
Contribution of Microhomology to Genome Instability: Connection between DNA Repair and Replication Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
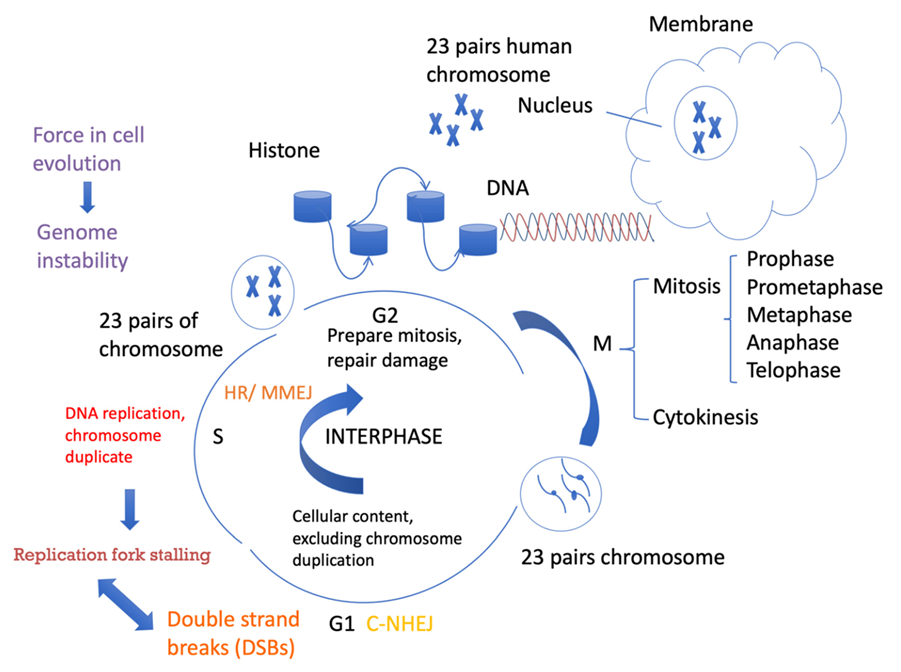
:1. Genome Instability and Cell Evolution
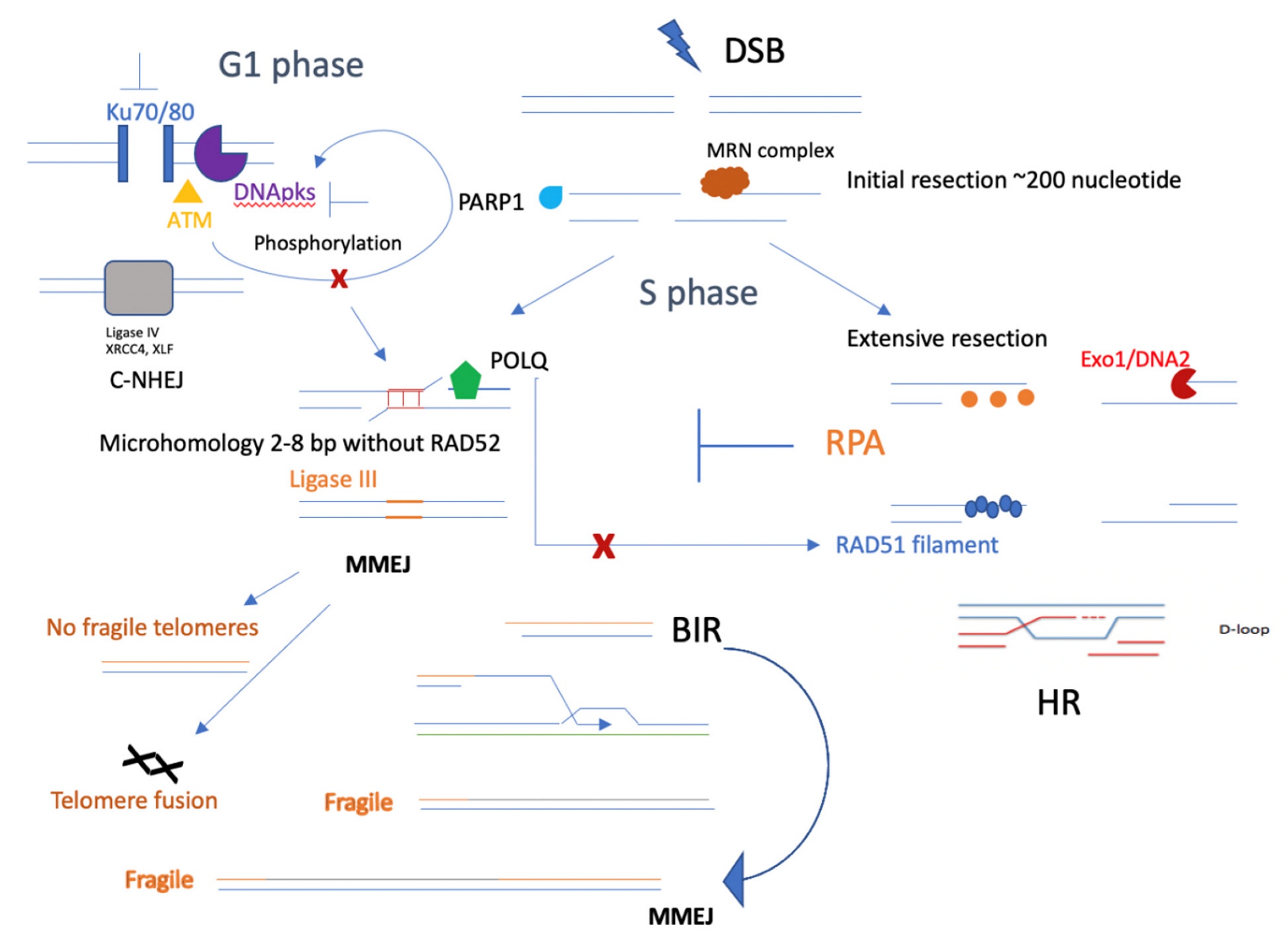
2. MMEJ and Genome Instability
3. Initial DNA End Resection Promotes MMEJ
4. Synthetic Lethal Role of MMEJ for Other DNA Repair Pathways
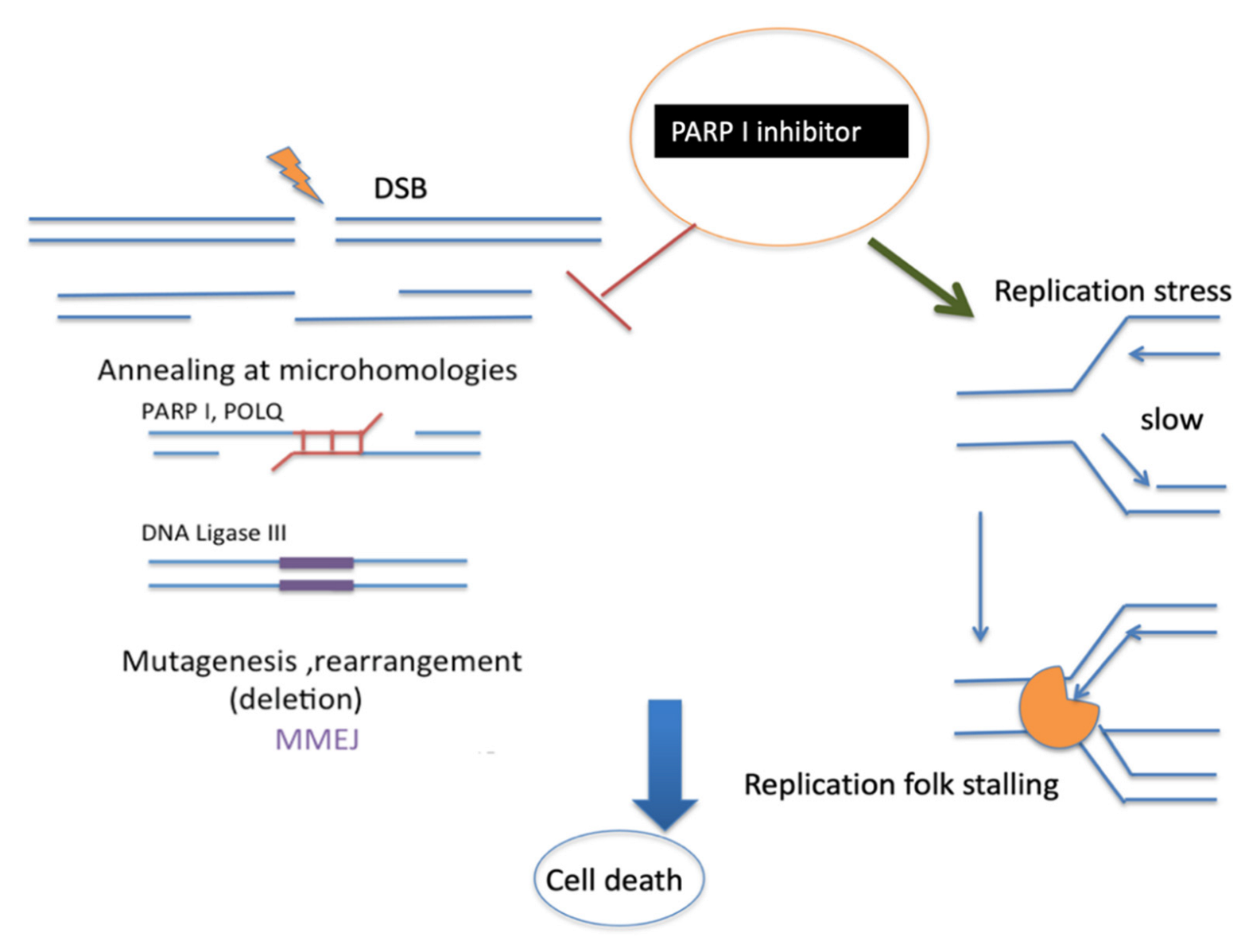
5. Connection between MMEJ and Replication Stress
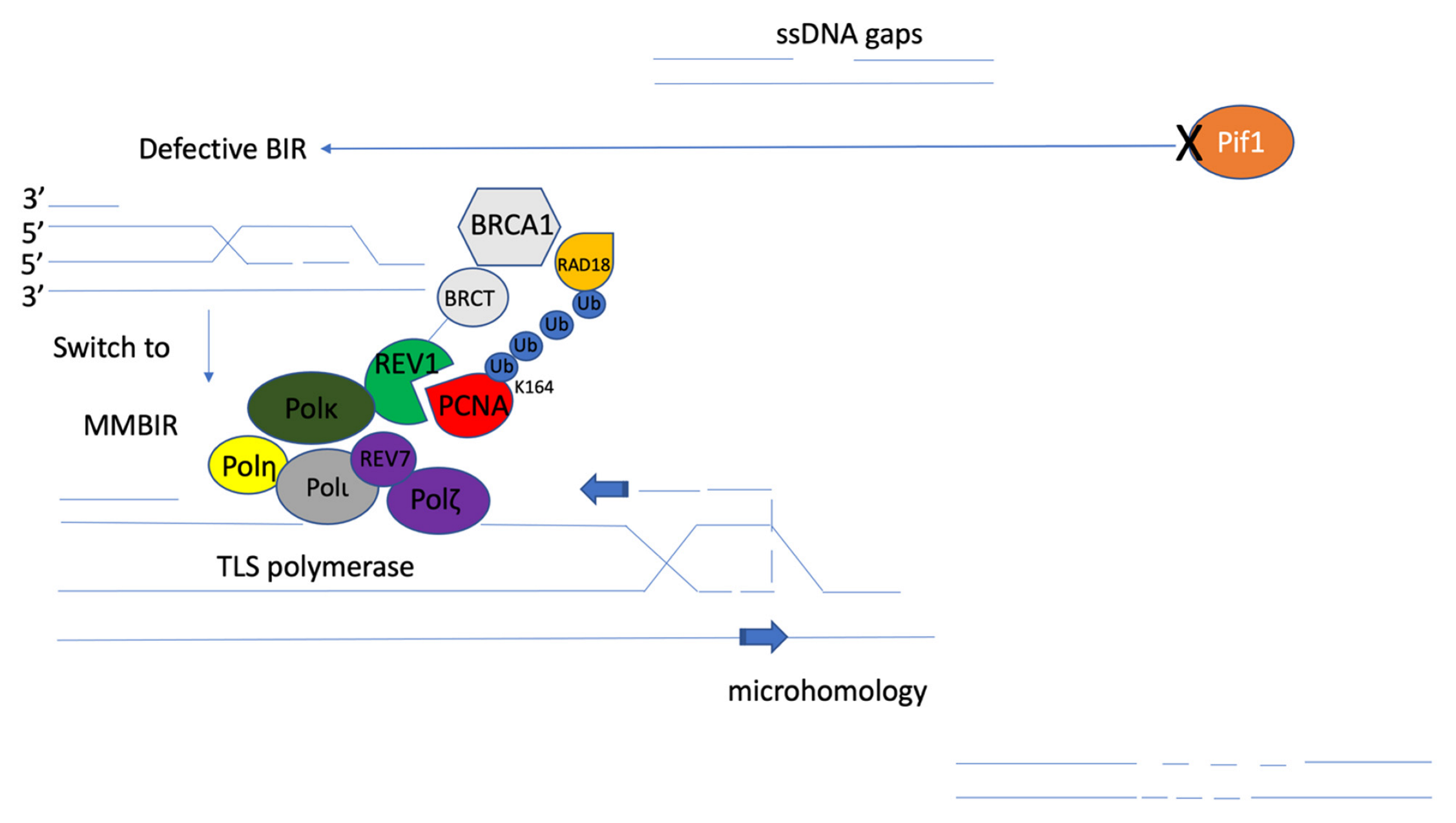
6. Microhomology-Mediated Break-Induced Replication (MMBIR)
7. Therapeutic Opportunities of New Regulators of MMEJ for Cancer Treatment
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, K.Y.; Yan, C.S.; Roan, H.Y.; Hsu, S.C.; Tseng, T.L.; Hsiao, C.D.; Hsu, C.P.; Chen, C.H. Skin cells undergo asynthetic fission to expand body surfaces in zebrafish. Nature 2022, 605, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Ledesma, F.; Aguilera, A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep. 2006, 7, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Pages, V.; Fuchs, R.P. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Berdichevsky, A.; Izhar, L.; Livneh, Z. Error-free recombinational repair predominates over mutagenic translesion replication in E. coli. Mol. Cell 2002, 10, 917–924. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polzeta maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chu, W.K. Potential Roles of the Retinoblastoma Protein in Regulating Genome Editing. Front. Cell Dev. Biol. 2018, 6, 81. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Vrtis, K.B.; Dewar, J.M.; Chistol, G.; Wu, R.A.; Graham, T.G.W.; Walter, J.C. Single-strand DNA breaks cause replisome disassembly. Mol. Cell 2021, 81, 1309–1318.e6. [Google Scholar] [CrossRef]
- Li, X.; Li, J.; Harrington, J.; Lieber, M.R.; Burgers, P.M. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem. 1995, 270, 22109–22112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Javadekar, S.M.; Pandey, M.; Srivastava, M.; Kumari, R.; Raghavan, S.C. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015, 6, e1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, K. The GINS complex: Structure and function. Subcell. Biochem. 2012, 62, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Kamimura, Y.; Okawa, M.; Muramatsu, S.; Sugino, A.; Araki, H. