
Computational Docking Reveals Co-Evolution of C4 Carbon Delivery Enzymes in Diverse Plants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
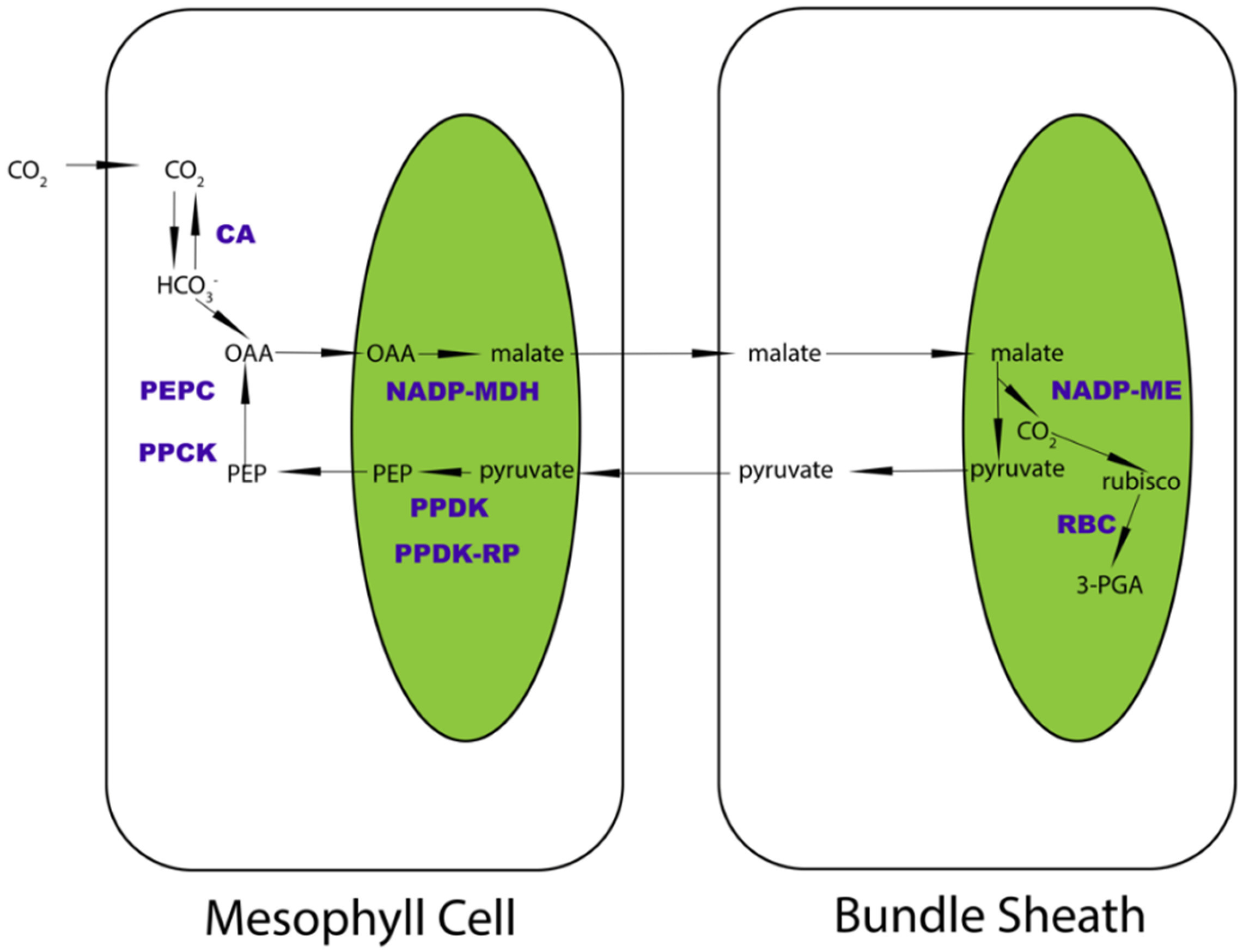
:1. Introduction
2. Results
2.1. Homologous Protein Identification between C3 and C4 Species Revealed Higher Homogeneity of Photosynthetic Enzymes in C4 Plants
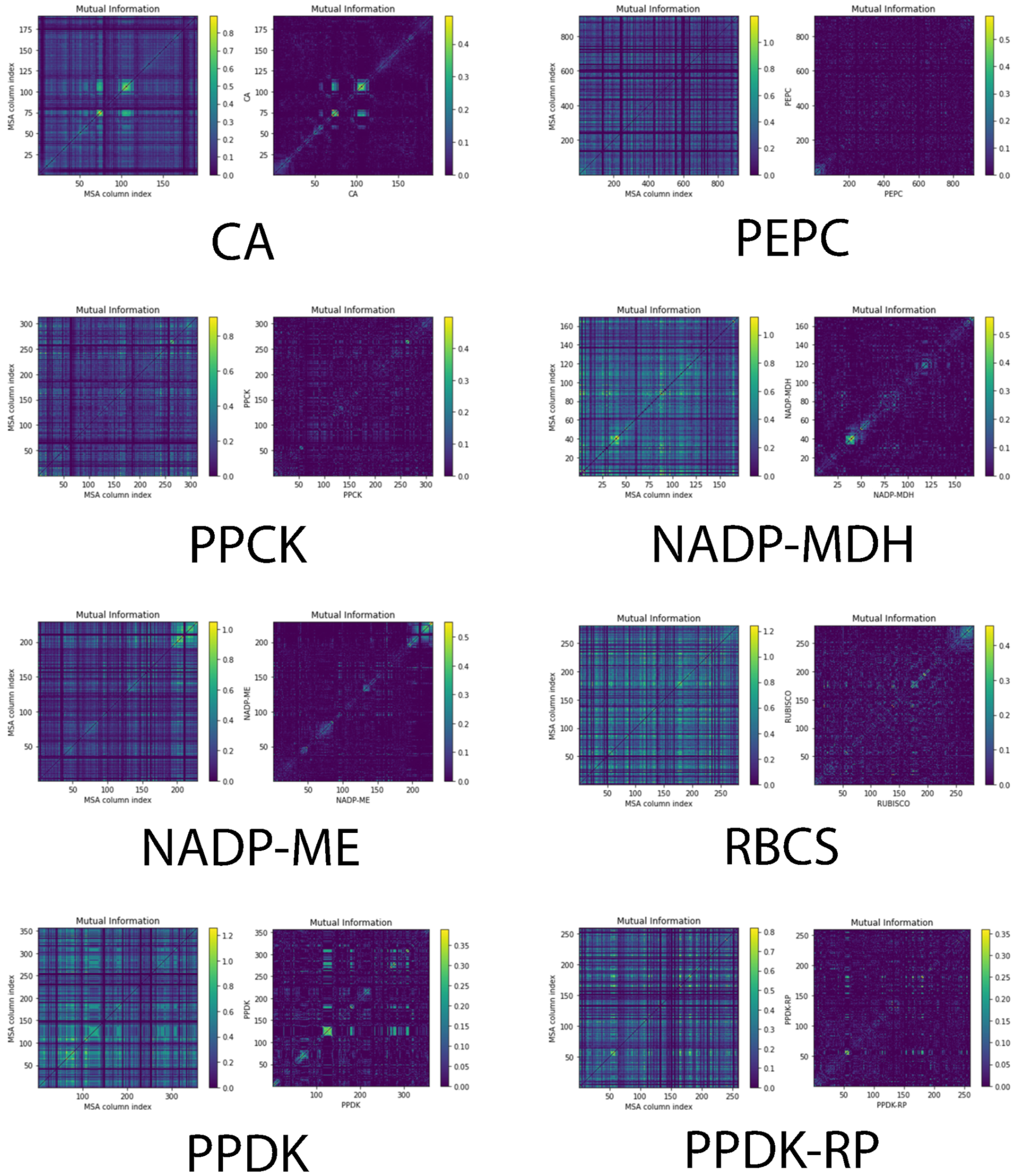
2.2. N-terminus Covarying Sites Occur Distinctively in Diverse Photosynthetic Genes
2.3. Co-evolution Is Not Necessary for PEPC and PPCK to Maintain Their Regulatory Relationship
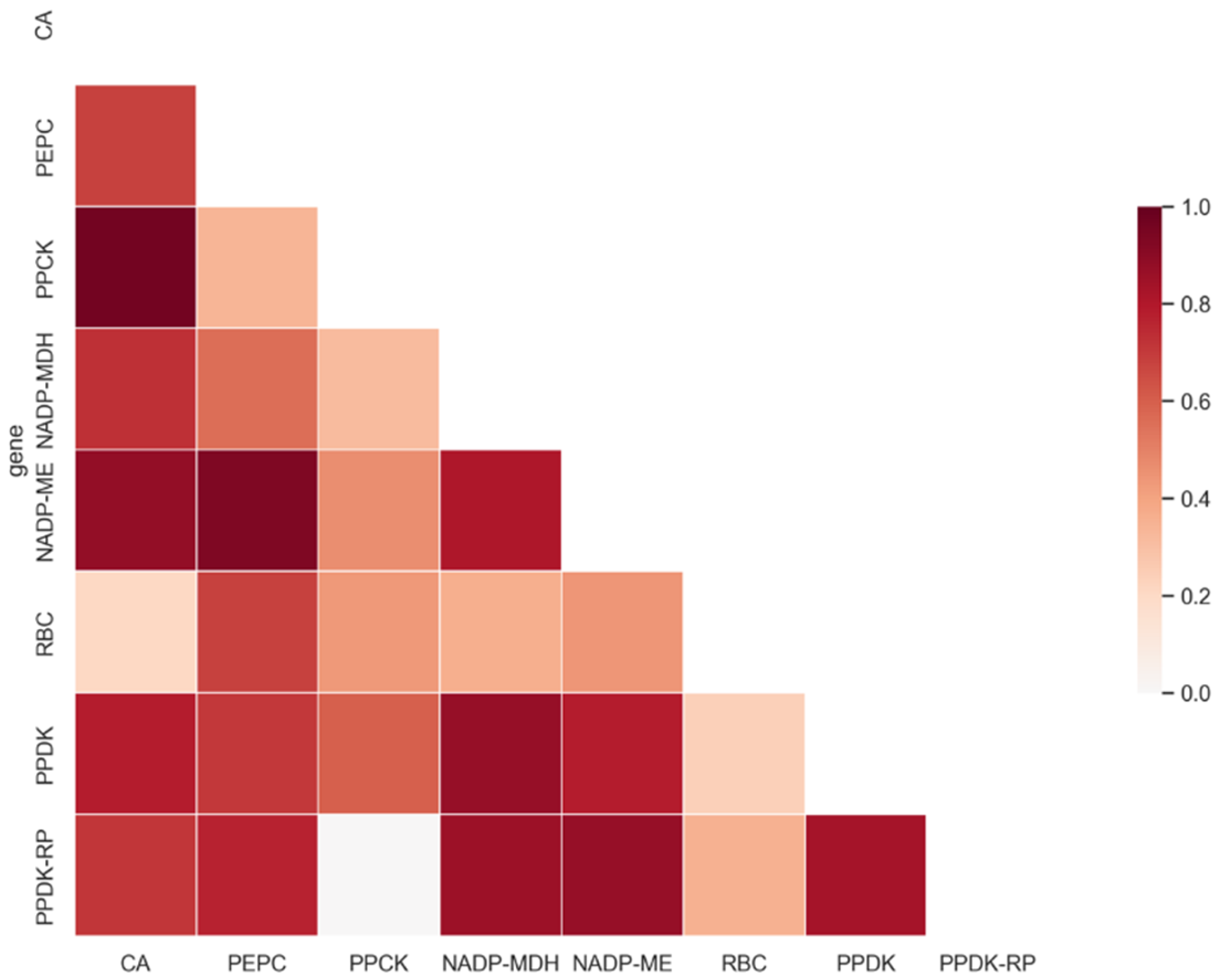
2.4. Global Co-varying Sites Identification in C4 Enzymes
2.5. Protein–Protein Interaction Prediction Revealed the Possible New Function of Photosynthetic Enzymes
2.6. Pocket Formation at the Interface Is Not Necessary for PEPC and PPCK Interaction
3. Discussion
4. Materials and Methods
4.1. Phylogeny Study of Eight Key Photosynthetic Enzymes
4.2. Co-Evolved Positions Identification
4.3. Global Tree Similarity Comparison between Eight Key Photosynthetic Enzymes
4.4. Selection Criteria of Key C4 Enzyme Candidates
4.5. Protein–Protein Interaction Prediction
4.6. Co-Varying Amino Acids Identification
4.7. Protein–Protein Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CA | carbonic anhydrase |
| PEPC | phosphoenolpyruvate carboxylase |
| PPCK | phosphoenolpyruvate carboxylase kinase |
| NADP-MDH | NAD(P)+-dependent malate dehydrogenase |
| NADP-ME | NADP-dependent malic enzyme |
| RBC | ribulose bisphosphate carboxylase |
| PPDK | pyruvate, phosphate dikinase |
| PPDK-RP | pyruvate, phosphate dikinase regulatory protein |
| OAA | oxaloacetic acid |
| PEP | phosphoenolpyruvate |
| 3-PGA | 3-phosphoglyceric acid |
| DEG | differentially expressed gene |
| CO2 | carbon dioxide |
| RMSD | root-mean-square deviation |
| AT | ARABIDOPSIS THALIANA |
| BD | Brachypodium distachyon |
| OS | Oryza sativa |
| SV | Setaria viridis |
| SB | Sorghum bicolor |
| ZM | Zea mays |
References
