
Hereditary Disorders of Manganese Metabolism: Pathophysiology of Childhood-Onset Dystonia-Parkinsonism in SLC39A14 Mutation Carriers and Genetic Animal Models
Abstract
:1. Manganese: A Double-Edge Sword—Essential Metal and Neurotoxicant
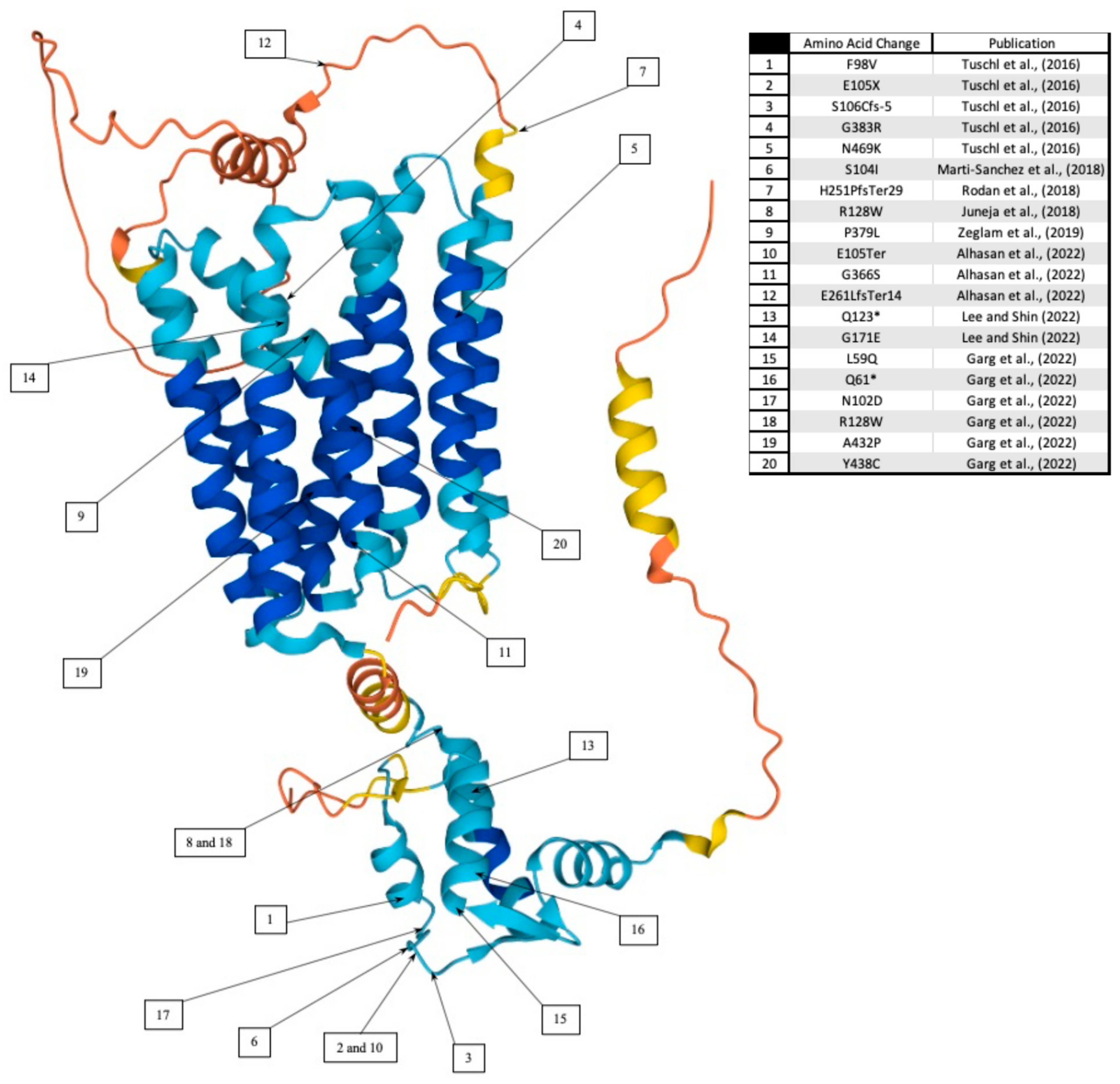
1.1. Childhood-Onset Dystonia-Parkinsonism in SLC39A14 Mutation Carriers
1.2. Clinical Presentation
1.3. Neuropathological Findings in Mutation Carriers
1.4. Current Therapeutic Approaches in SLC39A14 Mutation Carriers
2. Experimental Animal Model(s) Using Global Slc39a14 Gene Deletion
2.1. Behavioral Characterization
2.2. Pathophysiological Characterization of Slc39a14 Knockout Mice
2.3. Loss of slc39a14 in a Zebrafish Animal Model
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aschner, J.L.; Aschner, M. Nutritional aspects of manganese homeostasis. Mol. Aspects Med. 2005, 26, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Todd, G.D.; Roney, N.; Crawford, J.; Coles, C.; McClure, P.R.; Garey, J.D.; Zaccaria, K.; Citra, M. Toxicological Profile for Manganese; Federal Register: Atlanta, GA, USA, 2012.