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 2003, 17, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Loveland, A.B.; Habuchi, S.; Walter, J.C.; van Oijen, A.M. A general approach to break the concentration barrier in single-molecule imaging. Nat. Methods 2012, 9, 987–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Stucki, M. How Cells Respond to DNA Breaks in Mitosis. Trends Biochem. Sci. 2020, 45, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Li, S.; Watanabe, G.; Lieber, M.R. Non-homologous end joining often uses microhomology: Implications for alternative end joining. DNA Repair 2014, 17, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, R.; Sandhu, M.; Muk, S.; Lee, G.; Vaidehi, N.; Stark, J.M. C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nat. Commun. 2018, 9, 2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussenzweig, A.; Nussenzweig, M.C. A backup DNA repair pathway moves to the forefront. Cell 2007, 131, 223–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eki, R.; She, J.; Parlak, M.; Benamar, M.; Du, K.P.; Kumar, P.; Abbas, T. A robust CRISPR-Cas9-based fluorescent reporter assay for the detection and quantification of DNA double-strand break repair. Nucleic Acids Res. 2020, 48, e126. [Google Scholar] [CrossRef] [PubMed]
- Decottignies, A. Alternative end-joining mechanisms: A historical perspective. Front. Genet. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.T.; Boboila, C.; Souza, E.K.; Franco, S.; Hickernell, T.R.; Murphy, M.; Gumaste, S.; Geyer, M.; Zarrin, A.A.; Manis, J.P.; et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007, 449, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.T.; Letsolo, B.T.; Jones, R.E.; Rowson, J.; Pratt, G.; Hewamana, S.; Fegan, C.; Pepper, C.; Baird, D.M. Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: Evidence for a telomere crisis. Blood 2010, 116, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Letsolo, B.T.; Rowson, J.; Baird, D.M. Fusion of short telomeres in human cells is characterized by extensive deletion and microhomology, and can result in complex rearrangements. Nucleic Acids Res. 2010, 38, 1841–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Zheng, H.; He, H.; Luo, Y.; Multani, A.; Carpenter, P.B.; Chang, S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010, 29, 2598–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, Y.M.; Falabella, A.F.; Schachner, L.A. Management of epidermolysis bullosa in infants and children. Clin. Dermatol. 2003, 21, 278–282. [Google Scholar] [CrossRef]
