
Neutrophils’ Extracellular Trap Mechanisms: From Physiology to Pathology

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
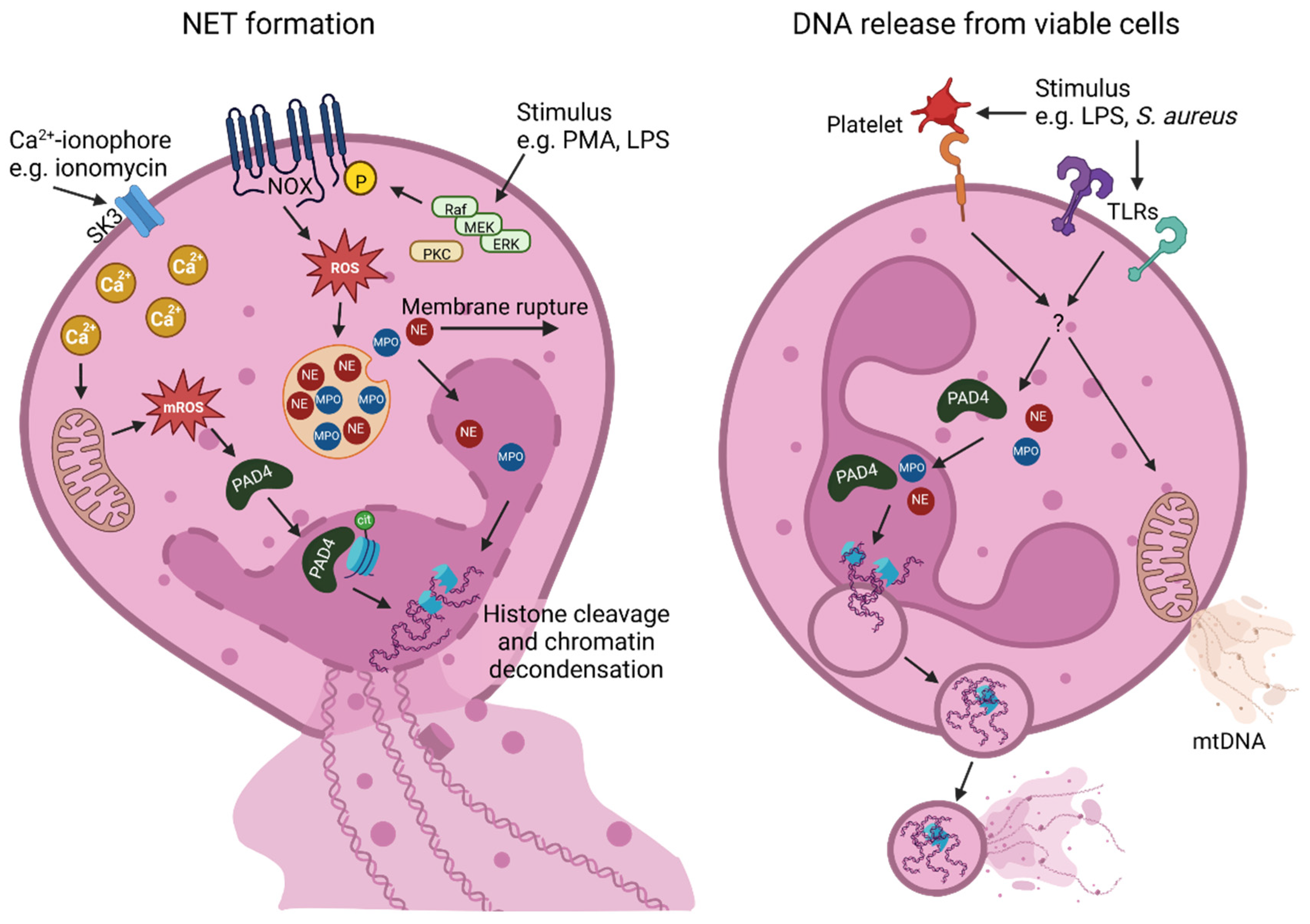
2. NET Formation Mechanisms and Their Components
2.1. NADPH Oxidase (NOX)-Dependent NET Formation
2.2. NOX-Independent NET Formation
2.3. DNA Release from Viable Cells
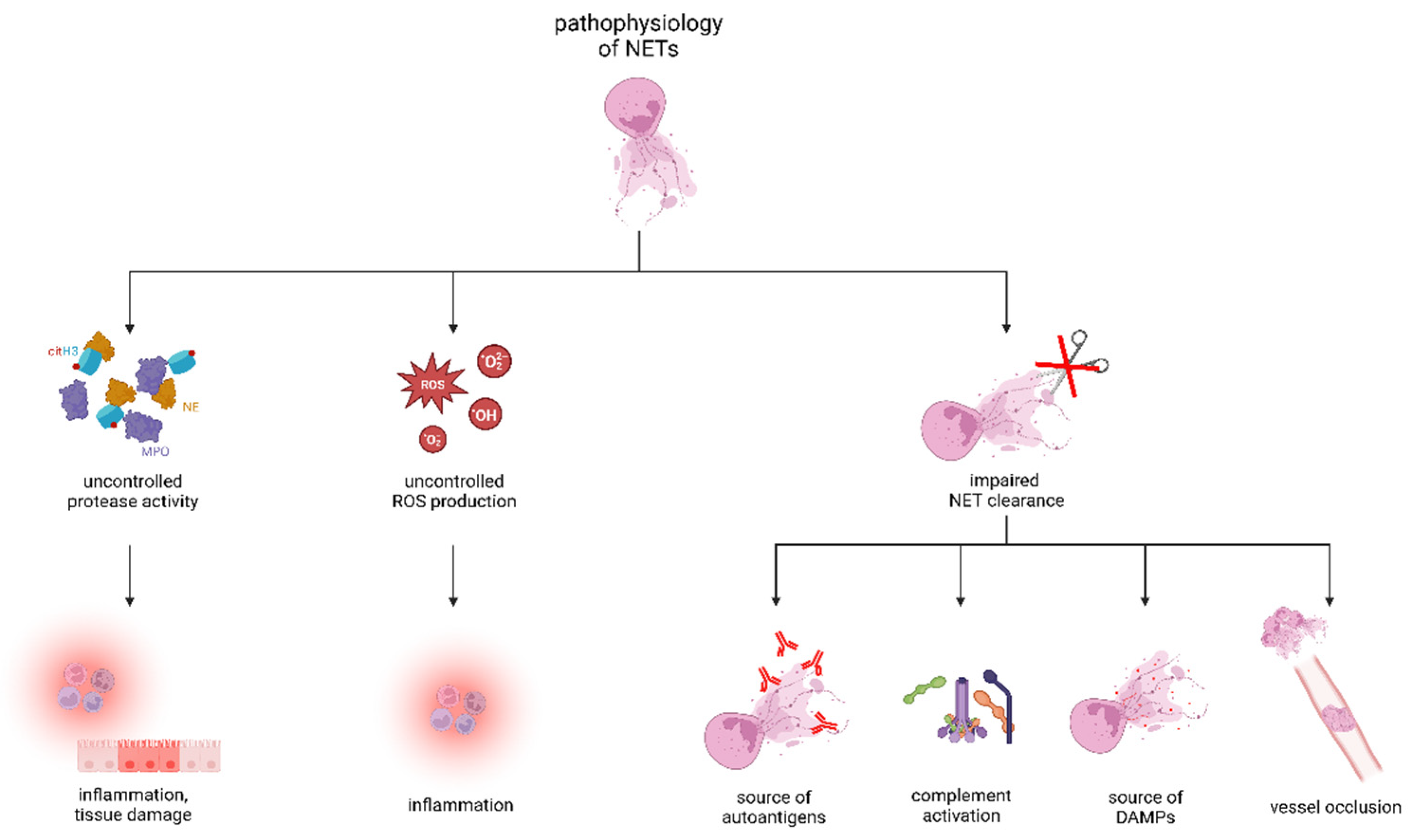
3. Physiological Functions of NETs and Removal of NETs Remnants
4. Pathophysiology of NETs in Diseases
5. Approaches to the Regulation of NET Formation
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perez-Figueroa, E.; Alvarez-Carrasco, P.; Ortega, E.; Maldonado-Bernal, C. Neutrophils: Many Ways to Die. Front. Immunol. 2021, 12, 631821. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Hsieh, S.C.; Liu, C.W.; Lu, C.S.; Wu, C.H.; Liao, H.T.; Chen, M.H.; Li, K.J.; Shen, C.Y.; Kuo, Y.M.; et al. Cross-Talk among Polymorphonuclear Neutrophils, Immune, and Non-Immune Cells via Released Cytokines, Granule Proteins, Microvesicles, and Neutrophil Extracellular Trap Formation: A Novel Concept of Biology and Pathobiology for Neutrophils. Int. J. Mol. Sci. 2021, 22, 3119. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Tackenberg, H.; Moller, S.; Filippi, M.D.; Laskay, T. The Small GTPase Cdc42 Negatively Regulates the Formation of Neutrophil Extracellular Traps by Engaging Mitochondria. Front. Immunol. 2021, 12, 564720. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Khan, M.A.; Bade, G.; Talwar, A. Orchestration of Neutrophil Extracellular Traps (Nets), a Unique Innate Immune Function during Chronic Obstructive Pulmonary Disease (COPD) Development. Biomedicines 2021, 9, 53. [Google Scholar] [CrossRef]
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef]
- Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; Migliorini, P.; et al. Neutrophil Extracellular Traps in the Autoimmunity Context. Front. Med. 2021, 8, 614829. [Google Scholar] [CrossRef]
- Yaykasli, K.O.; Schauer, C.; Munoz, L.E.; Mahajan, A.; Knopf, J.; Schett, G.; Herrmann, M. Neutrophil Extracellular Trap-Driven Occlusive Diseases. Cells 2021, 10, 2208. [Google Scholar] [CrossRef]
- Daniel, C.; Leppkes, M.; Munoz, L.E.; Schley, G.; Schett, G.; Herrmann, M. Extracellular DNA traps in inflammation, injury and healing. Nat. Rev. Nephrol. 2019, 15, 559–575. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Grinstein, S. Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death. Sci. STKE 2007, 2007, pe11. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NETs of neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, Z.; Diaz-Godinez, C.; Mora, N.; Aleman, O.R.; Uribe-Querol, E.; Carrero, J.C.; Rosales, C. Entamoeba histolytica Induce Signaling via Raf/MEK/ERK for Neutrophil Extracellular Trap (NET) Formation. Front. Cell Infect. Microbiol. 