1. Introduction
Biological systems in a cell are controlled by proteins, and while many proteins function independently, the majority work in conjunction with others to function properly. The term protein-protein interaction (PPI) refers to physical contact between two or more protein molecules arising from a combination of electrostatic forces, hydrogen bonds, and hydrophobic interactions. The residues of the sequence of proteins that facilitate the connection between them are referred to as interaction sites.
Detecting the interaction sites of proteins will help researchers understand the mechanism of various biological processes, disease development, and drug design [
1]. Protein-protein interactions play a preponderant role on the functionality of proteins, and it is because of this that some databases like PDB [
2] or Uniprot [
3] include information about the interactions and interaction sites. Other databases, such as PiSite [
4], contain exclusively protein interaction sites.
Interaction site prediction can be achieved experimentally [
5,
6] or computationally [
7,
8]. Experimental methods are expensive, time consuming, and label intensive. Therefore, computational methods are prevalent. Computational models can be further classified into sequence-based methods [
1,
9,
10], that rely only on the protein sequences, and those using also the protein structure as input [
11,
12,
13]. Considering the fact that the number of existing protein sequences outnumbers the number of available structures by two orders of magnitude [
14], sequence-based methods have the advantage of being much more widely applicable. For this reason, our new method, PITHIA, belongs to this category.
In recent years, machine learning methods and deep learning models have been extensively employed and have shown a great promise for applications in the field of protein interaction site prediction. Specifically, in sequence-based models, features like position-specific scoring matrix (PSSM), evolutionary conservation (ECO), putative relative solvent accessibility (RSA) have been used extensively in numerous models. With respect to machine learning models, support vector machine (SVM) classifiers [
15,
16], random forests [
17] or the combination of the two [
18] have been studied and have achieved good results. There exist multiple models that use recurrent neural networks (RNN) to improve the predictions, such as DLPred [
1] that uses SLSTM, a modified version of the long short-term memory architecture (LSTM). Although convolutional neural network (CNN) models are mostly used in the field of image recognition, a number of studies have demonstrated that it could be effective for prediction of interaction sites. In addition to using structural information as input, a hybrid method like DeepPPISP [
13] that combines a CNN and a multilayer perceptron (MLP), can further improve the performance of the model. Furthermore, this architecture processes entire sequences to enhance prediction. DELPHI [
9] combines both CNN and RNN to further improve the performance. Additionally, it also adds some new features to the model like HSP [
19], position information, and a reduced 3-mer amino acid embedding, ProtVec1D [
20] and exhibits their effectiveness. Finally, SCRIBER [
10] uses only logistic regression, yet provides a successful model due to its cross-prediction minimization approach.
Representing words as vectors in natural language processing has shown that their meaning can be captured in vector embeddings, such as those produced by word2vec [
21] or GloVe [
22]. Such embeddings can represent meaning in a very convenient way, since operations on vectors have a direct relationship with meaning; for example,
results in a vector closest to
[
21]. Fixed embeddings were soon replaced by the more effective contextual ones, such as BERT [
23] and ELMo [
24]. BERT uses the very efficient attention model of transformers [
25]. In recent years, there have been multiple models that tried to represent amino acids with embedding vectors employing the similar algorithms that exist in NLP [
26,
27,
28].
Sequence alignment is the most widely used procedure for extracting useful information from sequences. Multiple sequence alignment (MSA) can identify deeply hidden relations between genes and evolutionary patterns by detecting structural or functional similarities among proteins in the same family. In particular, it could be used for identifying interaction sites. The powerful MSA has been combined with the highly successful attention learning model of transformers [
25] to produce the MSA-transformer [
26].
We introduce in this paper our new model, PITHIA, that uses the embeddings computed by the MSA-transformer in combination with an attention-based deep learning architecture to produce the most efficient model to date. PITHIA surpasses the current state-of-the-art programs by a wide margin. We prove this by throughly testing and comparing the programs on several of the most widely used datasets. Along the way, we update the older datasets with more recent and complete interaction site information, as well as design a new dataset that is the largest and most suitable for testing.
