

Insulin Diminishes Superoxide Increase in Cytosol and Mitochondria of Cultured Cortical Neurons Treated with Toxic Glutamate



 ,
,  ,
,
Abstract
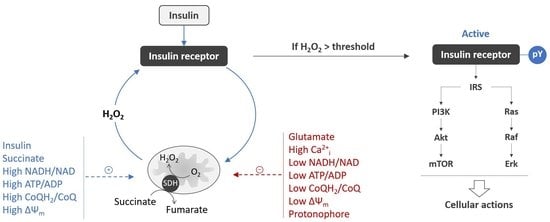
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Changes in the Viability of Neurons under Glutamate Excitotoxicity and Insulin Treatment
2.2. Changes in Intracellular Calcium [Ca2+]i and Superoxide
2.3. Changes in [Ca2+]i and the Mitochondrial Superoxide Production
2.4. Effects of Glutamate and Insulin on Oxygen Consumption Rates in Neurons
3. Discussion
4. Materials and Methods
4.1. Primary Culture of Rat Cortical Neurons
4.2. Detection of Living, Apoptotic and Necrotic Cells Using Vital Fluorescent Dyes
4.3. Measurement of [Ca2+]i and Superoxide
4.4. Measurement of Oxygen Consumption Rates
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morland, C.; Nordengen, K. N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease. Int. J. Mol. Sci. 2022, 23, 1268. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Arabaci, O.; Acari, A.; Ciftci, P.; Gozuacik, D. Glutamate Scavenging as a Neuroreparative Strategy in Ischemic Stroke. Front. Pharmacol. 2022, 13, 1018. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Budd, S.L. Mitochondria and Neuronal Survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed]
- Khodorov, B. Glutamate-Induced Deregulation of Calcium Homeostasis and Mitochondrial Dysfunction in Mammalian Central Neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and Stroke: Identifying Novel Targets for Neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sheng, M. NMDA Receptors in Nervous System Diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D. Mitochondrial Dysfunction and Glutamate Excitotoxicity Studied in Primary Neuronal Cultures. Curr. Mol. Med. 2005, 4, 149–177. [Google Scholar] [CrossRef]
- Vergun, O.; Keelan, J.; Khodorov, B.I.; Duchen, M.R. Glutamate-Induced Mitochondrial Depolarisation and Perturbation of Calcium Homeostasis in Cultured Rat Hippocampal Neurones. J. Physiol. 1999, 519, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Bakaeva, Z.; Goncharov, M.; Krasilnikova, I.; Zgodova, A.; Frolov, D.; Grebenik, E.; Timashev, P.; Pinelis, V.; Surin, A. Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome. Int. J. Mol. Sci. 2022, 23, 3858. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate Induces the Production of Reactive Oxygen Species in Cultured Forebrain Neurons Following NMDA Receptor Activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Sensi, S.L.; Canzoniero, L.M.T.; Handran, S.D.; Rothman, S.M.; Lin, T.S.; Goldberg, M.P.; Choi, D.W. Mitochondrial Production of Reactive Oxygen Species in Cortical Neurons Following Exposure to N-Methyl-D-Aspartate. J. Neurosci. 1995, 15, 6377–6388. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Tomoki; Nakamizo; Inoue, R.; Sawada, H.; Kihara, T.; Honda, K.; Akaike, A.; Shimohama, S. N-Methyl-D-Aspartate Receptor-Mediated Mitochondrial Ca2+ Overload in Acute Excitotoxic Motor Neuron Death: A Mechanism Distinct from Chronic Neurotoxicity after Ca2+ Influx. J. Neurosci. Res. 2001, 63, 377–387. [Google Scholar] [CrossRef]
- Duan, Y.; Gross, R.A.; Sheu, S.S. Ca2+-Dependent Generation of Mitochondrial Reactive Oxygen Species Serves as a Signal for Poly(ADP-Ribose) Polymerase-1 Activation during Glutamate Excitotoxicity. J. Physiol. 2007, 585, 741–758. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-Dependent Superoxide Production and Neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Culcasi, M.; Gaven, F.; Pietri, S.; Bockaert, J. Nitric Oxide, Superoxide and Peroxynitrite: Putative Mediators of NMDA-Induced Cell Death in Cerebellar Granule Cells. Neuropharmacology 1993, 32, 1259–1266. [Google Scholar] [CrossRef]
- Patel, M.; Day, B.J.; Crapo, J.D.; Fridovich, I.; McNamara, J.O. Requirement for Superoxide in Excitotoxic Cell Death. Neuron 1996, 16, 345–355. [Google Scholar] [CrossRef]