- Guilarte, T.R. Manganese neurotoxicity: New perspectives from behavioral, neuroimaging, and neuropathological studies in humans and non-human primates. Front. Aging Neurosci. 2013, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R.; Gonzales, K.K. Manganese-Induced Parkinsonism Is Not Idiopathic Parkinson’s Disease: Environmental and Genetic Evidence. Toxicol. Sci. 2015, 146, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Blanc, P.D. The early history of manganese and the recognition of its neurotoxicity, 1837–1936. Neurotoxicology 2018, 64, 5–11. [Google Scholar] [CrossRef] [PubMed]
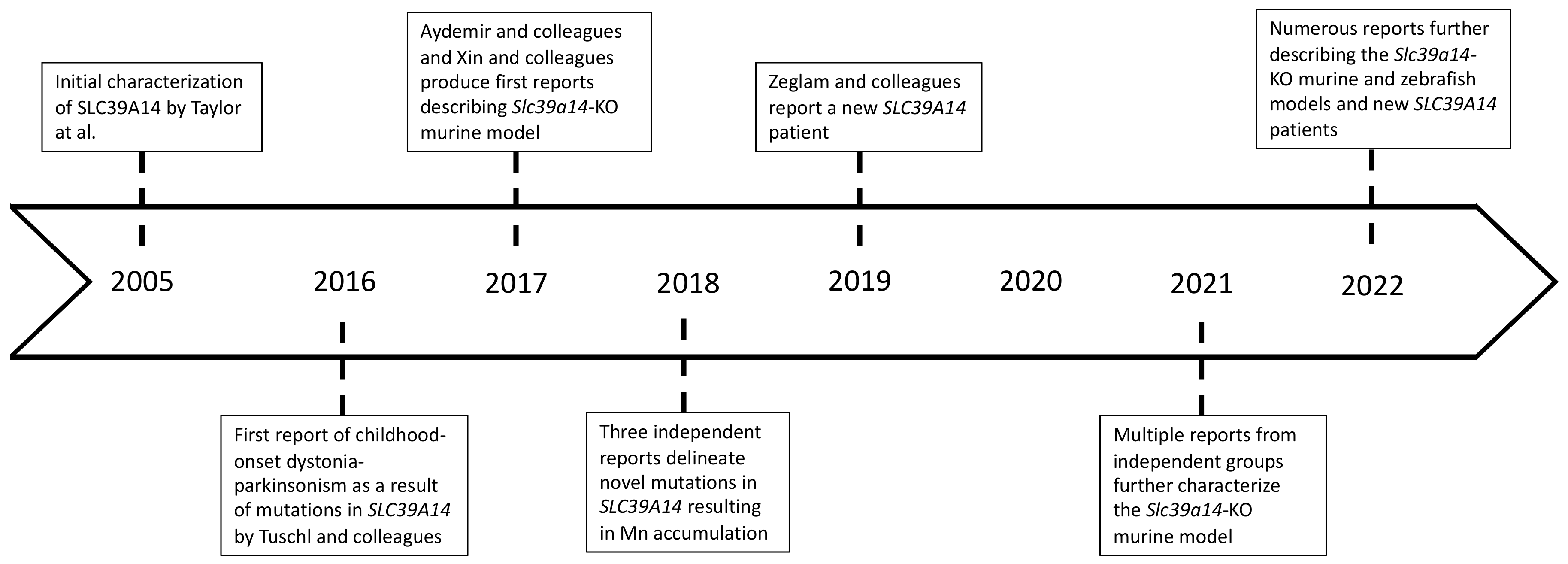
- Tuschl, K.; Meyer, E.; Valdivia, L.E.; Zhao, N.; Dadswell, C.; Abdul-Sada, A.; Hung, C.Y.; Simpson, M.A.; Chong, W.K.; Jacques, T.S.; et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 2016, 7, 11601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.C. Parkinsonism induced by chronic manganese intoxication--an experience in Taiwan. Chang. Gung. Med. J. 2007, 30, 385–395. [Google Scholar]
- Criswell, S.R.; Perlmutter, J.S.; Videen, T.O.; Moerlein, S.M.; Flores, H.P.; Birke, A.M.; Racette, B.A. Reduced uptake of [(1)(8)F]FDOPA PET in asymptomatic welders with occupational manganese exposure. Neurology 2011, 76, 1296–1301. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, S.L.; Zheng, W. Manganese Toxicity Upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Lucas, E.L.; Bertrand, P.; Guazzetti, S.; Donna, F.; Peli, M.; Jursa, T.P.; Lucchini, R.; Smith, D.R. Impact of ferromanganese alloy plants on household dust manganese levels: Implications for childhood exposure. Environ. Res. 2015, 138, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Sikk, K.; Haldre, S.; Aquilonius, S.M.; Taba, P. Manganese-Induced Parkinsonism due to Ephedrone Abuse. Parkinsons Dis. 2011, 2011, 865319. [Google Scholar] [CrossRef] [Green Version]
- Koksal, A.; Baybas, S.; Sozmen, V.; Koksal, N.S.; Altunkaynak, Y.; Dirican, A.; Mutluay, B.; Kucukoglu, H.; Keskinkilic, C. Chronic manganese toxicity due to substance abuse in Turkish patients. Neurol. India 2012, 60, 224–227. [Google Scholar]
- Sanotsky, Y.; Selikhova, M.; Fedoryshyn, L.; Kuzyk, P.; Matviyenko, Y.; Semeryak, O.; Dziewulska, D.; Holton, J.L.; Lees, A.J. Neuropathological Findings in Ephedrone Encephalopathy. Mov. Disord. 2020, 35, 1858–1863. [Google Scholar] [CrossRef]
- Hauser, R.A.; Zesiewicz, T.A.; Rosemurgy, A.S.; Martinez, C.; Olanow, C.W. Manganese intoxication and chronic liver failure. Ann. Neurol. 1994, 36, 871–875. [Google Scholar] [CrossRef]
- Cherian, A.; Priya, L.; Divya, K.P. “Cock-walk” gait and “horseshoe moustache” sign on MRI in inherited hypermanganesemia. Neurol. Sci. 2022, 43, 1441–1445. [Google Scholar] [CrossRef]