2018, 8, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douda, D.N.; Yip, L.; Khan, M.A.; Grasemann, H.; Palaniyar, N. Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood 2014, 123, 597–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, A.J.; Jin, Y.; Knudsen, G.M.; Perera, N.C.; Jenne, D.E.; Murphy, J.E.; Craik, C.S.; Hermiston, T.W. Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS ONE 2013, 8, e75141. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef]
- Behnen, M.; Moller, S.; Brozek, A.; Klinger, M.; Laskay, T. Extracellular Acidification Inhibits the ROS-Dependent Formation of Neutrophil Extracellular Traps. Front. Immunol. 2017, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Philip, L.M.; Cheung, G.; Vadakepeedika, S.; Grasemann, H.; Sweezey, N.; Palaniyar, N. Regulating NETosis: Increasing pH Promotes NADPH Oxidase-Dependent NETosis. Front. Med. 2018, 5, 19. [Google Scholar] [CrossRef]
- Maueroder, C.; Mahajan, A.; Paulus, S.; Gosswein, S.; Hahn, J.; Kienhofer, D.; Biermann, M.H.; Tripal, P.; Friedrich, R.P.; Munoz, L.E.; et al. Menage-a-Trois: The Ratio of Bicarbonate to CO2 and the pH Regulate the Capacity of Neutrophils to Form NETs. Front. Immunol. 2016, 7, 583. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [Green Version]
- Kenny, E.F.; Herzig, A.; Kruger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Bernuth, H.V.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Martin Monreal, M.T.; Rebak, A.S.; Massarenti, L.; Mondal, S.; Senolt, L.; Odum, N.; Nielsen, M.L.; Thompson, P.R.; Nielsen, C.H.; Damgaard, D. Applicability of Small-Molecule Inhibitors in the Study of Peptidyl Arginine Deiminase 2 (PAD2) and PAD4. Front. Immunol. 2021, 12, 716250. [Google Scholar] [CrossRef]
- Tian, Y.; Qu, S.; Alam, H.B.; Williams, A.M.; Wu, Z.; Deng, Q.; Pan, B.; Zhou, J.; Liu, B.; Duan, X.; et al. Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis. JCI Insight 2020, 5, e138873. [Google Scholar] [CrossRef]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.M.; Voorberg, J.; Nicolaes, G.A.F. PAD4 takes charge during neutrophil activation: Impact of PAD4 mediated NET formation on immune-mediated disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef]