3. Materials and Methods
3.1. Datasets
The majority of datasets which have been used for either training or testing are outdated due to numerous changes in the protein sequences and interaction sites. Consequently, it is critical to use updated information if we want to create a more accurate model. To update the older datasets and to create a new one, we used the most recent version (January 2019) of protein interaction residue chains provided by the PiSite database [
4].
The existing datasets we consider are Dset_72 [
32], Dset_164, Dset_186, [
33], and Dset_448 [
10]. Dset_448 is fairly recent but the others are not. We updated all datasets except Dset_448 with the most recent information in PiSite, to obtain the new versions: NDset_72, NDset_164, NDset_186. The first step was to search the PDB database for each sequence of the old datasets and match it with the provided ID. In case of multiple matches, we chose the ID of the sequence that has the highest similarity with the given sequence. After matching the IDs with their corresponding sequences, we searched PiSITE for the IDs and we updated the sequences and the interaction sites accordingly.
We also created a completely new testing dataset, Dset_500, as follows. We took all 22,654 proteins from PiSite and removed all sequences with no interaction residues or containing less than 50 amino acids; 14,203 sequences remained after this elimination. We then used PSI-CD-HIT [
34,
35] to detect and remove all sequences that have at least 25% similarity with any sequence in the training datasets of the main competing programs. After this we retain only the 2985 sequences that are alone in their clusters, from which we randomly selected 500 proteins to form Dset_500. Note that the sequences in Dset_500 not only have no similarities with any sequences in the other datasets but also with each other. This makes Dset_500 not only the largest but also the most dissimilar dataset, which makes it the most suitable for testing.
The process of creating a training dataset is described next. Starting with the 14,203 protein sequences obtained above, we use PSI-CD-HIT to remove any sequences that have any similarity above 25% with any of the testing datasets, NDset_72, NDset_164, NDset_186, Dset_448, Dset_500. The remaining proteins, 11,523, form PITHIA’s training dataset; this is further split into training (80%) and validation (20%) sets.
Table 3 gives an overview of all datasets.
3.2. Input Features
3.2.1. Embeddings
Recently, a lot of attention has been given to protein language models that can capture various protein properties from unsupervised learning on millions of sequences [
26,
36,
37]. The embeddings computed using transformer architectures outperform older methods in many applications. Transformers attention was coupled with the most widely used tool in bioinformatics, multiple sequence alignment, to produce the best protein folding prediction model, AlphaFold [
38]. However, this was completed in a supervised manner. The first unsupervised use of multiple sequence alignment with attention is in the MSA-transformer of [
39], which creates a 768-dimensional vector for each amino acids. These embeddings are the main feature used by our model.
The multiple sequence alignments are computed using HHblits [
40] on the UniRef-50 database [
41] dated 2020-03. Default settings are used except for HHblits’s number of search iterations (
-n), which is instead set to 4. With this change, the algorithm will be able to find more sequences which could be useful for those proteins that might not have enough sequences. At the same time, since the result is ordered based on the similarity, increasing the number of iterations does not affect the quality of the sequences for the majority of the proteins.
The MSA-transformer, by default, uses the first 128 sequences to create the embedding. We have tested and compared several values for the number of sequences: 32, 64, and 128. For those proteins which total number of sequences in their MSA were less than the number required by the algorithm, the embeddings were computed with the sequences available, in order to enable testing of the model on any protein sequence.
3.2.2. Other Features
The main feature of our model is the embeddings created by the MSA-transformer, but we study also the impact of traditionally used features, outlined below.
PSSM: Position-Specific Scoring Matrix (PSSM) represents protein sequences in an intuitive and highly informative way. As a result, PSSM-based feature descriptors have proven effective in improving the performance of a variety of protein attribute predictors. PSI-Blast [
42] is used to compute the PSSM matrices with the number of iterations set to 3. Using PSI-Blast, each input sequence is aligned multiple times against the non-redundant database.
Physicochemical characteristics: These characteristics include three aspects of each amino acid: atom count, electrostatic charge and hydrogen bond potential [
10].
Evolutionary Conservation (ECO): It reflects the fact that genes, regions of genes, or chromosome segments are conserved among species, not only demonstrating the common ancestry of species, but also implying a functional characteristic of the conserved element. The ECO score which is a one dimensional feature is computed with the same procedure that has been described in [
10].
