S., V.R., G.P., M.N., J.G., A.I. and B.I.; Project administration: A.I., D.I., J.Q., R.A., J.C. and B.I.; Supervision: A.I., D.I. and B.I.; Visualization: L.F.C., M.N., J.C. and J.G.; Writing original draft: L.F.C., D.I., J.C., A.I. and B.I.; Writing-review and editing: L.F.C., J.Q., R.A., D.I., J.C., J.G., A.I. and B.I. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by MINCIENCIAS (project No. 110680864255); Universidad del Valle (CI 71183) and Universidad del Norte (MINCIENCIAS-Project Number: 201477758055, contract. No. 777 de 2018), Colombia. The Spanish “Ministerio de Ciencia, Innovación, y Universidades” (R+D projects RTI2018-098560-BC22; co-financed by the FEDER funds of the European Union). Universidad de Jaén, Vicerrectorado de Investigación, PAIUJA “acción 1 plan 2019–2020 and 2021–2022”. Consejería de Innovación, Ciencia y Empresa (Junta de Andalucía), Spain.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the Developmental Therapeutics Program (DTP) of the National Cancer Institute of the United States for performing the anticancer screening of the compounds and Centro de Instrumentación Científico-Técnica of Universidad de Jaén for X-ray data collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Santucci, C.; Carioli, G.; Bertuccio, P.; Malvezzi, M.; Pastorino, U.; Boffetta, P.; Negri, E.; Bosetti, C.; La Vecchia, C. Progress in Cancer Mortality, Incidence, and Survival: A Global Overview. Eur. J. Cancer Prev. 2020, 29, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Shalini; Kumar, V. Have Molecular Hybrids Delivered Effective Anti-Cancer Treatments and What Should Future Drug Discovery Focus On? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef]
- Global Tuberculosis Report. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2021 (accessed on 31 August 2022).
- Peloquin, C.A.; Davies, G.R. The Treatment of Tuberculosis. Clin. Pharmacol. Ther. 2021, 110, 1455–1466. [Google Scholar] [CrossRef]
- Furin, J.; Cox, H.; Pai, M. Tuberculosis. Lancet 2019, 393, 1642–1656. [Google Scholar] [CrossRef]
- Natarajan, A.; Beena, P.M.; Devnikar, A.V.; Mali, S. A Systemic Review on Tuberculosis. Indian J. Tuberc. 2020, 67, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Apaydın, S.; Török, M. Sulfonamide Derivatives as Multi-Target Agents for Complex Diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Carta, F.; Supuran, C.T. Secondary and Tertiary Sulfonamides: A Patent Review. Expert Opin. Ther. Pat. 2013, 23, 203–213. [Google Scholar] [CrossRef]
- Zhao, C.; Rakesh, K.P.; Ravidar, L.; Fang, W.Y.; Qin, H.L. Pharmaceutical and Medicinal Significance of Sulfur (SVI)-Containing Motifs for Drug Discovery: A Critical Review. Eur. J. Med. Chem. 2019, 162, 679–734. [Google Scholar] [CrossRef]
- Catarro, M.; Serrano, J.L.; Ramos, S.S.; Silvestre, S.; Almeida, P. Nimesulide Analogues: From Anti-Inflammatory to Antitumor Agents. Bioorg. Chem. 2019, 88, 102966. [Google Scholar] [CrossRef]
- Roell, D.; Baniahmad, A. The Natural Compounds Atraric Acid and N-Butylbenzene-Sulfonamide as Antagonists of the Human Androgen Receptor and Inhibitors of Prostate Cancer Cell Growth. Mol. Cell. Endocrinol. 2011, 332, 1–8. [Google Scholar] [CrossRef]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Sulfonamides and Their Isosters as Carbonic Anhydrase Inhibitors. Future Med. Chem. 2014, 6, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Kazuhiko, I.; Takaya, N.; Yamaguchi, Y.; Kishii, R.; Kohno, Y.; Kurasaki, H. Sulfonamide-Based Non-Alkyne LpxC Inhibitors as Gram-Negative Antibacterial Agents. Bioorg. Med. Chem. Lett. 2017, 27, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Tassini, S.; Sun, L.; Lanko, K.; Crespan, E.; Langron, E.; Falchi, F.; Kissova, M.; Armijos-Rivera, J.I.; Delang, L.; Mirabelli, C.; et al. Discovery of Multitarget Agents Active as Broad-Spectrum Antivirals and Correctors of Cystic Fibrosis Transmembrane Conductance Regulator for Associated Pulmonary Diseases. J. Med. Chem. 2017, 60, 1400–1416. [Google Scholar] [CrossRef] [PubMed]
