
Breast Cancer with Increased Drug Resistance, Invasion Ability, and Cancer Stem Cell Properties through Metabolism Reprogramming

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
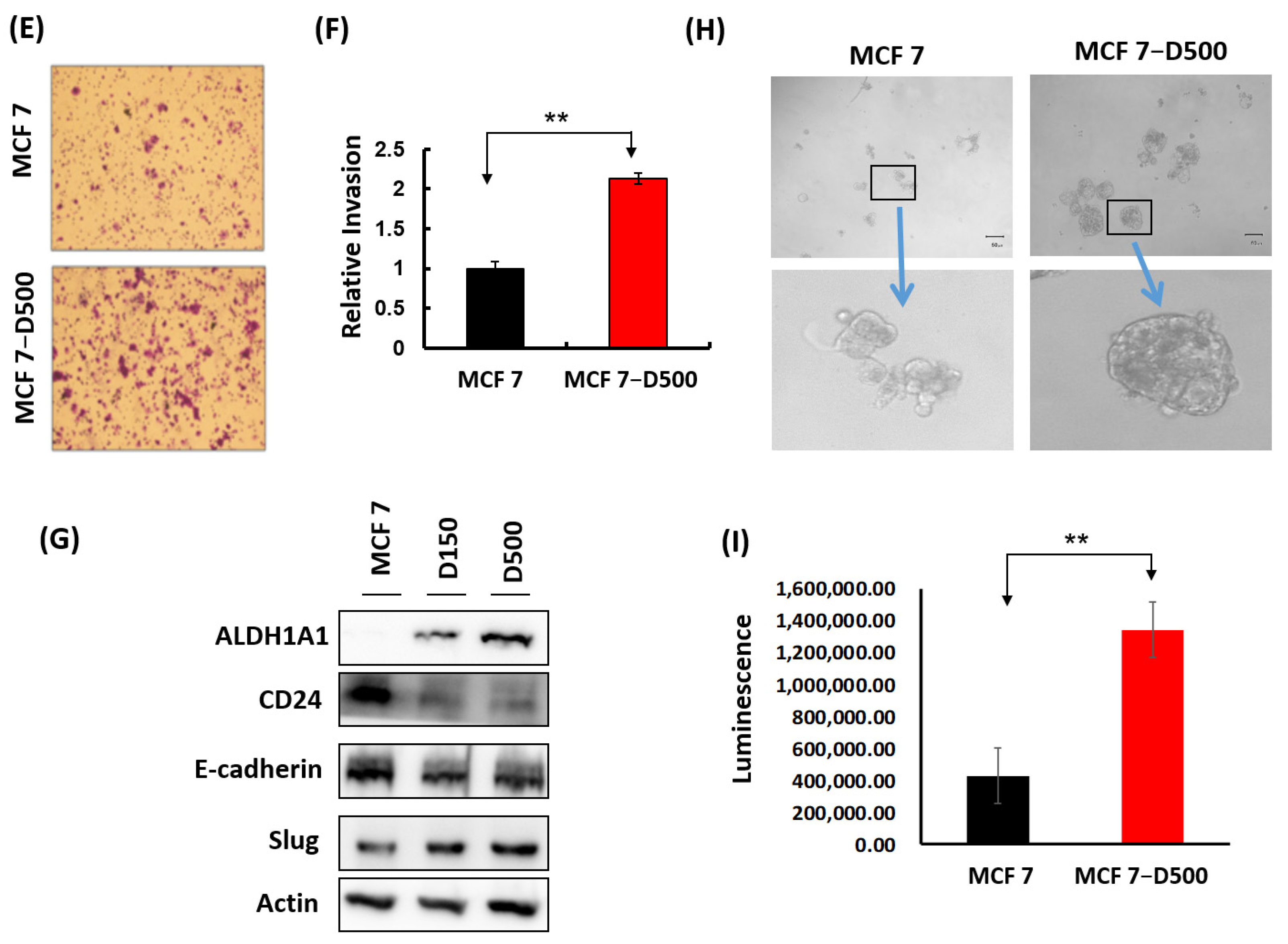
2.1. Generation and Characterization of MCF-7 Cells Resistant to Doxorubicin
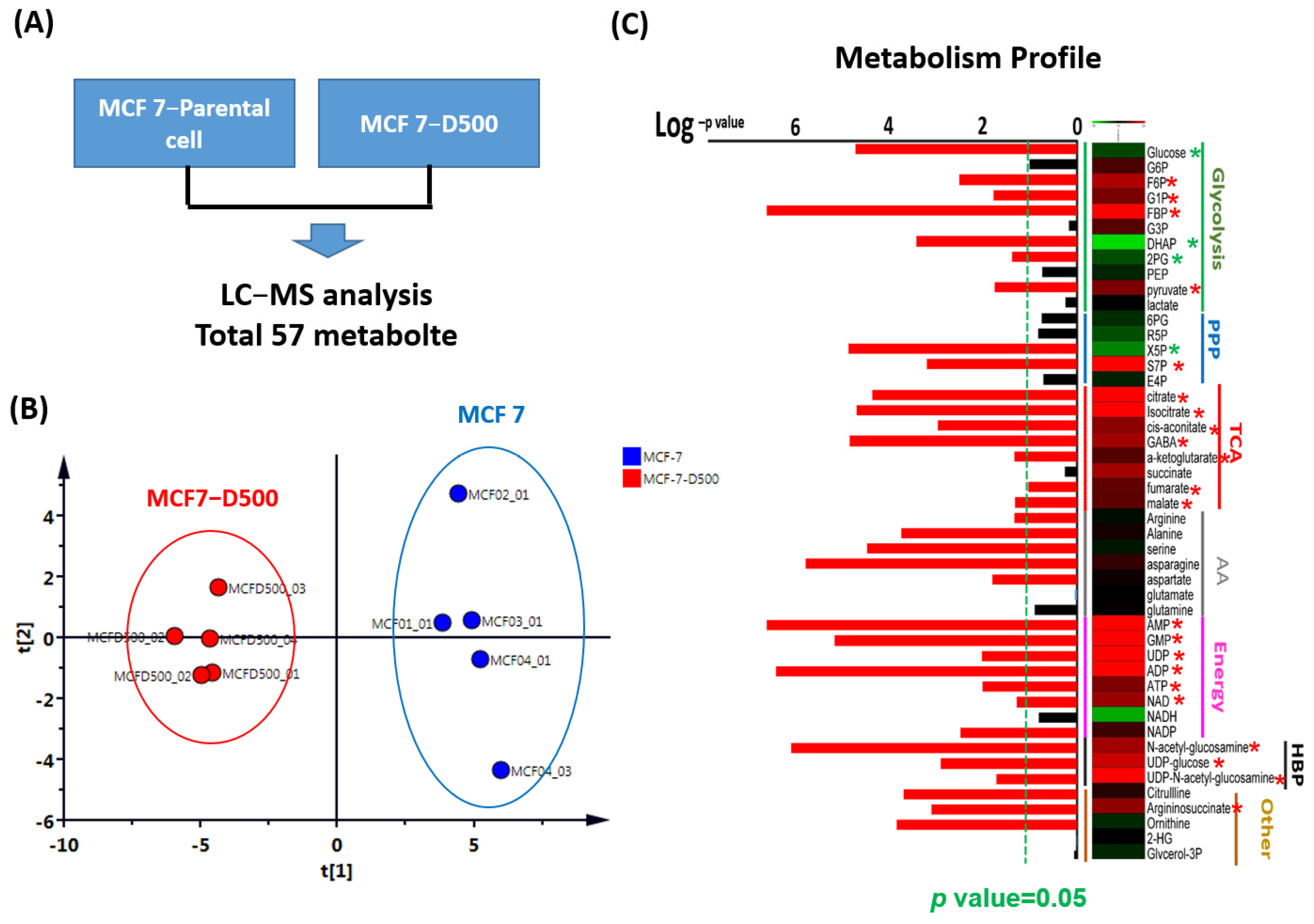
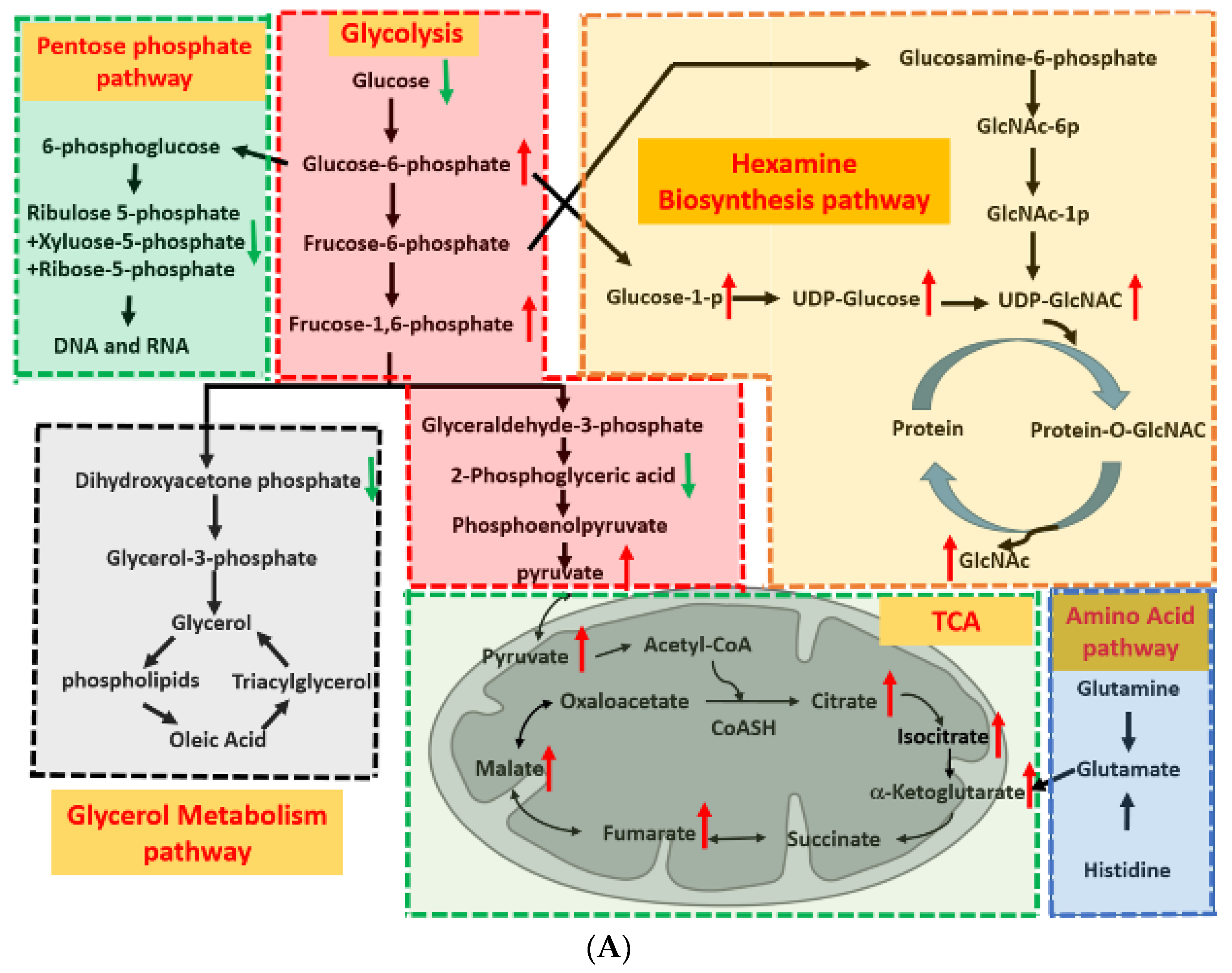
2.2. Metabolism Reprogramming Involved in the Drug Resistance of Breast Cancer Cells
2.3. Glycolysis-Associated Enzymes Promoted Glucose Consumption
2.4. Glycolysis and TCA Cycle Were More Active in Drug-Resistant Cells
2.5. HBP Activity in Drug-Resistant Cells
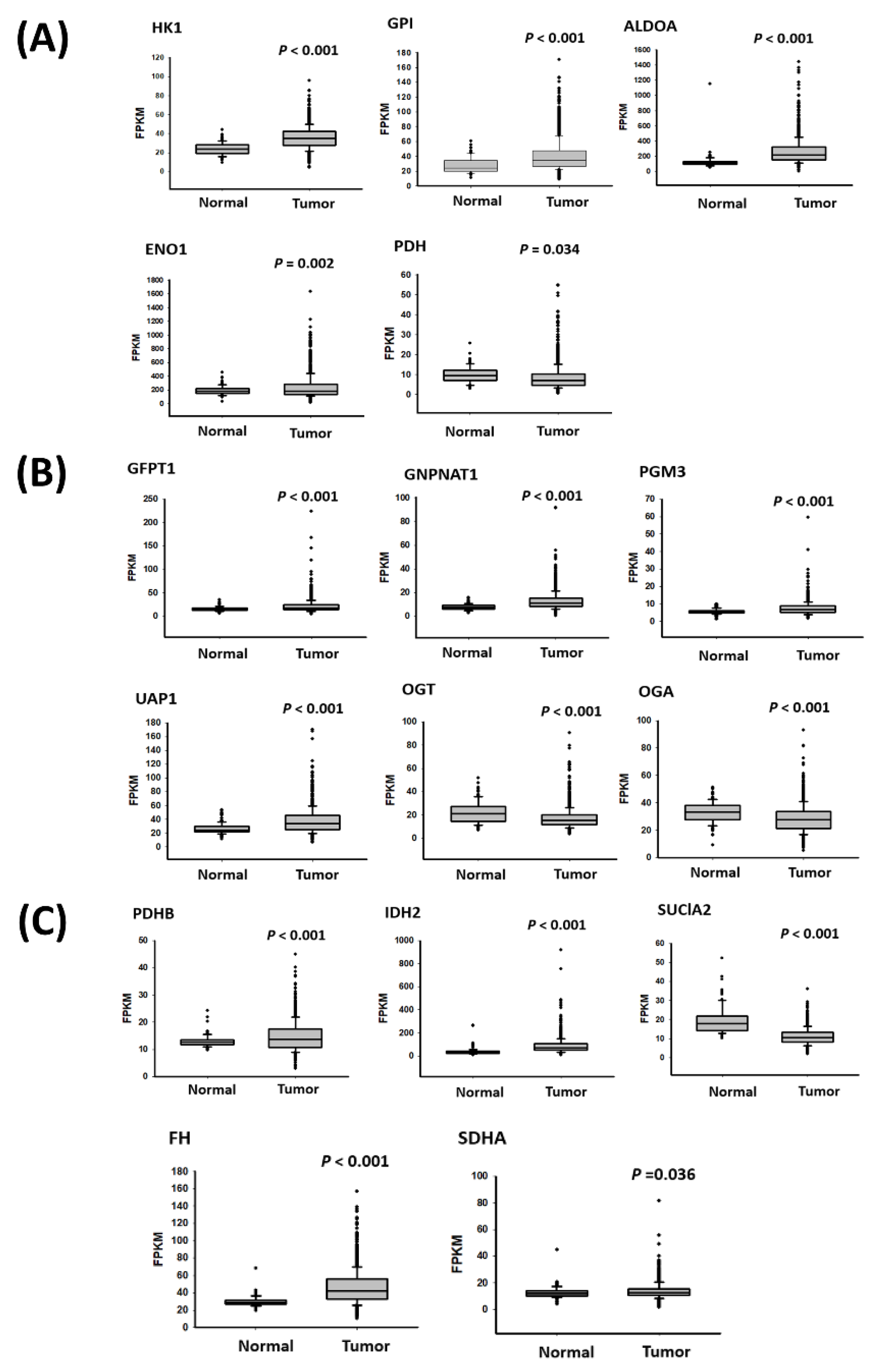
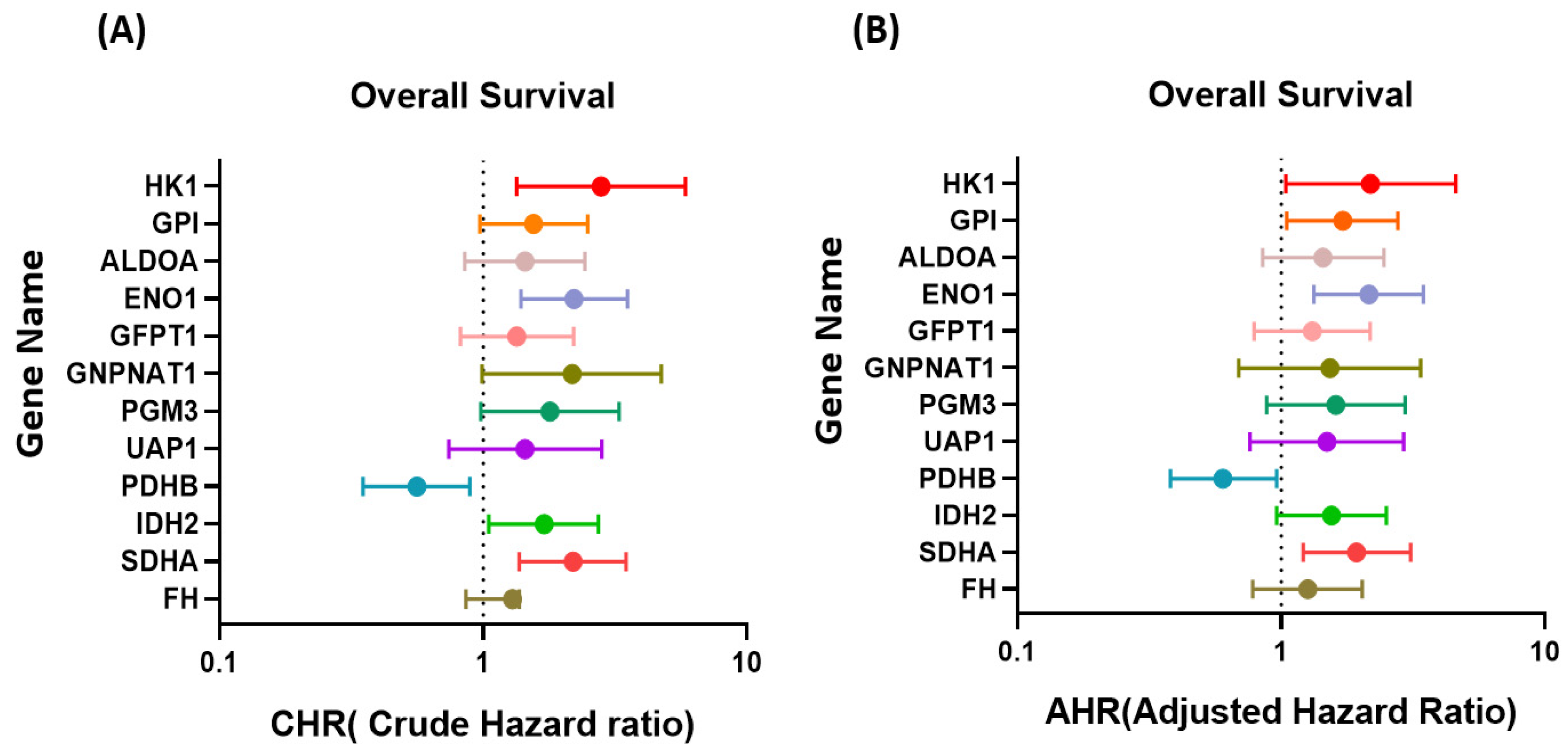
2.6. Metabolism-Related Gene Expression in Breast Cancer Cells with Drug Resistance
3. Discussion
4. Material and Methods
4.1. Cell Line and Doxorubicin Resistance of Breast Cancer Cells
4.2. Cell Proliferation and Migration Assay
4.3. Colony Formation Assay
4.4. RNA Extraction and Real-Time Polymerase Chain Reaction
4.5. Western Blotting
4.6. Metabolic Profiling by Liquid Chromatography–Mass Spectrometry
4.7. Expression Data from the Cancer Genome Atlas
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, L.J.; Shupe, M.P.; Schneble, E.J.; Flynt, F.L.; Clemenshaw, M.N.; Kirkpatrick, A.D.; Gallagher, C.; Nissan, A.; Henry, L.; Stojadinovic, A.; et al. Current approaches and challenges in monitoring treatment responses in breast cancer. J. Cancer 2014, 5, 58–68. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- DeMichele, A.; Yee, D.; Esserman, L. Mechanisms of Resistance to Neoadjuvant Chemotherapy in Breast Cancer. N. Engl. J. Med. 2017, 377, 2287–2289. [Google Scholar] [CrossRef]
- Mayor, S. Side-effects of cancer drugs are under-reported in trials. Lancet Oncol. 2015, 16, e107. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Cha, H.K.; Cheon, S.; Kim, H.; Lee, K.M.; Ryu, H.S.; Han, D. Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis. Molecules 2022, 27, 1762. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef]
- Calaf, G.M.; Zepeda, A.B.; Castillo, R.L.; Figueroa, C.A.; Arias, C.; Figueroa, E.; Farias, J.G. Molecular aspects of breast cancer resistance to drugs (Review). Int. J. Oncol. 2015, 47, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Taurin, S.; Alkhalifa, H. Breast cancers, mammary stem cells, and cancer stem cells, characteristics, and hypotheses. Neoplasia 2020, 22, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, X.; Guo, X.; Liu, H.; Ma, R.; Wang, Y.; Liang, Y.; Sun, Y.; Wang, M.; Zhao, R.; et al. Low Glucose-Induced Overexpression of HOXC-AS3 Promotes Metabolic Reprogramming of Breast Cancer. Cancer Res. 2022, 82, 805–818. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, X.; Ding, H.; Liu, X.; Cao, D.; Liu, Y.; Liu, J.; Lin, C.; Zhang, N.; Wang, G.; et al. Arginine and lysine methylation of MRPS23 promotes breast cancer metastasis through regulating OXPHOS. Oncogene 2021, 40, 3548–3563. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Paez Arango, N.; Cruz Pico, C.X.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef]