- Chavali, S.; Singh, A.K.; Santhanam, B.; Babu, M.M. Amino acid homorepeats in proteins. Nat. Rev. Chem. 2020, 4, 420–434. [Google Scholar] [CrossRef]
- De Juan, D.; Pazos, F.; Valencia, A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013, 14, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Colavin, A.; Atolia, E.; Bitbol, A.F.; Huang, K.C. Extracting phylogenetic dimensions of coevolution reveals hidden functional signals. Sci. Rep. 2022, 12, 820. [Google Scholar] [CrossRef] [PubMed]
- Green, A.G.; Elhabashy, H.; Brock, K.P.; Maddamsetti, R.; Kohlbacher, O.; Marks, D.S. Large-scale discovery of protein interactions at residue resolution using co-evolution calculated from genomic sequences. Nat. Commun. 2021, 12, 1396. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Green, A.G.; Wang, B.; Lite, T.V.; Weinstein, E.N.; Marks, D.S.; Laub, M.T. Co-evolution of interacting proteins through non-contacting and non-specific mutations. Nat. Ecol. Evol. 2022, 6, 590–603. [Google Scholar] [CrossRef]
- Hopf, T.A.; Scharfe, C.P.; Rodrigues, J.P.; Green, A.G.; Kohlbacher, O.; Sander, C.; Bonvin, A.M.; Marks, D.S. Sequence co-evolution gives 3D contacts and structures of protein complexes. eLife 2014, 3, e03430. [Google Scholar] [CrossRef]
- Mukherjee, I.; Chakrabarti, S. Co-evolutionary landscape at the interface and non-interface regions of protein-protein interaction complexes. Comput. Struct. Biotechnol. J. 2021, 19, 3779–3795. [Google Scholar] [CrossRef]
- Ochoa, D.; Pazos, F. Practical aspects of protein co-evolution. Front. Cell. Dev. Biol. 2014, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Zhu, J.; Shao, B.; Kong, L.; Liu, T.Y.; Zheng, W.M.; Bu, D. CopulaNet: Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction. Nat. Commun. 2021, 12, 2535. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Panchenko, A.R. Coevolution in defining the functional specificity. Proteins 2009, 75, 231–240. [Google Scholar] [CrossRef]