- Thareja, S.; Kokil, G.R.; Aggarwal, S.; Bhardwaj, T.R.; Kumar, M. Sulphonamides as Inhibitors of Protein Tyrosine Phosphatase 1B: A Three-Dimensional Quantitative Structure-Activity Relationship Study Using Self-Organizing Molecular Field Analysis Approach. Chem. Pharm. Bull. 2010, 58, 526–532. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. The η-Class Carbonic Anhydrases as Drug Targets for Antimalarial Agents. Expert Opin. Ther. Targets 2015, 19, 551–563. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.R.; Jiang, K.; Yang, J.L.; Shi, Y.P. Flavonoids as Key Bioactive Components of Oxytropis Falcata Bunge, a Traditional Anti-Inflammatory and Analgesic Tibetan Medicine. Nat. Prod. Res. 2020, 34, 3335–3352. [Google Scholar] [CrossRef]
- Sadula, A.; Peddaboina, U.R.; Prameela Subhashini, N.J. Synthesis and Characterization of Novel Chalcone Linked Imidazolones as Potential Antimicrobial and Antioxidant Agents. Med. Chem. Res. 2015, 24, 851–859. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Zhang, Y.; Chen, R.; Cui, Y.; Wang, Q. Anti-Inflammatory Chalcone-Isoflavone Dimers and Chalcone Dimers from Caragana Jubata. J. Nat. Prod. 2019, 82, 2761–2767. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Mantzanidou, M.; Hadjipavlou-Litina, D. Chalcones and Their Potential Role in Inflammation. Mini-Rev. Med. Chem. 2008, 8, 1224–1242. [Google Scholar] [CrossRef]
- Mateeva, N.; Eyunni, S.V.K.; Redda, K.K.; Ononuju, U.; Hansberry, T.D.; Aikens, C.; Nag, A. Functional Evaluation of Synthetic Flavonoids and Chalcones for Potential Antiviral and Anticancer Properties. Bioorg. Med. Chem. Lett. 2017, 27, 2350–2356. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K. Therapeutic Potential of Chalcones as Cardiovascular Agents. Life Sci. 2016, 148, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Chalcones and Their Therapeutic Targets for the Management of Diabetes: Structural and Pharmacological Perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Braga, R.C.; Grzelak, E.M.; Neves, B.J.; Muratov, E.; Ma, R.; Klein, L.L.; Cho, S.; Oliveira, G.R.; Franzblau, S.G.; et al. QSAR-Driven Design, Synthesis and Discovery of Potent Chalcone Derivatives with Antitubercular Activity. Eur. J. Med. Chem. 2017, 137, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, S.A.; Saeed, A.; Siddique, M.N.; un Nisa, Z.; Khan, S.; Lecka, J.; Sévigny, J.; Iqbal, J. Synthesis, Characterization and Biological Evaluation of Novel Chalcone Sulfonamide Hybrids as Potent Intestinal Alkaline Phosphatase Inhibitors. Bioorg. Chem. 2017, 70, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Bashir, R.; Ovais, S.; Yaseen, S.; Hamid, H.; Alam, M.S.; Samim, M.; Singh, S.; Javed, K. Synthesis of Some New 1,3,5-Trisubstituted Pyrazolines Bearing Benzene Sulfonamide as Anticancer and Anti-Inflammatory Agents. Bioorg. Med. Chem. Lett. 2011, 21, 4301–4305. [Google Scholar] [CrossRef]
- Tucker, J.W.; Chenard, L.; Young, J.M. Selective Access to Heterocyclic Sulfonamides and Sulfonyl Fluorides via a Parallel Medicinal Chemistry Enabled Method. ACS Comb. Sci. 2015, 17, 653–657. [Google Scholar] [CrossRef]