- Naffah de Souza, C.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Camara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline pH Promotes NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation: A Matter of Mitochondrial Reactive Oxygen Species Generation and Citrullination and Cleavage of Histone. Front. Immunol. 2017, 8, 1849. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fay, A.J.; Qian, X.; Jan, Y.N.; Jan, L.Y. SK channels mediate NADPH oxidase-independent reactive oxygen species production and apoptosis in granulocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 17548–17553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Hamam, H.J.; Palaniyar, N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules 2019, 9, 369. [Google Scholar] [CrossRef] [Green Version]
- Kearney, P.L.; Bhatia, M.; Jones, N.G.; Yuan, L.; Glascock, M.C.; Catchings, K.L.; Yamada, M.; Thompson, P.R. Kinetic characterization of protein arginine deiminase 4: A transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005, 44, 10570–10582. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.D. The Role of Reactive Oxygen Species (ROS) in the Formation of Extracellular Traps (ETs) in Humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Neubert, E.; Meyer, D.; Kruss, S.; Erpenbeck, L. The power from within—Understanding the driving forces of neutrophil extracellular trap formation. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; McKee, T.; Bosshard, C.; Durual, S.; Matthes, T.; Myit, S.; Donze, O.; Frossard, C.; Chizzolini, C.; Favre, C.; et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J. Clin. Investig. 2008, 118, 2887–2895. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.; Karimi, N.; Morovati, S.; Alizadeh, K.; Kakish, J.E.; Vanderkamp, S.; Fazel, F.; Napoleoni, C.; Alizadeh, K.; Mehrani, Y.; et al. The Roles of Neutrophils in Cytokine Storms. Viruses 2021, 13, 2318. [Google Scholar] [CrossRef]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12952. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal. 2019, 17, 147. [Google Scholar] [CrossRef] [Green Version]
- Niedzwiedzka-Rystwej, P.; Repka, W.; Tokarz-Deptula, B.; Deptula, W. “In sickness and in health”—how neutrophil extracellular trap (NET) works in infections, selected diseases and pregnancy. J. Inflamm. 2019, 16, 15. [Google Scholar] [CrossRef]
- Fernandez-Dominguez, I.J.; Manzo-Merino, J.; Taja-Chayeb, L.; Duenas-Gonzalez, A.; Perez-Cardenas, E.; Trejo-Becerril, C. The role of extracellular DNA (exDNA) in cellular processes. Cancer Biol. Ther. 2021, 22, 267–278. [Google Scholar] [CrossRef]