- Quadri, M.; Federico, A.; Zhao, T.; Breedveld, G.J.; Battisti, C.; Delnooz, C.; Severijnen, L.A.; Di Toro Mammarella, L.; Mignarri, A.; Monti, L.; et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 2012, 90, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Tuschl, K.; Clayton, P.T.; Gospe, S.M., Jr.; Gulab, S.; Ibrahim, S.; Singhi, P.; Aulakh, R.; Ribeiro, R.T.; Barsottini, O.G.; Zaki, M.S.; et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 2012, 90, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Stamelou, M.; Tuschl, K.; Chong, W.K.; Burroughs, A.K.; Mills, P.B.; Bhatia, K.P.; Clayton, P.T. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: A new treatable disorder. Mov. Disord. 2012, 27, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Lechpammer, M.; Clegg, M.S.; Muzar, Z.; Huebner, P.A.; Jin, L.W.; Gospe, S.M., Jr. Pathology of inherited manganese transporter deficiency. Ann. Neurol. 2014, 75, 608–612. [Google Scholar] [CrossRef]
- Quadri, M.; Kamate, M.; Sharma, S.; Olgiati, S.; Graafland, J.; Breedveld, G.J.; Kori, I.; Hattiholi, V.; Jain, P.; Aneja, S.; et al. Manganese transport disorder: Novel SLC30A10 mutations and early phenotypes. Mov. Disord. 2015, 30, 996–1001. [Google Scholar] [CrossRef]
- Zaki, M.S.; Issa, M.Y.; Elbendary, H.M.; El-Karaksy, H.; Hosny, H.; Ghobrial, C.; El Safty, A.; El-Hennawy, A.; Oraby, A.; Selim, L.; et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: Six novel SLC30A10 mutations and further phenotype delineation. Clin. Genet. 2018, 93, 905–912. [Google Scholar] [CrossRef]
- Balint, B.; Bhatia, K.P. SLC39A14 mutations expand the spectrum of manganese transporter defects causing parkinsonism-dystonia. Mov. Disord. 2016, 31, 1630. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, K.; Gregory, A.; Meyer, E.; Clayton, P.T.; Hayflick, S.J.; Mills, P.B.; Kurian, M.A. SLC39A14 Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; NCBI: Seattle, WA, USA, 2017. [Google Scholar]
- Juneja, M.; Shamim, U.; Joshi, A.; Mathur, A.; Uppili, B.; Sairam, S.; Ambawat, S.; Dixit, R.; Faruq, M. A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J. Gene. Med. 2018, 20, e3012. [Google Scholar] [CrossRef] [PubMed]
- Marti-Sanchez, L.; Ortigoza-Escobar, J.D.; Darling, A.; Villaronga, M.; Baide, H.; Molero-Luis, M.; Batllori, M.; Vanegas, M.I.; Muchart, J.; Aquino, L.; et al. Hypermanganesemia due to mutations in SLC39A14: Further insights into Mn deposition in the central nervous system. Orphanet J. Rare Dis. 2018, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Rodan, L.H.; Hauptman, M.; D’Gama, A.M.; Qualls, A.E.; Cao, S.; Tuschl, K.; Al-Jasmi, F.; Hertecant, J.; Hayflick, S.J.; Wessling-Resnick, M.; et al. Novel founder intronic variant in SLC39A14 in two families causing Manganism and potential treatment strategies. Mol. Genet. Metab. 2018, 124, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Zeglam, A.; Abugrara, A.; Kabuka, M. Autosomal-recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene SLC39A14. Acta Neurol. Belg. 2019, 119, 379–384. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, J.H. Effect of Chelation Therapy on a Korean Patient With Brain Manganese Deposition Resulting From a Compound Heterozygous Mutation in the SLC39A14 Gene. J. Mov. Disord. 2022, 15, 171–174. [Google Scholar] [CrossRef]