- Kucukoglu, K.; Oral, F.; Aydin, T.; Yamali, C.; Algul, O.; Sakagami, H.; Gulcin, I.; Supuran, C.T.; Gul, H.I. Synthesis, Cytotoxicity and Carbonic Anhydrase Inhibitory Activities of New Pyrazolines. J. Enzyme Inhib. Med. Chem. 2016, 31, 20–24. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Kazaz, C.; Levent, S.; Gulcin, I. Synthesis, Structure Elucidation, and in Vitro Pharmacological Evaluation of Novel Polyfluoro Substituted Pyrazoline Type Sulfonamides as Multi-Target Agents for Inhibition of Acetylcholinesterase and Carbonic Anhydrase I and II Enzymes. Bioorg. Chem. 2020, 96, 103627. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, M.R.; Mayhoub, A.S.; Farag, A.M. Recent Advances in the Therapeutic Applications of Pyrazolines. Expert Opin. Ther. Pat. 2012, 22, 253–291. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.T.; Bhavsar, Z.A.; Jethava, D.J.; Patel, D.B.; Patel, H.D. A Review on Development of Bio-Active Thiosemicarbazide Derivatives: Recent Advances. J. Mol. Struct. 2021, 1226, 129268. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Meyer-Almes, F.J.; Loiodice, F.; Anisimova, N.Y.; Spirina, T.S.; Sokolova, D.V.; Smirnova, G.B.; Choe, J.Y.; Pokrovsky, V.S.; et al. Synthesis and Biological Evaluation of Pyrazoline and Pyrrolidine-2,5-Dione Hybrids as Potential Antitumor Agents. ChemMedChem 2020, 15, 1813–1825. [Google Scholar] [CrossRef]
- Montoya, A.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J.; Insuasty, B. Synthesis and in Vitro Antitumor Activity of a Novel Series of 2-Pyrazoline Derivatives Bearing the 4-Aryloxy-7-Chloroquinoline Fragment. Molecules 2014, 19, 18656–18675. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.L.; Fan, J.; Ma, X.; Qin, Y.J.; Zhu, H.L. Novel Nicotinoyl Pyrazoline Derivates Bearing N-Methyl Indole Moiety as Antitumor Agents: Design, Synthesis and Evaluation. Eur. J. Med. Chem. 2018, 156, 722–737. [Google Scholar] [CrossRef]
- Nehra, B.; Rulhania, S.; Jaswal, S.; Kumar, B.; Singh, G.; Monga, V. Recent Advancements in the Development of Bioactive Pyrazoline Derivatives. Eur. J. Med. Chem. 2020, 205, 112666. [Google Scholar] [CrossRef]
- Iñiguez, M.A.; Punzón, C.; Cacheiro-Llaguno, C.; Díaz-Muñoz, M.D.; Duque, J.; Cuberes, R.; Alvarez, I.; Andrés, E.M.; Buxens, J.; Buschmann, H.; et al. Cyclooxygenase-Independent Inhibitory Effects on T Cell Activation of Novel 4,5-Dihydro-3 Trifluoromethyl Pyrazole Cyclooxygenase-2 Inhibitors. Int. Immunopharmacol. 2010, 10, 1295–1304. [Google Scholar] [CrossRef]
- Kharbanda, C.; Alam, M.S.; Hamid, H.; Javed, K.; Bano, S.; Dhulap, A.; Ali, Y.; Nazreen, S.; Haider, S. Synthesis and Evaluation of Pyrazolines Bearing Benzothiazole as Anti-Inflammatory Agents. Bioorg. Med. Chem. 2014, 22, 5804–5812. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Al-Mohammadi, M.M.; Alamry, K.A.; Khan, K.A. Synthesis and Biological Evaluation of Fluoropyrazolesulfonylurea and Thiourea Derivatives as Possible Antidiabetic Agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 157–163. [Google Scholar] [CrossRef]
- Özdemir, A.; Altintop, M.D.; Kaplancikli, Z.A.; Can, Ö.D.; Özkay, Ü.D.; Turan-Zitouni, G. Synthesis and Evaluation of New 1,5-Diaryl-3-[4-(Methylsulfonyl)Phenyl]-4,5-Dihydro-1 H-Pyrazole Derivatives as Potential Antidepressant Agents. Molecules 2015, 20, 2668–2684. [Google Scholar] [CrossRef] [PubMed]
- Insuasty, B.; Montoya, A.; Becerra, D.; Quiroga, J.; Abonia, R.; Robledo, S.; Vélez, I.D.; Upegui, Y.; Nogueras, M.; Cobo, J. Synthesis of Novel Analogs of 2-Pyrazoline Obtained from [(7-Chloroquinolin-4-Yl)Amino]Chalcones and Hydrazine as Potential Antitumor and Antimalarial Agents. Eur. J. Med. Chem. 2013, 67, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, R.; Desideri, N.; Carta, A.; Atzori, E.M.; Delogu, I.; Collu, G.; Loddo, R. Inhibitors of Yellow Fever Virus Replication Based on 1,3,5-Triphenyl-4,5-Dihydropyrazole Scaffold: Design, Synthesis and Antiviral Evaluation. Eur. J. Med. Chem. 2017, 141, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; Rahman, D.E.A.; Saleh, D.O.; Jaleel, G.A.R.A. Benzofuran-Morpholinomethyl-Pyrazoline Hybrids as a New Class of Vasorelaxant Agents: Synthesis and Quantitative Structure-Activity Relationship Study. Chem. Pharm. Bull. 2014, 62, 1238–1251. [Google Scholar] [CrossRef]