- Brambati, A.; Barry, R.M.; Sfeir, A. DNA polymerase theta (Poltheta)—An error-prone polymerase necessary for genome stability. Curr. Opin. Genet. Dev. 2020, 60, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef]
- Lee, K.; Ji, J.H.; Yoon, K.; Che, J.; Seol, J.H.; Lee, S.E.; Shim, E.Y. Microhomology Selection for Microhomology Mediated End Joining in Saccharomyces cerevisiae. Genes 2019, 10, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, P.; Greenberg, R.A. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016, 30, 1138–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Yamada, S.; Keeney, S. A global view of meiotic double-strand break end resection. Science 2017, 355, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [Green Version]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Shi, L.Z.; Wong, C.C.; Han, X.; Hwang, P.Y.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.S.; Menolfi, D.; Wu-Baer, F.; Fangazio, M.; Meyer, S.N.; Shao, Z.; Wang, Y.; Zhu, Y.; Lee, B.J.; Estes, V.M.; et al. DNA damage-induced phosphorylation of CtIP at a conserved ATM/ATR site T855 promotes lymphomagenesis in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2105440118. [Google Scholar] [CrossRef] [PubMed]
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, A.; Lopez-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Du, Z.; Wang, Y.; Feng, Z.; Fan, P.; Yan, C.; Willers, H.; Zhang, J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015, 43, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Qiu, Z.; Liu, B.; Wu, Y.; Ren, J.; Liu, Y.; Zhao, Y.; Wang, Y.; Hao, S.; Li, Z.; et al. PLK1 targets CtIP to promote microhomology-mediated end joining. Nucleic Acids Res. 2018, 46, 10724–10739. [Google Scholar] [CrossRef]
- Jiang, Y.; Yam, J.C.; Tham, C.C.; Pang, C.P.; Chu, W.K. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. Int. J. Mol. Sci. 2020, 21, 9176. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.J.; Jackson, S.P. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996, 15, 5093–5103. [Google Scholar] [CrossRef] [PubMed]
- Aniukwu, J.; Glickman, M.S.; Shuman, S. The pathways and outcomes of mycobacterial NHEJ depend on the structure of the broken DNA ends. Genes Dev. 2008, 22, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.H.; Yu, A.M.; McVey, M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 2010, 6, e1001005. [Google Scholar] [CrossRef] [Green Version]