- Alhasan, K.A.; Alshuaibi, W.; Hamad, M.H.; Salim, S.; Jamjoom, D.Z.; Alhashim, A.H.; AlGhamdi, M.A.; Kentab, A.Y.; Bashiri, F.A. Hypermanganesemia with Dystonia Type 2: A Potentially Treatable Neurodegenerative Disorder: A Case Series in a Tertiary University Hospital. Children 2022, 9, 1335. [Google Scholar] [CrossRef]
- Garg, D.; Yoganathan, S.; Shamim, U.; Mankad, K.; Gulati, P.; Bonifati, V.; Botre, A.; Kalane, U.; Saini, A.G.; Sankhyan, N.; et al. Clinical Profile and Treatment Outcomes of Hypermanganesemia with Dystonia 1 and 2 among 27 Indian Children. Mov. Disord. Clin. Prac. 2022, 9, 886–899. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Taylor, K.M.; Morgan, H.E.; Johnson, A.; Nicholson, R.I. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005, 579, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Perl, D.P.; Olanow, C.W. The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol. 2007, 66, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Uchino, A.; Noguchi, T.; Nomiyama, K.; Takase, Y.; Nakazono, T.; Nojiri, J.; Kudo, S. Manganese accumulation in the brain: MR imaging. Neuroradiology 2007, 49, 715–720. [Google Scholar] [CrossRef]
- Hornykiewicz, O. A brief history of levodopa. J. Neurol. 2010, 257, S249–S252. [Google Scholar] [CrossRef]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef]
- Tuschl, K.; White, R.J.; Trivedi, C.; Valdivia, L.E.; Niklaus, S.; Bianco, I.H.; Dadswell, C.; Gonzalez-Mendez, R.; Sealy, I.M.; Neuhauss, S.C.F.; et al. Loss of slc39a14 causes simultaneous manganese hypersensitivity and deficiency in zebrafish. Dis. Model. Mech. 2022, 15, dmm044594. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Kim, M.H.; Kim, J.; Colon-Perez, L.M.; Banan, G.; Mareci, T.H.; Febo, M.; Cousins, R.J. Metal Transporter Zip14 (Slc39a14) Deletion in Mice Increases Manganese Deposition and Produces Neurotoxic Signatures and Diminished Motor Activity. J. Neurosci. 2017, 37, 5996–6006. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Gao, H.; Wang, J.; Qiang, Y.; Imam, M.U.; Li, Y.; Wang, J.; Zhang, R.; Zhang, H.; Yu, Y.; et al. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 2017, 3, 17025. [Google Scholar] [CrossRef] [Green Version]
- Jenkitkasemwong, S.; Akinyode, A.; Paulus, E.; Weiskirchen, R.; Hojyo, S.; Fukada, T.; Giraldo, G.; Schrier, J.; Garcia, A.; Janus, C.; et al. SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E1769–E1778. [Google Scholar] [CrossRef] [Green Version]
- Rodichkin, A.N.; Edler, M.K.; McGlothan, J.L.; Guilarte, T.R. Behavioral and neurochemical studies of inherited manganese-induced dystonia-parkinsonism in Slc39a14-knockout mice. Neurobiol. Dis. 2021, 158, 105467. [Google Scholar] [CrossRef]