- Becherini, P.; Caffa, I.; Piacente, F.; Damonte, P.; Vellone, V.G.; Passalacqua, M.; Benzi, A.; Bonfiglio, T.; Reverberi, D.; Khalifa, A.; et al. SIRT6 enhances oxidative phosphorylation in breast cancer and promotes mammary tumorigenesis in mice. Cancer Metab. 2021, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Cohen, J.S.; Faustino, P.J.; Megnin, F.; Myers, C.E. Glucose metabolism in drug-sensitive and drug-resistant human breast cancer cells monitored by magnetic resonance spectroscopy. Cancer Res. 1988, 48, 870–877. [Google Scholar] [PubMed]
- Damaghi, M.; West, J.; Robertson-Tessi, M.; Xu, L.; Ferrall-Fairbanks, M.C.; Stewart, P.A.; Persi, E.; Fridley, B.L.; Altrock, P.M.; Gatenby, R.A.; et al. The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2011342118. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 573. [Google Scholar] [CrossRef]
- Ma, L.; Zong, X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front. Oncol. 2020, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Sen, G.S.; Mohanty, S.; Hossain, D.M.; Bhattacharyya, S.; Banerjee, S.; Chakraborty, J.; Saha, S.; Ray, P.; Bhattacharjee, P.; Mandal, D.; et al. Curcumin enhances the efficacy of chemotherapy by tailoring p65NFkappaB-p300 cross-talk in favor of p53-p300 