- Domer, D.; Walther, T.; Moller, S.; Behnen, M.; Laskay, T. Neutrophil Extracellular Traps Activate Proinflammatory Functions of Human Neutrophils. Front. Immunol. 2021, 12, 636954. [Google Scholar] [CrossRef]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhulsdonk, L.; Mannherz, H.G.; Napirei, M. Comparison of the secretory murine DNase1 family members expressed in Pichia pastoris. PLoS ONE 2021, 16, e0253476. [Google Scholar] [CrossRef] [PubMed]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahorec, R. Neutrophil-to-lymphocyte ratio, past, present and future perspectives. Bratisl. Lek. Listy. 2021, 122, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Lowsby, R.; Gomes, C.; Jarman, I.; Lisboa, P.; Nee, P.A.; Vardhan, M.; Eckersley, T.; Saleh, R.; Mills, H. Neutrophil to lymphocyte count ratio as an early indicator of blood stream infection in the emergency department. Emerg. Med. J. 2015, 32, 531–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, B.; Guo, L.; Shen, R.; Cao, C.; Xie, R.; Jiang, W.; Wen, L.; Bi, X.; Shi, H.; Zheng, S.; et al. Prognostic Significance of NLR About NETosis and Lymphocytes Perturbations in Localized Renal Cell Carcinoma With Tumor Thrombus. Front. Oncol. 2021, 11, 771545. [Google Scholar] [CrossRef] [PubMed]
- Larco, J.A.; Abbasi, M.; Madhani, S.I.; Mereuta, M.O.; Liu, Y.; Dai, D.; Kadirvel, R.; Savastano, L.; Kallmes, D.F.; Brinjikji, W. Correlation of Neutrophil to Lymphocyte Ratio with Expression of Neutrophil Extracellular Traps Within Stroke Emboli. Interv. Neuroradiol. 2021, 8, 15910199211065530. [Google Scholar] [CrossRef]
- Hidalgo, A.; Libby, P.; Soehnlein, O.; Aramburu, I.V.; Papayannopoulos, V.; Silvestre-Roig, C. Neutrophil extracellular traps: From physiology to pathology. Cardiovasc. Res. 2022, 118, 2737–2753. [Google Scholar] [CrossRef]
- Hosari, D.; Hosari, S.; Mahajan, A.; Oreski, L.; Schoen, J.; Shan, X.; Singh, J.; Wang, H.; Yaykasli, K.O.; Knopf, J.; et al. Neutrophils and Nets as Orchestrators of Inflammatory Diseases. Clin. Exp. Rheumatol. 2021, 39, 1151–1152. [Google Scholar]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Mattey, D.L.; Nixon, N.B.; Dawes, P.T. Association of circulating levels of MMP-8 with mortality from respiratory disease in patients with rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, K.M.; Lo, C.Y.; Summers, S.A.; Elgass, K.D.; McMillan, P.J.; Longano, A.; Ford, S.L.; Gan, P.Y.; Kerr, P.G.; Kitching, A.R.; et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015, 88, 1030–1046. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Zhang, Y.; Yin, S.W.; Gao, X.J.; Shi, W.W.; Wang, Y.; Huang, X.; Wang, L.; Zou, L.Y.; Zhao, J.H.; et al. Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin. Exp. Immunol. 2015, 180, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Yaniv, G.; Twig, G.; Shor, D.B.; Furer, A.; Sherer, Y.; Mozes, O.; Komisar, O.; Slonimsky, E.; Klang, E.; Lotan, E.; et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: A diversity of 180 different antibodies found in SLE patients. Autoimmun. Rev. 2015, 14, 75–79. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Gripenberg, M.; Helve, T.; Kurki, P. Profiles of antibodies to histones, DNA and IgG in patients with systemic rheumatic diseases determined by ELISA. J. Rheumatol. 1985, 12, 934–939. [Google Scholar]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef]
- Mansour, R.B.; Lassoued, S.; Gargouri, B.; El Gaid, A.; Attia, H.; Fakhfakh, F. Increased levels of autoantibodies against catalase and superoxide dismutase associated with oxidative stress in patients with rheumatoid arthritis and systemic lupus erythematosus. Scand. J. Rheumatol. 2008, 37, 103–108. [Google Scholar] [CrossRef]
- Iaccarino, L.; Ghirardello, A.; Canova, M.; Zen, M.; Bettio, S.; Nalotto, L.; Punzi, L.; Doria, A. Anti-annexins autoantibodies: Their role as biomarkers of autoimmune diseases. Autoimmun. Rev. 2011, 10, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Kretz, C.C.; Norpo, M.; Abeler-Dorner, L.; Linke, B.; Haust, M.; Edler, L.; Krammer, P.H.; Kuhn, A. Anti-annexin 1 antibodies: A new diagnostic marker in the serum of patients with discoid lupus erythematosus. Exp. Dermatol. 2010, 19, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grayson, P.C.; Kaplan, M.J. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J. Leukoc. Biol. 2016, 99, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, P.; Nakabo, S.; O’Neil, L.; Goel, R.R.; Jiang, K.; Carmona-Rivera, C.; Gupta, S.; Chan, D.W.; Carlucci, P.M.; Wang, X.; et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2019, 116, 25222–25228. [Google Scholar] [CrossRef]
- Rahman, S.; Sagar, D.; Hanna, R.N.; Lightfoot, Y.L.; Mistry, P.; Smith, C.K.; Manna, Z.; Hasni, S.; Siegel, R.M.; Sanjuan, M.A.; et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 957–966. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef]
- Grayson, P.C.; Carmona-Rivera, C.; Xu, L.; Lim, N.; Gao, Z.; Asare, A.L.; Specks, U.; Stone, J.H.; Seo, P.; Spiera, R.F.; et al. Neutrophil-Related Gene Expression and Low-Density Granulocytes Associated With Disease Activity and Response to Treatment in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2015, 67, 1922–1932. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.W.; Woodruff, T.M. Complement: Bridging the innate and adaptive immune systems in sterile inflammation. J. Leukoc. Biol. 2020, 108, 339–351. [Google Scholar] [CrossRef]