- Giraldo, G.; Janus, C. Phenotypic evaluation of a childhood-onset parkinsonism-dystonia mouse model with inherent postural abnormalities. Brain Res. Bull. 2021, 166, 54–63. [Google Scholar] [CrossRef]
- Rodichkin, A.N.; Edler, M.K.; McGlothan, J.L.; Guilarte, T.R. Pathophysiological studies of aging Slc39a14 knockout mice to assess the progression of manganese-induced dystonia-parkinsonism. Neurotoxicology 2022, 93, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Chen, M.K.; McGlothan, J.L.; Verina, T.; Wong, D.F.; Zhou, Y.; Alexander, M.; Rohde, C.A.; Syversen, T.; Decamp, E.; et al. Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp. Neurol. 2006, 202, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Burton, N.C.; McGlothan, J.L.; Verina, T.; Zhou, Y.; Alexander, M.; Pham, L.; Griswold, M.; Wong, D.F.; Syversen, T.; et al. Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): Implications to manganese-induced parkinsonism. J. Neurochem. 2008, 107, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, M.; Aoun, R.A.; Mathews, T.A. Altered striatal dopamine release following a sub-acute exposure to manganese. J. Neurosci. Methods 2011, 202, 182–191. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Publication | Neurobehavioral Assessment | Outcome |
|---|---|---|
| [38] | Spontaneous open field locomotor activity | KO animals present with a decrease in open field locomotor activity when compared to WT controls |
| Balance beam traverse | KO animals required more time to cross the balance beam with more errors | |
| Pole descent test | KO animals exhibited an increase in the amount of time required to descend the pole | |
| Hind limb clasping reflex | Impairment of the hind limb clasping reflex function in KO animals | |
| Inverted grid assay | No difference detected between WT and KO animals | |
| [39] | Balance beam traverse | KO animals present with balance beam traverse performance beginning at 11 weeks of age |
| Rotarod | Significant decrease in maximum speed when compared to WT controls | |
| CatWalk Gait Analysis | Irregularities in gait patterns with longer swing speeds in right forelimb and left hindlimb | |
| [40] | Pole descent test | KO animals showed an increase in the overall latency to descend. Low diet Mn treatment did not have a significant effect on PDT performance |
| Balance beam traverse | KO mice slipped and/or fell at a higher frequency than the WT counterparts. Overall time to cross the beams was not significantly different between genotypes | |
| Rotarod | KO animals exhibited shorter latencies to fall off the rotating rod, relative to weight-, sex- and age-matched WT controls. Low-Mn diet did not rescue the phenotype | |
| SHIRPA Screen | No differences were detected between genotypes using the following parameters: tremors, respiration, transfer arousal, palpebral closure, piloerection, pinna reflex, limb grasping, wire maneuver, negative geotaxis. Diet did not have a significant effect on the outcomes. | |