- Nei, M.; Rogozin, I.B.; Piontkivska, H. Purifying selection and birth-and-death ecolution in the ubiquitin gene family. Proc. Natl. Acad. Sci. USA 2000, 97, 10866–10871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, F.; Shibata, Y.; Serrano-Vega, M.J.; Tate, C.G. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 10744–10749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaban, T.; Varga, J.K.; Avraham, O.; Ben-Aharon, Z.; Khramushin, A.; Schueler-Furman, O. Harnessing protein folding neural networks for peptide-protein docking. Nat. Commun. 2022, 13, 176. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Weitzner, B.D.; Jeliazkov, J.R.; Lyskov, S.; Marze, N.; Kuroda, D.; Frick, R.; Adolf-Bryfogle, J.; Biswas, N.; Dunbrack, R.L., Jr.; Gray, J.J. Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 2017, 12, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Moreira, I.S.; Koukos, P.I.; Melo, R.; Almeida, J.G.; Preto, A.J.; Schaarschmidt, J.; Trellet, M.; Gumus, Z.H.; Costa, J.; Bonvin, A. SpotOn: High Accuracy Identification of Protein-Protein Interface Hot-Spots. Sci. Rep. 2017, 7, 8007. [Google Scholar] [CrossRef] [Green Version]
- Paulus, J.K.; Schlieper, D.; Groth, G. Greater efficiency of photosynthetic carbon fixation due to single amino-acid substitution. Nat. Commun. 2013, 4, 1518. [Google Scholar] [CrossRef] [Green Version]
- Brutnell, T.P.; Wang, L.; Swartwood, K.; Goldschmidt, A.; Jackson, D.; Zhu, X.-G.; Kellogg, E.; Van Eck, J. Setaria viridis: A Model for C4 Photosynthesis. Plant Cell 2010, 22, 2537–2544. [Google Scholar] [CrossRef]
- Christin, P.-A.; Salamin, N.; Kellogg, E.A.; Vicentini, A.; Besnard, G. Integrating Phylogeny into Studies of C4 Variation in the Grasses. Plant Physiol. 2009, 149, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermakova, M.; Danila, F.R.; Furbank, R.T.; von Caemmerer, S. On the road to C4 rice: Advances and perspectives. Plant J. 2020, 101, 940–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibberd, J.M.; Sheehy, J.E.; Langdale, J.A. Using C4 photosynthesis to increase the yield of rice—Rationale and feasibility. Curr. Opin. Plant Biol. 2008, 11, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Reeves, G.; Grange-Guermente, M.J.; Hibberd, J.M. Regulatory gateways for cell-specific gene expression in C4 leaves with Kranz anatomy. J. Exp. Bot. 2017, 68, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Sage, R.F. The evolution of C 4 photosynthesis. New Phytol. 