RSA: For determining protein folding and stability, the accessible surface area (ASA) of proteins has long been regarded as one of the main factors. ASA is commonly expressed in terms of RSA (Relative Solvent Accessibility). This is a one dimensional feature which is predicted using ASAquick [
43].
RAA: An AA interaction propensity is defined by the relative abundance of a given AA type in comparison to the corresponding noninteraction residues on the protein surface. This is a one dimensional feature which is computed with the same formula presented in [
44].
Hydropathy index: An amino acid’s hydropathy index is a number that indicates whether its sidechain is hydrophobic or hydrophilic [
45]. Generally, the larger the number, the more hydrophobic the amino acid.
PKx: Dissociation constants measure the propensity of a molecule to dissociate into its constituent parts [
46]. There are three different dissociation constants: PK
a is the negative of the logarithm of the dissociation constant for the -COOH group, PK
b is the negative of the logarithm of the dissociation constant for the -NH3 group, and PK
x is the negative of the logarithm of the dissociation constant for any other group in the molecule. This paper follows the same patterns that other papers follow and it only considers PK
x as a side feature.
Putative protein-interaction disorder: Functional elements of disordered proteins play a significant role in protein-protein interactions. This is a one dimensional feature which is computed using the ANCHOR program [
47].
Physical properties: Each amino acid type is assigned a 7D property. Among them are graph shape index, polarizability, volume (normalized van der Waals volume), hydrophobicity, isoelectric point, helix probability, and sheet probability. Pre-computed values are taken from [
10].
To improve the convergence of the model, the values of the feature vectors are normalized, after they have been computed, using the min-max normalization formula:
Many papers have evaluated the aforementioned features, and all of them found that these features were able to increase prediction accuracy. ECO, PSSM, and multiple sequence alignment, on the other hand, use HHbilts as their core layer. Consequently, it is expected that the embeddings created by the MSA-transformer already incorporate ECO and PSSM information to some extent. Furthermore, ref. [
37] shows that the embeddings are capable of understanding the physicochemical and physical characteristic of the protein residues. It is therefore interesting to investigate how much information these features can add to the MSA-transformer embeddings.