in breast cancer. J. Biol. Chem. 2011, 286, 42232–42247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M. Understanding the causes of multidrug resistance in cancer: A comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Li, M.; Zha, W.; Zhao, Q.; Gu, R.; Liu, L.; Shi, J.; Zhou, J.; Zhou, F.; Wu, X.; et al. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 2013, 9, 960–973. [Google Scholar] [CrossRef] [Green Version]
- Panda, M.; Biswal, B.K. Cell signaling and cancer: A mechanistic insight into drug resistance. Mol. Biol. Rep. 2019, 46, 5645–5659. [Google Scholar] [CrossRef]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.P.; Caron, M.; Savage, P.; Labbe, D.P.; Begin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRalpha mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef] [Green Version]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Usuba, W.; Takahashi, R.U.; Shimomura, I.; Sasaki, H.; Ochiya, T.; Yamamoto, Y. Single-Cell Analysis Reveals a Preexisting Drug-Resistant Subpopulation in the Luminal Breast Cancer Subtype. Cancer Res. 2019, 79, 4412–4425. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Liskova, A.; Samec, M.; Kubatka, P.; Busselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, 2252. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Ahmadpour, S.T.; Desquiret-Dumas, V.; Yikilmaz, U.; Dartier, J.; Domingo, I.; Wetterwald, C.; Orre, C.; Gueguen, N.; Brisson, L.; Maheo, K.; et al. Doxorubicin-Induced Autophagolysosome Formation Is Partly Prevented by Mitochondrial ROS Elimination in DOX-Resistant Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 9283. [Google Scholar] [CrossRef]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Effects of Doxorubicin, Cisplatin, and Tamoxifen on the Metabolic Profile of Human Breast Cancer MCF-7 Cells As Determined by (1)H High-Resolution Magic Angle Spinning Nuclear Magnetic Resonance. Biochemistry 2017, 56, 2219–2224. [Google Scholar] [CrossRef] [Green Version]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using (1)H HR-MAS NMR spectroscopy. J. Pharm. Biomed. Anal. 2017, 146, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Cai, T.; Huang, C.; Zang, X.; Sun, L.; Guo, S.; Wang, Q.; Chen, Z.; Zhao, Y.; Han, Z.; et al. G6PD-NF-kappaB-HGF Signal in Gastric Cancer-Associated Mesenchymal Stem Cells Promotes the Proliferation and Metastasis of Gastric Cancer Cells by Upregulating the Expression of HK2. Front. Oncol. 2021, 11, 648706. [Google Scholar] [CrossRef]
- Tang, Y.C.; Hsiao, J.R.; Jiang, S.S.; Chang, J.Y.; Chu, P.Y.; Liu, K.J.; Fang, H.L.; Lin, L.M.; Chen, H.H.; Huang, Y.W.; et al. c-MYC-directed NRF2 drives malignant progression of head and neck cancer via glucose-6-phosphate dehydrogenase and transketolase activation. Theranostics 2021, 11, 5232–5247. [Google Scholar] [CrossRef]