- Ramsey-Goldman, R.; Alexander, R.V.; Massarotti, E.M.; Wallace, D.J.; Narain, S.; Arriens, C.; Collins, C.E.; Saxena, A.; Putterman, C.; Kalunian, K.C.; et al. Complement Activation in Patients With Probable Systemic Lupus Erythematosus and Ability to Predict Progression to American College of Rheumatology-Classified Systemic Lupus Erythematosus. Arthritis Rheumatol. 2020, 72, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Gaipl, U.S.; Beyer, T.D.; Heyder, P.; Kuenkele, S.; Bottcher, A.; Voll, R.E.; Kalden, J.R.; Herrmann, M. Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum. 2004, 50, 640–649. [Google Scholar] [CrossRef]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Massenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef] [Green Version]
- van Dam, L.S.; Kraaij, T.; Kamerling, S.W.A.; Bakker, J.A.; Scherer, U.H.; Rabelink, T.J.; van Kooten, C.; Teng, Y.K.O. Intrinsically Distinct Role of Neutrophil Extracellular Trap Formation in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Compared to Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 2047–2058. [Google Scholar]
- Whittall-Garcia, L.P.; Torres-Ruiz, J.; Zentella-Dehesa, A.; Tapia-Rodriguez, M.; Alcocer-Varela, J.; Mendez-Huerta, N.; Gomez-Martin, D. Neutrophil extracellular traps are a source of extracellular HMGB1 in lupus nephritis: Associations with clinical and histopathological features. Lupus 2019, 28, 1549–1557. [Google Scholar] [CrossRef]
- Ma, Y.H.; Ma, T.T.; Wang, C.; Wang, H.; Chang, D.Y.; Chen, M.; Zhao, M.H. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res. Ther. 2016, 18, 2. [Google Scholar] [CrossRef] [Green Version]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014, 73, 1854–1863. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1alpha and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [Green Version]
- Egorina, E.M.; Sovershaev, M.A.; Olsen, J.O.; Osterud, B. Granulocytes do not express but acquire monocyte-derived tissue factor in whole blood: Evidence for a direct transfer. Blood 2008, 111, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Brambilla, M.; Camera, M.; Carbone, A.; Tremoli, E.; Donati, M.B.; de Gaetano, G.; Cerletti, C. Human polymorphonuclear leukocytes produce and express functional tissue factor upon stimulation. J. Thromb. Haemost. 2006, 4, 1323–1330. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Kambas, K.; Mitroulis, I.; Ritis, K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front. Immunol. 2012, 3, 385. [Google Scholar] [CrossRef] [Green Version]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis: A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Li, T.; Jin, J.; Liu, Y.; Li, B.; Sun, Q.; Tian, J.; Zhao, H.; Liu, Z.; Ma, S.; et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine 2020, 53, 102671. [Google Scholar] [CrossRef]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Sturzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef]