- Shin, S.Y.; Ahn, S.; Yoon, H.; Jung, H.; Jung, Y.; Koh, D.; Lee, Y.H.; Lim, Y. Colorectal Anticancer Activities of Polymethoxylated 3-Naphthyl-5-Phenylpyrazoline-Carbothioamides. Bioorg. Med. Chem. Lett. 2016, 26, 4301–4309. [Google Scholar] [CrossRef]
- Kim, B.S.; Shin, S.Y.; Ahn, S.; Koh, D.; Lee, Y.H.; Lim, Y. Biological Evaluation of 2-Pyrazolinyl-1-Carbothioamide Derivatives against HCT116 Human Colorectal Cancer Cell Lines and Elucidation on QSAR and Molecular Binding Modes. Bioorg. Med. Chem. 2017, 25, 5423–5432. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, B.S.; Ahn, S.; Koh, D.; Lee, Y.H.; Shin, S.Y.; Lim, Y. Anticancer and Structure-Activity Relationship Evaluation of 3-(Naphthalen-2-Yl)-N,5-Diphenyl-Pyrazoline-1-Carbothioamide Analogs of Chalcone. Bioorg. Chem. 2016, 68, 166–176. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, J.; Xu, W.; Chen, C.; Wei, D.; Ni, W.; Pan, Y. A New Series of Cytotoxic Pyrazoline Derivatives as Potential Anticancer Agents That Induce Cell Cycle Arrest and Apoptosis. Molecules 2017, 22, 1635. [Google Scholar] [CrossRef]
- Zaki, Y.H.; Al-Gendey, M.S.; Abdelhamid, A.O. A Facile Synthesis, and Antimicrobial and Anticancer Activities of Some Pyridines, Thioamides, Thiazole, Urea, Quinazoline, β-Naphthyl Carbamate, and Pyrano[2,3-d]Thiazole Derivatives. Chem. Cent. J. 2018, 12, 1–14. [Google Scholar] [CrossRef]
- Siddiqui, N.; Alam, P.; Ahsan, W. Design, Synthesis, and In-Vivo Pharmacological Screening of N,3-(Substituted Diphenyl)-5-Phenyl-1 H-Pyrazoline-1- Carbothioamide Derivatives. Arch. Pharm. 2009, 342, 173–181. [Google Scholar] [CrossRef]
- Mathew, B.; Ahsan, M.J. Carboxamide/Carbothioamide Anticonvulsant Analogues Towards GABA-Aminotransferase- An in Silico Approach. Cent. Nerv. Syst. Agents Med. Chem. 2014, 14, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Jain, P. Synthetic and Biological Studies of Pyrazolines and Related Heterocyclic Compounds. Arab. J. Chem. 2014, 7, 553–596. [Google Scholar] [CrossRef]
- Abdelhamid, A.O.; El Sayed, I.E.; Hussein, M.Z.; Mangoud, M.M. Synthesis and Antimicrobial Activity of Some New Thiadiazoles, Thioamides, 5-Arylazothiazoles and Pyrimido[4,5-d][1,2,4]Triazolo[4,3-a]Pyrimidines. Molecules 2016, 21, 1072. [Google Scholar] [CrossRef] [PubMed]
- Alex, J.M.; Kumar, R. 4,5-Dihydro-1H-Pyrazole: An Indispensable Scaffold. J. Enzyme Inhib. Med. Chem. 2014, 29, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Castaño, L.F.; Cuartas, V.; Bernal, A.; Insuasty, A.; Guzman, J.; Vidal, O.; Rubio, V.; Puerto, G.; Lukáč, P.; Vimberg, V.; et al. New Chalcone-Sulfonamide Hybrids Exhibiting Anticancer and Antituberculosis Activity. Eur. J. Med. Chem. 2019, 176, 50–60. [Google Scholar] [CrossRef]
- Riaz, S.; Khan, I.U.; Bajda, M.; Ashraf, M.; Shaukat, A.; Rehman, T.U.; Mutahir, S.; Hussain, S.; Yar, M.; Khan, I.U.; et al. Pyridine Sulfonamide as a Small Key Organic Molecule for the Potential Treatment of Type-II Diabetes Mellitus and Alzheimer’s Disease: In Vitro Studies against Yeast α-Glucosidase, Acetylcholinesterase and Butyrylcholinesterase. Bioorg. Chem. 2015, 63, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mótyán, G.; Kovács, F.; Wölfling, J.; Gyovai, A.; Zupkó, I.; Frank, É. Microwave-Assisted Stereoselective Approach to Novel Steroidal Ring D-Fused 2-Pyrazolines and an Evaluation of Their Cell-Growth Inhibitory Effects in Vitro. Steroids 2016, 112, 36–46. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Cao, D.; Chen, T.; Wang, Q.; Li, Y.; Xu, Y.; Zhang, N.; Wang, X.; Chen, D.; et al. Fragment-Based Drug Discovery of 2-Thiazolidinones as BRD4 Inhibitors: Structure-Based Optimization. J. Med. Chem. 2015, 58, 1281–1297. [Google Scholar] [CrossRef]