2004, 161, 341–370. [Google Scholar] [CrossRef]
- Wang, P.; Fouracre, J.; Kelly, S.; Karki, S.; Gowik, U.; Aubry, S.; Shaw, M.K.; Westhoff, P.; Slamet-Loedin, I.H.; Quick, W.P.; et al. Evolution of GOLDEN2-LIKE gene function in C(3) and C(4) plants. Planta 2013, 237, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gowik, U.; Tang, H.; Bowers, J.E.; Westhoff, P.; Paterson, A.H. Comparative genomic analysis of C4 photosynthetic pathway evolution in grasses. Genome Biol. 2009, 10, R68. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Weissmann, S.; Wang, M.; Du, B.; Huang, L.; Wang, L.; Tu, X.; Zhong, S.; Myers, C.; Brutnell, T.P.; et al. Identification of Photosynthesis-Associated C4 Candidate Genes through Comparative Leaf Gradient Transcriptome in Multiple Lineages of C3 and C4 Species. PLoS ONE 2015, 10, e0140629. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Lee, C.; Lim, S.W.; Adhikari, A.; Andring, J.T.; McKenna, R.; Ghim, C.M.; Kim, C.U. Elucidating the role of metal ions in carbonic anhydrase catalysis. Nat. Commun. 2020, 11, 4557. [Google Scholar] [CrossRef]
- Minges, A.; Ciupka, D.; Winkler, C.; Hoppner, A.; Gohlke, H.; Groth, G. Structural intermediates and directionality of the swiveling motion of Pyruvate Phosphate Dikinase. Sci. Rep. 2017, 7, 45389. [Google Scholar] [CrossRef]
- Rao, X.; Dixon, R.A. The Differences between NAD-ME and NADP-ME Subtypes of C4 Photosynthesis: More than Decarboxylating Enzymes. Front. Plant Sci. 2016, 7, 1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; George-Jaeggli, B.; Bouteille-Pallas, M.; Tai, S.; Cruickshank, A.; Jordan, D.; Mace, E. Genetic Diversity of C4 Photosynthesis Pathway Genes in Sorghum bicolor (L.). Genes 2020, 11, 806. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Struik, P.C. C3 and C4 photosynthesis models: An overview from the perspective of crop modelling. NJAS -Wagening. J. Life Sci. 2009, 57, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Liao, D. Concerted evolution: Molecular mechanism and biological implications. Am. J. Hum. Genet. 1999, 64, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Lehti-Shiu, M.D.; Shiu, S.H. Diversity, classification and function of the plant protein kinase superfamily. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2619–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas, V.H.; Ranganathan, R. Coevolution-based inference of amino acid interactions underlying protein function. eLife 2018, 7, e34300. [Google Scholar] [CrossRef]
- Rosen, C.B.; Francis, M.B. Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol. 2017, 13, 697–705. [Google Scholar] [CrossRef]
- Yeom, J.; Ju, S.; Choi, Y.; Paek, E.; Lee, C. Comprehensive analysis of human protein N-termini enables assessment of various protein forms. Sci. Rep. 2017, 7, 6599. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.D.; Wahl, L.M.; Gloor, G.B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 2008, 24, 333–340. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, S.; Deane, C. Co-evolution techniques are reshaping the way we do structural bioinformatics. F1000Research 2017, 6, 1224. [Google Scholar] [CrossRef]
- John, C.R.; Smith-Unna, R.D.; Woodfield, H.; Covshoff, S.; Hibberd, J.M. Evolutionary convergence of cell-specific gene expression in independent lineages of C4 grasses. Plant Physiol. 2014, 165, 62–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010, 42, 1060–1067. [Google Scholar] [CrossRef]
- Mamidi, S.; Healey, A.; Huang, P.; Grimwood, J.; Jenkins, J.; Barry, K.; Sreedasyam, A.; Shu, S.; Lovell, J.T.; Feldman, M.; et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 2020, 38, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bunemann, M.; Kolb, P. The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef] [PubMed]