- Leppkes, M.; Maueroder, C.; Hirth, S.; Nowecki, S.; Gunther, C.; Billmeier, U.; Paulus, S.; Biermann, M.; Munoz, L.E.; Hoffmann, M.; et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat. Commun. 2016, 7, 10973. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Hasikova, L.; Hampel, U.; Gruneboom, A.; Shan, X.; Herrmann, I.; Garreis, F.; Bock, F.; Knopf, J.; Singh, J.; et al. Aggregated neutrophil extracellular traps occlude Meibomian glands during ocular surface inflammation. Ocul. Surf. 2021, 20, 1–12. [Google Scholar] [CrossRef]
- Schapher, M.; Koch, M.; Weidner, D.; Scholz, M.; Wirtz, S.; Mahajan, A.; Herrmann, I.; Singh, J.; Knopf, J.; Leppkes, M.; et al. Neutrophil Extracellular Traps Promote the Development and Growth of Human Salivary Stones. Cells 2020, 9, 2139. [Google Scholar] [CrossRef]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef]
- Leung, H.H.L.; Perdomo, J.; Ahmadi, Z.; Yan, F.; McKenzie, S.E.; Chong, B.H. Inhibition of NADPH oxidase blocks NETosis and reduces thrombosis in heparin-induced thrombocytopenia. Blood Adv. 2021, 5, 5439–5451. [Google Scholar] [CrossRef]
- Hair, P.S.; Enos, A.I.; Krishna, N.K.; Cunnion, K.M. Inhibition of Immune Complex Complement Activation and Neutrophil Extracellular Trap Formation by Peptide Inhibitor of Complement C1. Front. Immunol. 2018, 9, 558. [Google Scholar] [CrossRef]
- Zheng, W.; Warner, R.; Ruggeri, R.; Su, C.; Cortes, C.; Skoura, A.; Ward, J.; Ahn, K.; Kalgutkar, A.; Sun, D.; et al. PF-1355, a mechanism-based myeloperoxidase inhibitor, prevents immune complex vasculitis and anti-glomerular basement membrane glomerulonephritis. J. Pharmacol. Exp. Ther. 2015, 353, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Zhang, Y.; Wang, S. Histone citrullination: A new target for tumors. Mol. Cancer 2021, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Massie, J.; Robinson, P.J.; Cooper, P.J. The story of cystic fibrosis 1965–2015. J. Paediatr. Child Health 2016, 52, 991–994. [Google Scholar] [CrossRef]
- Angeletti, A.; Volpi, S.; Bruschi, M.; Lugani, F.; Vaglio, A.; Prunotto, M.; Gattorno, M.; Schena, F.; Verrina, E.; Ravelli, A.; et al. Neutrophil Extracellular Traps-DNase Balance and Autoimmunity. Cells 2021, 10, 2667. [Google Scholar] [CrossRef]