- Evangelopoulos, D.; Bhakta, S. Rapid methods for testing inhibitors of mycobacterial growth. in Antibiotic Resistance Protocols. Methods Mol. Biol. 2010, 642, 193–201. [Google Scholar] [CrossRef]
- Brudey, K.; Driscoll, J.R.; Rigouts, L.; Prodinger, W.M.; Gori, A.; Al-Hajoj, S.A.; Allix, C.; Aristimuño, L.; Arora, J.; Baumanis, V.; et al. Mycobacterium tuberculosis complex genetic diversity: Mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006, 6, 1–17. [Google Scholar] [CrossRef]
- Puerto, G.; Erazo, L.; Wintaco, M.; Castro, C.; Ribón, W.; Guerrero, M.I. Mycobacterium tuberculosis Genotypes Determined by Spoligotyping to Be Circulating in Colombia between 1999 and 2012 and Their Possible Associations with Transmission and Susceptibility to First-Line Drugs. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Realpe, T.; Correa, N.; Rozo, J.C.; Ferro, B.E.; Gómez, V.; Zapata, E.; Wellman, R.; Puerta, G.; Castro, C.; Nieto, L.M.; et al. Population Structure among Mycobacterium tuberculosis Isolates from Pulmonary Tuberculosis Patients in Colombia. PLoS ONE 2014, 9, e93848. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, Y.; Millet, J.; Rastogi, N. Genetic Structuration, Demography and Evolutionary History of Mycobacterium tuberculosis LAM9 Sublineage in the Americas as Two Distinct Subpopulations Revealed by Bayesian Analyses. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef]
- Cubillos, A.; Sandoval, A.; Ritacco, V.; López, B.; Robledo, J.; Correa, N.; Hernandez-Neuta, I.; Zambrano, M.M.; Del Portillo, P. Genomic signatures of the Haarlem lineage of Mycobacterium tuberculosis: Implications of strain genetic variation in drug and vaccine development. J. Clin. Microbiol. 2010, 48, 3614–3623. [Google Scholar] [CrossRef]
- National Cancer Insititute. DCTD Division of Cancer Treatment & Diagnosis. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 27 June 2022).
- Guzman, J.D.; Mortazavi, P.N.; Munshi, T.; Evangelopoulos, D.; McHugh, T.D.; Gibbons, S.; Malkinson, J.; Bhakta, S. 2-Hydroxy-Substituted Cinnamic Acids and Acetanilides Are Selective Growth Inhibitors of Mycobacterium Tuberculosis. Med. Chem. Comm. 2014, 5, 47–50. [Google Scholar] [CrossRef]
- Chen, T.; Li, Q.; Guo, L.; Yu, L.; Li, Z.; Guo, H.; Li, H.; Zhao, M.; Chen, L.; Chen, X.; et al. Lower cytotoxicity, high stability, and long-term antibacterial activity of a poly (methacrylic acid)/isoniazid/rifampin nanogel against multidrug-resistant intestinal Mycobacterium tuberculosis. Mater. Sci. Eng. C 2016, 58, 659–665. [Google Scholar] [CrossRef]
- Kabongo, P.N.; Eloff, J.N.; Obi, C.L.; Mcgaw, L.J. Antimycobacterial Activity and Low Cytotoxicity of Leaf Extracts of Some African Anacardiaceae Tree Species. Phytother. Res. 2016, 30, 2001–2011. [Google Scholar] [CrossRef]
- Ying, R.; Huang, X.; Gao, Y.; Wang, J.; Liu, Y.; Sha, W.; Yang, H. In vitro Synergism of Six Antituberculosis Agents Against Drug-Resistant Mycobacterium tuberculosis Isolated from Retreatment Tuberculosis Patients. Infect. Drug Resist. 2021, 14, 3729–3736. [Google Scholar] [CrossRef]
- Caleffi, K.R.; Maltempe, F.G.; Siqueira, V.L.D.; Cardoso, R.F. Fast detection of drug interaction in Mycobacterium tuberculosis by a checkerboard resazurin method. Tuberculosis 2013, 93, 660–663. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).