3.3. Model Architecture
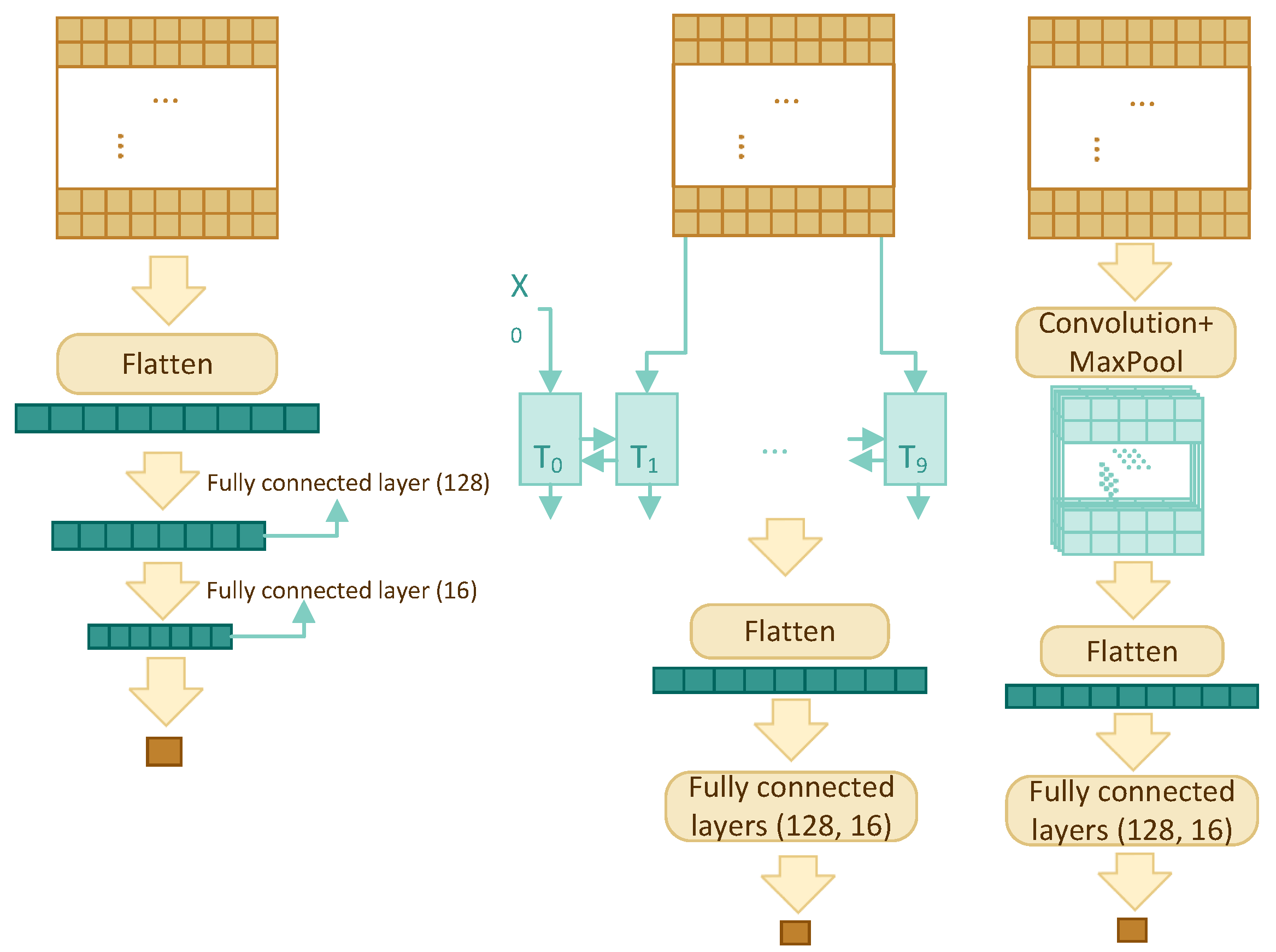
Four different architectures have been compared in order to find the best model: multilayer perceptron (MLP), recurrent neural network (RNN), convolutional neural network (CNN), and transformer self-attention (TF). The architectures are shown schematically in
Figure 2, for the first three architectures, and
Figure 3, for TF, and discussed in detail below.
All architectures accept a 2-dimensional input of size , where w is window size, and output a single value between 0 and 1, which is the predicted interaction likelihood of the residue residing in the centre of the window.
3.3.1. Multilayer Perceptron
This is the simplest yet one of the most effective models in this paper. It uses the sliding window and the concept of many-to-one. The model consists of a flatten layer and three fully connected layers with dropout for regularization. The input is flattened and fed into a fully connected layer to reduce its size to 128, then everything passes through two more fully connected layers to reduce the dimension to one.
3.3.2. RNN
The RNN component consists of a bidirectional GRU layer, a flatten layer, and two fully connected layers. A bidirectional GRU layer is applied to the feature profile with the intention of storing the dependency and relationship among the w residues. Instead of returning a single value, we have set the GRU layer to return an entire sequence. Flattened results are fed into two fully connected layers with dropout to reduce the dimension to one.
3.3.3. CNN
The two-dimensional (2D) CNN model has one convolution layer, one max pooling layer, one flatten layer, and two fully connected layers with dropout. The sigmoid activation function is used in the last fully connected layer, so that the output is a single number between 0 and 1.
In contrast to two-dimensional CNN’s which are mostly used for images, one-dimensional (1D) CNN’s are used for text and one-dimensional signals. The architecture of one-dimensional CNN is similar to the 2D CNN; it has one convolution layer, one max pooling layer, one flatten layer, and two fully connected layers.
3.3.4. Self-Attention
One of the important parts of Transformers is self-attention. The TF model consists of a multihead attention layer, a flatten layer, and three fully connected layers with dropout. If side features are used (other than embeddings), then the size of input is reduced to before concatenating the side features.