- Min, H.Y.; Lee, H.J.; Suh, Y.A.; Pei, H.; Kwon, H.; Jang, H.J.; Yun, H.J.; Moon, H.G.; Lee, H.Y. Targeting epidermal growth factor receptor in paclitaxel-resistant human breast and lung cancer cells with upregulated glucose-6-phosphate dehydrogenase. Br. J. Cancer 2022, 127, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fu, A.; Wu, R.; Wei, N.; Song, K.; Lim, S.; Luo, K.Q. High Expression of G6PD Increases Doxorubicin Resistance in Triple Negative Breast Cancer Cells by Maintaining GSH Level. Int. J. Biol. Sci. 2022, 18, 1120–1133. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Tsogka, K.; Nikolaou, E.; Liakopoulos, V.; Stefanidis, I. A unifying model of glucotoxicity in human renal proximal tubular epithelial cells and the effect of the SGLT2 inhibitor dapagliflozin. Int. Urol. Nephrol. 2020, 52, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Peng, P.; Li, L.; Shao, M.; Zhao, J.; Wang, L.; Duan, F.; Song, S.; Wu, H.; Zhang, J.; et al. High expression of GFAT1 predicts poor prognosis in patients with pancreatic cancer. Sci. Rep. 2016, 6, 39044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Hart, G.W. Targeting O-GlcNAcylation to develop novel therapeutics. Mol. Asp. Med. 2021, 79, 100885. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Pan, X.; Shi, M.; Wu, Q.; Huang, T.; Jiang, H.; Li, W.; Zhang, J. O-GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis. 2018, 9, 485. [Google Scholar] [CrossRef]