- Podolska, M.J.; Mahajan, A.; Hahn, J.; Knopf, J.; Maueroder, C.; Petru, L.; Ullmann, M.; Schett, G.; Leppkes, M.; Herrmann, M.; et al. Treatment with DNases rescues hidden neutrophil elastase from aggregated NETs. J. Leukoc. Biol. 2019, 106, 1359–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, T.; Fisher, J.; Bakochi, A.; Neumann, A.; Cardoso, J.F.P.; Karlsson, C.A.Q.; Pavan, C.; Lundgaard, I.; Nilson, B.; Reinstrup, P.; et al. Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat. Commun. 2019, 10, 1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, W.; Paunel-Gorgulu, A.; Flohe, S.; Witte, I.; Schadel-Hopfner, M.; Windolf, J.; Logters, T.T. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediat. Inflamm. 2012, 2012, 149560. [Google Scholar] [CrossRef]
- Cedervall, J.; Zhang, Y.; Huang, H.; Zhang, L.; Femel, J.; Dimberg, A.; Olsson, A.K. Neutrophil Extracellular Traps Accumulate in Peripheral Blood Vessels and Compromise Organ Function in Tumor-Bearing Animals. Cancer Res. 2015, 75, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- Albadawi, H.; Oklu, R.; Raacke Malley, R.E.; O’Keefe, R.M.; Uong, T.P.; Cormier, N.R.; Watkins, M.T. Effect of DNase I treatment and neutrophil depletion on acute limb ischemia-reperfusion injury in mice. J. Vasc. Surg. 2016, 64, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Macanovic, M.; Sinicropi, D.; Shak, S.; Baughman, S.; Thiru, S.; Lachmann, P.J. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin. Exp. Immunol. 1996, 106, 243–252. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Liao, T.L.; Chen, Y.M.; Tang, K.T.; Chen, P.K.; Liu, H.J.; Chen, D.Y. MicroRNA-223 inhibits neutrophil extracellular traps formation through regulating calcium influx and small extracellular vesicles transmission. Sci. Rep. 2021, 11, 15676. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoen, J.; Euler, M.; Schauer, C.; Schett, G.; Herrmann, M.; Knopf, J.; Yaykasli, K.O. Neutrophils’ Extracellular Trap Mechanisms: From Physiology to Pathology. Int. J. Mol. Sci. 2022, 23, 12855. https://doi.org/10.3390/ijms232112855
Schoen J, Euler M, Schauer C, Schett G, Herrmann M, Knopf J, Yaykasli KO. Neutrophils’ Extracellular Trap Mechanisms: From Physiology to Pathology. International Journal of Molecular Sciences. 2022; 23(21):12855. https://doi.org/10.3390/ijms232112855
Chicago/Turabian StyleSchoen, Janina, Maximilien Euler, Christine Schauer, Georg Schett, Martin Herrmann, Jasmin Knopf, and Kursat Oguz Yaykasli. 2022. "Neutrophils’ Extracellular Trap Mechanisms: From Physiology to Pathology" International Journal of Molecular Sciences 23, no. 21: 12855. https://doi.org/10.3390/ijms232112855
APA StyleSchoen, J., Euler, M., Schauer, C., Schett, G., Herrmann, M., Knopf, J., & Yaykasli, K. O. (2022). Neutrophils’ Extracellular Trap Mechanisms: From Physiology to Pathology. International Journal of Molecular Sciences, 23(21), 12855. https://doi.org/10.3390/ijms232112855