- Koole, W.; van Schendel, R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kregten, M.; de Pater, S.; Romeijn, R.; van Schendel, R.; Hooykaas, P.J.; Tijsterman, M. T-DNA integration in plants results from polymerase-theta-mediated DNA repair. Nat. Plants 2016, 2, 16164. [Google Scholar] [CrossRef] [PubMed]
- Thyme, S.B.; Schier, A.F. Polq-Mediated End Joining Is Essential for Surviving DNA Double-Strand Breaks during Early Zebrafish Development. Cell Rep. 2016, 15, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Corneo, B.; Wendland, R.L.; Deriano, L.; Cui, X.; Klein, I.A.; Wong, S.Y.; Arnal, S.; Holub, A.J.; Weller, G.R.; Pancake, B.A.; et al. Rag mutations reveal robust alternative end joining. Nature 2007, 449, 483–486. [Google Scholar] [CrossRef]
- Simsek, D.; Brunet, E.; Wong, S.Y.; Katyal, S.; Gao, Y.; McKinnon, P.J.; Lou, J.; Zhang, L.; Li, J.; Rebar, E.J.; et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011, 7, e1002080. [Google Scholar] [CrossRef]
- Cisneros-Aguirre, M.; Ping, X.; Stark, J.M. To indel or not to indel: Factors influencing mutagenesis during chromosomal break end joining. DNA Repair 2022, 118, 103380. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soniat, M.M.; Myler, L.R.; Kuo, H.C.; Paull, T.T.; Finkelstein, I.J. RPA Phosphorylation Inhibits DNA Resection. Mol. Cell 2019, 75, 145–153.e5. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Kent, T.; Deng, S.K.; McDevitt, S.; Kashkina, E.; Hoang, T.M.; Pomerantz, R.T.; Sfeir, A. The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 2017, 24, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yucel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Kramara, J.; Osia, B.; Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Elango, R.; Sheng, Z.; Jackson, J.; DeCata, J.; Ibrahim, Y.; Pham, N.T.; Liang, D.H.; Sakofsky, C.J.; Vindigni, A.; Lobachev, K.S.; et al. Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat. Commun. 2017, 8, 1790. [Google Scholar] [CrossRef] [Green Version]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef]