- Prehn, J.H.M. NMDA-Induced Superoxide Production and Neurotoxicity in Cultured Rat Hippocampal Neurons: Role of Mitochondria. Eur. J. Neurosci. 1998, 10, 1903–1910. [Google Scholar] [CrossRef]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A New Vicious Cycle Involving Glutamate Excitotoxicity, Oxidative Stress and Mitochondrial Dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef]
- Li, Q.Y.; Pedersen, C.; Day, B.J.; Patel, M. Dependence of Excitotoxic Neurodegeneration on Mitochondrial Aconitase Inactivation. J. Neurochem. 2001, 78, 746–755. [Google Scholar] [CrossRef]
- Sang, W.S.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic Neuronal Death Is Triggered by Glucose Reperfusion and Activation of Neuronal NADPH Oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef]
- Girouard, H.; Wang, G.; Gallo, E.F.; Anrather, J.; Zhou, P.; Pickel, V.M.; Iadecola, C. NMDA Receptor Activation Increases Free Radical Production through Nitric Oxide and NOX2. J. Neurosci. 2009, 29, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Won Suh, S.; Joon Won, S.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH Oxidase Is the Primary Source of Superoxide Induced by NMDA Receptor Activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Minnella, A.M.; Shen, Y.; Swanson, R.A. Phosphoinositide 3-Kinase Couples NMDA Receptors to Superoxide Release in Excitotoxic Neuronal Death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef] [PubMed]
- Minnella, A.M.; Zhao, J.X.; Jiang, X.; Jakobsen, E.; Lu, F.; Wu, L.; El-Benna, J.; Gray, J.A.; Swanson, R.A. Excitotoxic Superoxide Production and Neuronal Death Require Both Ionotropic and Non-Ionotropic NMDA Receptor Signaling. Sci. Rep. 2018, 8, 17522. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Stocker, R.; Seredenina, T.; Holmdahl, R.; Aguzzi, A.; Chio, A.; Depaulis, A.; Heitz, F.; Olofsson, P.; Olsson, T.; et al. NADPH Oxidases as Drug Targets and Biomarkers in Neurodegenerative Diseases: What Is the Evidence? Free Radic. Biol. Med. 2017, 112, 387–396. [Google Scholar] [CrossRef]
- Glass, M.J.; Huang, J.; Oselkin, M.; Tarsitano, M.J.; Wang, G.; Iadecola, C.; Pickel, V.M. Subcellular Localization of Nicotinamide Adenine Dinucleotide Phosphate Oxidase Subunits in Neurons and Astroglia of the Rat Medial Nucleus Tractus Solitarius: Relationship with Tyrosine Hydroxylase Immunoreactive Neurons. Neuroscience 2006, 143, 547–564. [Google Scholar] [CrossRef][Green Version]
- Krasil’nikova, I.; Surin, A.; Sorokina, E.; Fisenko, A.; Boyarkin, D.; Balyasin, M.; Demchenko, A.; Pomytkin, I.; Pinelis, V. Insulin Protects Cortical Neurons Against Glutamate Excitotoxicity. Front. Neurosci. 2019, 13, 1027. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikova, I.; Pomytkin, I.; Pinelis, V.; Surin, A. Insulin Normalizes Ionic Homeostasis and the State of Mitochondria after a Mechanical Damage to the Culture of Brain Neurons. Biochem. (Moscow) Suppl. Ser. A Membr. Cell Biol. 2021, 15, 365–371. [Google Scholar] [CrossRef]
- Maimaiti, S.; Frazier, H.N.; Anderson, K.L.; Ghoweri, A.O.; Brewer, L.D.; Porter, N.M.; Thibault, O. Novel Calcium-Related Targets of Insulin in Hippocampal Neurons. Neuroscience 2017, 364, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Nampoothiri, M.; Reddy, N.D.; John, J.; Kumar, N.; Kutty Nampurath, G.; Rao Chamallamudi, M. Insulin Blocks Glutamate-Induced Neurotoxicity in Differentiated SH-SY5Y Neuronal Cells. Behav. Neurol. 2014, 2014, 674164. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.W.; Zhang, L.; Zhu, S.J.; Chen, W.C.; Mei, B. Excitotoxicity Effects of Glutamate on Human Neuroblastoma SH-SY5Y Cells via Oxidative Damage. Neurosci. Bull. 2010, 26, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Cadwell, L.I.; Jekabsons, M.B.; Wang, A.; Polster, B.M.; Nicholls, D.G. “Mild Uncoupling” Does Not Decrease Mitochondrial Superoxide Levels in Cultured Cerebellar Granule Neurons but Decreases Spare Respiratory Capacity and Increases Toxicity to Glutamate and Oxidative Stress. J. Neurochem. 2007, 101, 1619–1631. [Google Scholar] [CrossRef]
- Zhao, H.; Kalivendi, S.; Zhang, H.; Joseph, J.; Nithipatikom, K.; Vásquez-Vivar, J.; Kalyanaraman, B. Superoxide Reacts with Hydroethidine but Forms a Fluorescent Product That Is Distinctly Different from Ethidium: Potential Implications in Intracellular Fluorescence Detection of Superoxide. Free Radic. Biol. Med. 2003, 34, 1359–1368. [Google Scholar] [CrossRef]