- Jubb, H.; Blundell, T.L.; Ascher, D.B. Flexibility and small pockets at protein-protein interfaces: New insights into druggability. Prog. Biophys. Mol. Biol. 2015, 119, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sijbesma, E.; Visser, E.; Plitzko, K.; Thiel, P.; Milroy, L.G.; Kaiser, M.; Brunsveld, L.; Ottmann, C. Structure-based evolution of a promiscuous inhibitor to a selective stabilizer of protein-protein interactions. Nat. Commun. 2020, 11, 3954. [Google Scholar] [CrossRef] [PubMed]
- Panjkovich, A.; Daura, X. Assessing the structural conservation of protein pockets to study functional and allosteric sites: Implications for drug discovery. BMC Struct. Biol. 2010, 10, 9. [Google Scholar]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Gouridis, G.; Muthahari, Y.A.; de Boer, M.; Griffith, D.A.; Tsirigotaki, A.; Tassis, K.; Zijlstra, N.; Xu, R.; Eleftheriadis, N.; Sugijo, Y.; et al. Structural dynamics in the evolution of a bilobed protein scaffold. Proc. Natl. Acad. Sci. USA 2021, 118, e2026165118. [Google Scholar] [CrossRef]
- Martin, L.C.; Gloor, G.B.; Dunn, S.D.; Wahl, L.M. Using information theory to search for co-evolving residues in proteins. Bioinformatics 2005, 21, 4116–4124. [Google Scholar] [CrossRef] [Green Version]
- Gloor, G.B.; Martin, L.C.; Wahl, L.M.; Dunn, S.D. Mutual Information in Protein Multiple Sequence Alignments Reveals Two Classes of Coevolving Positions. Biochemistry 2005, 44, 7156–7165. [Google Scholar] [CrossRef]
- Kim, D.H.; Hwang, I. Direct targeting of proteins from the cytosol to organelles: The ER versus endosymbiotic organelles. Traffic 2013, 14, 613–621. [Google Scholar] [CrossRef]
- Kunze, M.; Berger, J. The similarity between N-terminal targeting signals for protein import into different organelles and its evolutionary relevance. Front. Physiol. 2015, 6, 259. [Google Scholar] [CrossRef] [Green Version]
- Kajala, K.; Covshoff, S.; Karki, S.; Woodfield, H.; Tolley, B.J.; Dionora, M.J.; Mogul, R.T.; Mabilangan, A.E.; Danila, F.R.; Hibberd, J.M.; et al. Strategies for engineering a two-celled C(4) photosynthetic pathway into rice. J. Exp. Bot. 2011, 62, 3001–3010. [Google Scholar] [CrossRef]