| Qualitative Observations | KO animals appeared to be less active than the WT counterparts when compared to the WT controls in a large, open cage. | |
| [42] | SHIRPA Screen | Presence of torticollis in KO animals did not affect their scoring on the SHIRPA Screen, when compared to KO counterparts with no torticollis |
| Pole descent test | Torticollis in KO animals did not have a significant effect on PDT performance | |
| Balance beam traverse | Torticollis in KO animals did not have a significant effect on balance beam traverse performance | |
| Rotarod | Torticollis in KO animals did not have a significant effect on latency to fall on the rotarod | |
| [41] | Open field locomotor activity | KO animals presented with a decrease in overall distance travelled, speed and rearing activity when compared to age- and sex-matched WT controls. Resting time was increased. |
| Rotarod | Latency to fall was decreased in KO animals when compared to WT controls | |
| Tail suspension test | On average, KO animals exhibited more instances of hind and front limb clasping, truncal and cervical torsion. Together these behaviors are interpreted as dystonia-like movements. | |
| Qualitative Observations | At post-natal day 21, KO a proportion of KO animals exhibited backward walking, which persisted until post-natal day 60. | |
| [43] | Open field locomotor activity | KO animals presented with a decrease in overall distance travelled, speed and rearing activity when compared to age and sex matched WT controls. Resting time was increased. |
| Rotarod | Latency to fall was not significantly different from age and sex matched WT controls. However, this is likely due to a decrease in performance of the WT animals. | |
| Tail suspension test | On average, KO animals exhibited more instances of hind and front limb clasping, truncal and cervical torsion. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodichkin, A.N.; Guilarte, T.R. Hereditary Disorders of Manganese Metabolism: Pathophysiology of Childhood-Onset Dystonia-Parkinsonism in SLC39A14 Mutation Carriers and Genetic Animal Models. Int. J. Mol. Sci. 2022, 23, 12833. https://doi.org/10.3390/ijms232112833
Rodichkin AN, Guilarte TR. Hereditary Disorders of Manganese Metabolism: Pathophysiology of Childhood-Onset Dystonia-Parkinsonism in SLC39A14 Mutation Carriers and Genetic Animal Models. International Journal of Molecular Sciences. 2022; 23(21):12833. https://doi.org/10.3390/ijms232112833
Chicago/Turabian StyleRodichkin, Alexander N., and Tomás R. Guilarte. 2022. "Hereditary Disorders of Manganese Metabolism: Pathophysiology of Childhood-Onset Dystonia-Parkinsonism in SLC39A14 Mutation Carriers and Genetic Animal Models" International Journal of Molecular Sciences 23, no. 21: 12833. https://doi.org/10.3390/ijms232112833