As illustrated in
Figure 3, the input is of size
and the attention layer, depending on the number of heads (three, in the figure), creates multiple sets of Query, Value, and Key matrices. These matrices are the results of multiplication between embeddings and different weight matrices whose values are tuned through training. The model then uses the following equation to capture information from each head:
The results from all heads are then concatenated and multiplied with a weight matrix that is trained jointly with the model; the final output is flattened and is fed to three fully connected layers with dropout before obtaining the final result.
3.4. Implementation
The program is written in Keras [
48] (Python 3.6.2) with TensorFlow GPU [
49] as a backend. In order to process the sliding windows for each amino acid in a sequence, since each amino acid is embedded into a vector of size 768, it would have required around 700 GB of space to be able to fit the whole dataset in the RAM. In order to mitigate this problem, we used generators to build up batches on the fly. Generators are special types of functions that return a sequence of values instead of an individual value. They allow the program to load only the amount of data necessary for the current epoch, instead of loading the entire dataset. Although this method decreases the required space in memory, it will increase the I/O and it will not let the program use the architecture to its full potential. One of the best practices in deep learning is to shuffle the whole dataset before feeding it to the architecture. For our specific problem, this is crucial due to the nature of the data. When each window is extracted using the sliding window, adjacent windows are very similar—only the first and last residues are different. Shuffling the entire training dataset makes each batch more heterogeneous. This shuffling might create a batch that each of its instances comes from a different protein. Since the input file expected to be a FASTA format and with the generators the program only reads small parts of the input file, we could not use global shuffling, instead, we used local shuffling. We have increased the batch size to 1024 so that, considering that most proteins have fewer than 400 amino acids in their sequence, using a batch size of 1024 makes it possible to have multiple proteins in each batch.
In the end, we have managed to both reduce the memory and properly prepare the data for learning. With this method, it was possible to generate the dataset on multiple cores in real time and feed it into the deep learning model. Our architecture is able to be run on a server with 64 Gigabytes of RAM, 12 CPU cores, and one GPU (model T4 with 16 Gigabytes of VRAM). It takes about 24 h to train the model for 100 epochs. Testing takes about 10 s per sequence if embeddings are available, and about 10–20 min to compute the embeddings.
3.5. Selecting the PITHIA Model
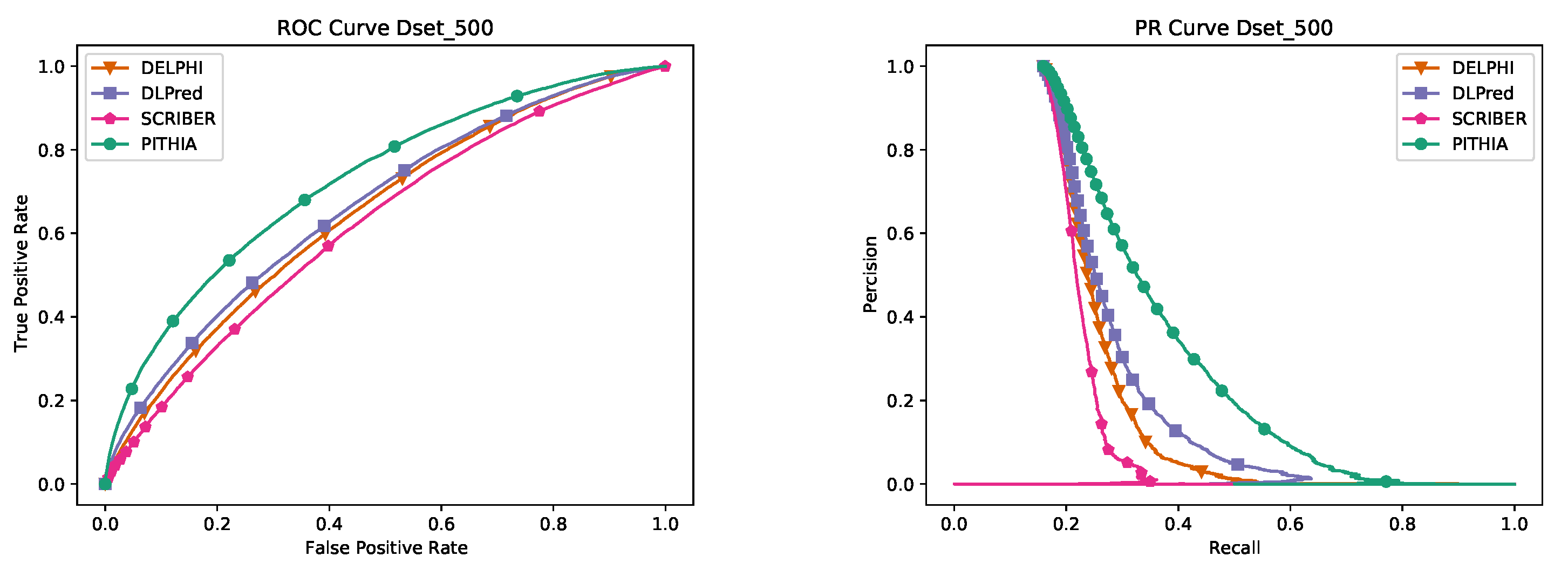
A number of models have been built and compared using the training dataset; each model was trained on 80% of the data and then its performance was measured on the 20% validation data. The model with the best performance, measured as the area under the PR curve, on the validation data was chosen as the final PITHIA model. (No testing data was used in training any of the models discussed.)