- Donnianni, R.A.; Symington, L.S. Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 13475–13480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruff, P.; Donnianni, R.A.; Glancy, E.; Oh, J.; Symington, L.S. RPA Stabilization of Single-Stranded DNA Is Critical for Break-Induced Replication. Cell Rep. 2016, 17, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.P.; Lovett, S.T.; Haber, J.E. Break-induced DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010397. [Google Scholar] [CrossRef] [Green Version]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef]
- Deem, A.; Keszthelyi, A.; Blackgrove, T.; Vayl, A.; Coffey, B.; Mathur, R.; Chabes, A.; Malkova, A. Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9, e1000594. [Google Scholar] [CrossRef] [Green Version]
- Hum, Y.F.; Jinks-Robertson, S. Mismatch recognition and subsequent processing have distinct effects on mitotic recombination intermediates and outcomes in yeast. Nucleic Acids Res. 2019, 47, 4554–4568. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Takai, K.K.; Lovejoy, C.A.; de Lange, T. Break-induced replication promotes fragile telomere formation. Genes Dev. 2020, 34, 1392–1405. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Godon, C.; Cordelieres, F.P.; Biard, D.; Giocanti, N.; Megnin-Chanet, F.; Hall, J.; Favaudon, V. PARP inhibition versus PARP-1 silencing: Different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008, 36, 4454–4464. [Google Scholar] [CrossRef] [Green Version]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Im, S.A.; Yoon, Y.K.; Song, S.H.; Nam, H.J.; Hur, H.S.; Kim, H.P.; Lee, K.H.; Han, S.W.; Oh, D.Y.; et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol. Cancer Ther. 2013, 12, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Paes Dias, M.; Jonkers, J. Filling in the gaps in PARP inhibitor-induced synthetic lethality. Mol. Cell. Oncol. 2021, 8, 2010512. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef]
- Lovett, S.T.; Hurley, R.L.; Sutera, V.A., Jr.; Aubuchon, R.H.; Lebedeva, M.A. Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics 2002, 160, 851–859. [Google Scholar] [CrossRef]
- Liskay, R.M.; Letsou, A.; Stachelek, J.L. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics 1987, 115, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Hastings, P.J.; Nelis, E.; De Jonghe, P.; Van Broeckhoven, C.; Lupski, J.R. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 1998, 62, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wang, H.; Jehi, S.; Li, J.; Liu, S.; Wang, Z.; Truong, L.; Chiba, T.; Wang, Z.; Wu, X. PIF1 helicase promotes break-induced replication in mammalian cells. EMBO J. 2021, 40, e104509. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Ayyar, S.; Deem, A.K.; Chung, W.H.; Ira, G.; Malkova, A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol. Cell 2015, 60, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Ulrich, H.D.; Walden, H. Ubiquitin signalling in DNA replication and repair. Nat. Rev. Mol. Cell Biol. 2010, 11, 479–489. [Google Scholar] [CrossRef]