- Wang, M.; Kapralov, M.V.; Anisimova, M. Coevolution of amino acid residues in the key photosynthetic enzyme Rubisco. BMC Ecol. Evol. 2011, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, G. Protein–Protein Interaction Interfaces and their Functional Implications. R. Soc. Chem. 2021, 78. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, D.; Pazos, F. Studying the co-evolution of protein families with the Mirrortree web server. Bioinformatics 2010, 26, 1370–1371. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Jiang, H.; Xiong, L.; Zan, J.; Liu, J.; Yang, M.; Zheng, K.; Wang, Z.; Nie, L. Detecting coevolution of positively selected in turtles sperm-egg fusion proteins. Mech. Dev. 2019, 156, 1–7. [Google Scholar] [CrossRef]
- Zhou, H.; Jakobsson, E. Predicting protein-protein interaction by the mirrortree method: Possibilities and limitations. PLoS ONE 2013, 8, e81100. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Baker, F.N.; Porollo, A. CoeViz: A web-based tool for coevolution analysis of protein residues. BMC Bioinform. 2016, 17, 119. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Guo, D. Computational Docking Reveals Co-Evolution of C4 Carbon Delivery Enzymes in Diverse Plants. Int. J. Mol. Sci. 2022, 23, 12688. https://doi.org/10.3390/ijms232012688
Wu C, Guo D. Computational Docking Reveals Co-Evolution of C4 Carbon Delivery Enzymes in Diverse Plants. International Journal of Molecular Sciences. 2022; 23(20):12688. https://doi.org/10.3390/ijms232012688
Chicago/Turabian StyleWu, Chao, and Dianjing Guo. 2022. "Computational Docking Reveals Co-Evolution of C4 Carbon Delivery Enzymes in Diverse Plants" International Journal of Molecular Sciences 23, no. 20: 12688. https://doi.org/10.3390/ijms232012688
APA StyleWu, C., & Guo, D. (2022). Computational Docking Reveals Co-Evolution of C4 Carbon Delivery Enzymes in Diverse Plants. International Journal of Molecular Sciences, 23(20), 12688. https://doi.org/10.3390/ijms232012688