- Hyrc, K.L.; Bownik, J.M.; Goldberg, M.P. Ionic Selectivity of Low-Affinity Ratiometric Calcium Indicators: Mag-Fura-2, Fura-2FF and BTC. Cell Calcium 2000, 27, 75–86. [Google Scholar] [CrossRef]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective Fluorescent Imaging of Superoxide in Vivo Using Ethidium-Based Probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rogers, S.C.; Kavdia, M. Analysis of Kinetics of Dihydroethidium Fluorescence with Superoxide Using Xanthine Oxidase and Hypoxanthine Assay. Ann. Biomed. Eng. 2013, 41, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Bessman, S.P.; Mohan, C.; Zaidise, I. Intracellular Site of Insulin Action: Mitochondrial Krebs Cycle. Proc. Natl. Acad. Sci. USA 1986, 83, 5067–5070. [Google Scholar] [CrossRef]
- Pomytkin, I.; Pinelis, V. Insulin Receptors and Intracellular Ca2+ Form a Double-Negative Regulatory Feedback Loop Controlling Insulin Sensitivity. F1000Research 2020, 9, 598. [Google Scholar] [CrossRef]
- Forkink, M.; Smeitink, J.A.M.; Brock, R.; Willems, P.H.G.M.; Koopman, W.J.H. Detection and Manipulation of Mitochondrial Reactive Oxygen Species in Mammalian Cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed Calcium Dysregulation in Neurons Requires Both the NMDA Receptor and the Reverse Na+/Ca2+ Exchanger. Neurobiol. Dis. 2012, 46, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M.; Nicholls, D.G.; Ge, S.X.; Roelofs, B.A. Use of Potentiometric Fluorophores in the Measurement of Mitochondrial Reactive Oxygen Species, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 547. [Google Scholar] [CrossRef]
- Carafoli, E.; Santella, L.; Branca, D.; Brini, M. Generation, Control, and Processing of Cellular Calcium Signals. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 107–260. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.W.; Carter, K.; Bhatt, A.; Pang, Y. Rapid Transport of Insulin to the Brain Following Intranasal Administration in Rats. Neural Regen. Res. 2019, 14, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal Insulin Therapy for Alzheimer Disease and Amnestic Mild Cognitive Impairment: A Pilot Clinical Trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef]
- Brabazon, F.; Wilson, C.M.; Jaiswal, S.; Reed, J.; Frey, W.H.; Byrnes, K.R. Intranasal Insulin Treatment of an Experimental Model of Moderate Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2017, 37, 3203–3218. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial Complex II Is a Source of the Reserve Respiratory Capacity That Is Regulated by Metabolic Sensors and Promotes Cell Survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinelis, V.; Krasilnikova, I.; Bakaeva, Z.; Surin, A.; Boyarkin, D.; Fisenko, A.; Krasilnikova, O.; Pomytkin, I. Insulin Diminishes Superoxide Increase in Cytosol and Mitochondria of Cultured Cortical Neurons Treated with Toxic Glutamate. Int. J. Mol. Sci. 2022, 23, 12593. https://doi.org/10.3390/ijms232012593
Pinelis V, Krasilnikova I, Bakaeva Z, Surin A, Boyarkin D, Fisenko A, Krasilnikova O, Pomytkin I. Insulin Diminishes Superoxide Increase in Cytosol and Mitochondria of Cultured Cortical Neurons Treated with Toxic Glutamate. International Journal of Molecular Sciences. 2022; 23(20):12593. https://doi.org/10.3390/ijms232012593
Chicago/Turabian StylePinelis, Vsevolod, Irina Krasilnikova, Zanda Bakaeva, Alexander Surin, Dmitrii Boyarkin, Andrei Fisenko, Olga Krasilnikova, and Igor Pomytkin. 2022. "Insulin Diminishes Superoxide Increase in Cytosol and Mitochondria of Cultured Cortical Neurons Treated with Toxic Glutamate" International Journal of Molecular Sciences 23, no. 20: 12593. https://doi.org/10.3390/ijms232012593
APA StylePinelis, V., Krasilnikova, I., Bakaeva, Z., Surin, A., Boyarkin, D., Fisenko, A., Krasilnikova, O., & Pomytkin, I. (2022). Insulin Diminishes Superoxide Increase in Cytosol and Mitochondria of Cultured Cortical Neurons Treated with Toxic Glutamate. International Journal of Molecular Sciences, 23(20), 12593. https://doi.org/10.3390/ijms232012593