- Echeverria, G.V.; Ge, Z.; Seth, S.; Zhang, X.; Jeter-Jones, S.; Zhou, X.; Cai, S.; Tu, Y.; McCoy, A.; Peoples, M.; et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci. Transl. Med. 2019, 11, eaav0936. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e637. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, K.-H.; Chang, Y.-J.; Hsu, W.-H.; Tu, Y.-T.; Chen, Y.-R.; Lee, M.-C.; Tsai, K.-W. Breast Cancer with Increased Drug Resistance, Invasion Ability, and Cancer Stem Cell Properties through Metabolism Reprogramming. Int. J. Mol. Sci. 2022, 23, 12875. https://doi.org/10.3390/ijms232112875
Chong K-H, Chang Y-J, Hsu W-H, Tu Y-T, Chen Y-R, Lee M-C, Tsai K-W. Breast Cancer with Increased Drug Resistance, Invasion Ability, and Cancer Stem Cell Properties through Metabolism Reprogramming. International Journal of Molecular Sciences. 2022; 23(21):12875. https://doi.org/10.3390/ijms232112875
Chicago/Turabian StyleChong, Kian-Hwee, Yao-Jen Chang, Wei-Hsin Hsu, Ya-Ting Tu, Yi-Ru Chen, Ming-Cheng Lee, and Kuo-Wang Tsai. 2022. "Breast Cancer with Increased Drug Resistance, Invasion Ability, and Cancer Stem Cell Properties through Metabolism Reprogramming" International Journal of Molecular Sciences 23, no. 21: 12875. https://doi.org/10.3390/ijms232112875
APA StyleChong, K.-H., Chang, Y.-J., Hsu, W.-H., Tu, Y.-T., Chen, Y.-R., Lee, M.-C., & Tsai, K.-W. (2022). Breast Cancer with Increased Drug Resistance, Invasion Ability, and Cancer Stem Cell Properties through Metabolism Reprogramming. International Journal of Molecular Sciences, 23(21), 12875. https://doi.org/10.3390/ijms232112875