We considered the four architectures mentioned above, with various parameter combinations from
Table 4, as well as several multiple alignment sizes. The final PITHIA model was the transformer self-attention model with one head, using a sliding window of size
over a sequence with zero padding at the ends, whose
input consists of the 768-dimensional MSA-Transformer embeddings corresponding to the 9 residues in the sliding window.
3.5.1. Model Robustness
After establishing our model using the validation data, we check its robustness by comparing a number of models on the Dset_500 dataset. The results are shown in
Table 5 where various architectures, window sizes, number of heads, and multiple alignment sizes have been considered. The final PITHIA model outperforms all the other candidates. It is interesting to remark that while the original paper [
26] suggests using 128 sequences would create the best predictions for their purpose, our best model was obtained using 64 sequences.
Interestingly, the second best model in
Table 5 is the MLP with the same window size as PITHIA. We have performed another test comparing only these two models on a different dataset, Dset_448. The results in
Table 6 show that our PITHIA model outperforms more clearly the MLP contender.
3.5.2. Side Features
We have tested the impact of adding some or all of the other features, in addition to the MSA-transformer embeddings. Some of the results are shown in
Table 7. It is clear from the results, particularly in the PR column, that there is practically no improvement due to adding these features. Therefore, for simplicity, as well as avoiding the need to compute these features, we have decided to keep as input features the 768-dimensional embeddings alone. We note that, in agreement with [
39], the embeddings appear to already contain the information provided by all the other features investigated.
Due to the large difference between the amino acid embedding size of 768 and the small dimension of a side feature—e.g., PSSM has dimension 20—a fully connected layer is added to the model after the self-attention layer in order to reduce the 768 dimension to 128 before concatenation with the side features.
4. Discussion
In this paper we propose a novel approach to the problem of protein interaction site prediction using embeddings produced by the MSA-transformer and a self-attention-based architecture. Our new model, PITHIA, clearly outperforms the state-of-the-art methods. We show this by extensive testing on multiple datasets, updated and a newly created one, largest and most relevant for testing.
In view of the above results, the old wisdom that says: “one or two homologous sequences whisper […] a full multiple alignment shouts out loud” [
50], receives confirmation into yet another dimension: a choir of MSAs sings much louder than any single alignment.
Protein interaction site prediction is a fundamental problem and there is still a lot of room for improvement. Our new model makes use of very powerful concepts, multiple sequence alignments and attention, which make it work much better than the existing methods. The ideas used here should be useful for improving the prediction of proteins with other molecules, such as DNA, RNA, and other ligands.
Availability
The web server is available at
pithia.csd.uwo.ca (accessed on 21 September 2022). It has been developed using Python Flask 2.1, Celery 5.2, and Redis 7.0 on Ubuntu 18.04 operating system. The user inputs protein sequences and receive the prediction results via e-mail. The average computation time for a protein of length 500 on the web server is 15 min.








{kind=link}
{kind=link}
{kind=link}
{kind=link}