- de Groote, F.H.; Jansen, J.G.; Masuda, Y.; Shah, D.M.; Kamiya, K.; de Wind, N.; Siegal, G. The Rev1 translesion synthesis polymerase has multiple distinct DNA binding modes. DNA Repair 2011, 10, 915–925. [Google Scholar] [CrossRef]
- Guo, C.; Sonoda, E.; Tang, T.S.; Parker, J.L.; Bielen, A.B.; Takeda, S.; Ulrich, H.D.; Friedberg, E.C. REV1 protein interacts with PCNA: Significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell 2006, 23, 265–271. [Google Scholar] [CrossRef]
- Liu, D.; Ryu, K.S.; Ko, J.; Sun, D.; Lim, K.; Lee, J.O.; Hwang, J.; Lee, Z.W.; Choi, B.S. Insights into the regulation of human Rev1 for translesion synthesis polymerases revealed by the structural studies on its polymerase-interacting domain. J. Mol. Cell Biol. 2013, 5, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozhidaeva, A.; Pustovalova, Y.; D’Souza, S.; Bezsonova, I.; Walker, G.C.; Korzhnev, D.M. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase eta. Biochemistry 2012, 51, 5506–5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.F.; de la Vega, M.B.; Calzetta, N.L.; Siri, S.O.; Gottifredi, V. CDK-Independent and PCNA-Dependent Functions of p21 in DNA Replication. Genes 2020, 11, 593. [Google Scholar] [CrossRef]
- Lancey, C.; Tehseen, M.; Bakshi, S.; Percival, M.; Takahashi, M.; Sobhy, M.A.; Raducanu, V.S.; Blair, K.; Muskett, F.W.; Ragan, T.J.; et al. Cryo-EM structure of human Pol kappa bound to DNA and mono-ubiquitylated PCNA. Nat. Commun. 2021, 12, 6095. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. Regulation of Mammalian DNA Replication via the Ubiquitin-Proteasome System. Adv. Exp. Med. Biol. 2017, 1042, 421–454. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.L.; Beagan, K.; Orr-Weaver, T.L.; McVey, M. Multiple mechanisms contribute to double-strand break repair at rereplication forks in Drosophila follicle cells. Proc. Natl. Acad. Sci. USA 2016, 113, 13809–13814. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.N.; Li, Y.; Sun, E.; Ang, K.; Hwang, P.Y.; Wu, X. Homologous recombination is a primary pathway to repair DNA double-strand breaks generated during DNA rereplication. J. Biol. Chem. 2014, 289, 28910–28923. [Google Scholar] [CrossRef] [Green Version]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase theta in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Jiang, Y.; Yam, J.C.; Chu, W.K. Poly ADP Ribose Polymerase Inhibitor Olaparib Targeting Microhomology End Joining in Retinoblastoma Protein Defective Cancer: Analysis of the Retinoblastoma Cell-Killing Effects by Olaparib after Inducing Double-Strand Breaks. Int. J. Mol. Sci. 2021, 22, 10687. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L.; Lee, S.E.; Dawson, L.K.; Marlow, L.A.; Edenfield, B.H.; Durham, W.F.; Flotte, T.J.; Thompson, M.; Small, D.L.; Synnott, A.J.; et al. Targeting the cyclin dependent kinase and retinoblastoma axis overcomes standard of care resistance in BRAF (V600E) -mutant melanoma. Oncotarget 2018, 9, 10905–10919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Yuan, Y.; Ren, H.; Yue, H.; Xu, B.; Qian, J. SUMOylation regulates Rb hyperphosphorylation and inactivation in uveal melanoma. Cancer Sci. 2021, 113, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [Green Version]
- Zoumpoulidou, G.; Alvarez-Mendoza, C.; Mancusi, C.; Ahmed, R.M.; Denman, M.; Steele, C.D.; Tarabichi, M.; Roy, E.; Davies, L.R.; Manji, J.; et al. Therapeutic vulnerability to PARP1,2 inhibition in RB1-mutant osteosarcoma. Nat. Commun. 2021, 12, 7064. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2020, 10, 623679. [Google Scholar] [CrossRef]
- Schwab, M.; Varmus, H.E.; Bishop, J.M.; Grzeschik, K.H.; Naylor, S.L.; Sakaguchi, A.Y.; Brodeur, G.; Trent, J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature 1984, 308, 288–291. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef]
- Swartling, F.J.; Grimmer, M.R.; Hackett, C.S.; Northcott, P.A.; Fan, Q.W.; Goldenberg, D.D.; Lau, J.; Masic, S.; Nguyen, K.; Yakovenko, S.; et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010, 24, 1059–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.H.; Murphree, A.L.; Benedict, W.F. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature 1984, 309, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nau, M.M.; Brooks, B.J., Jr.; Carney, D.N.; Gazdar, A.F.; Battey, J.F.; Sausville, E.A.; Minna, J.D. Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1092–1096. [Google Scholar] [CrossRef] [Green Version]
- Freier, K.; Flechtenmacher, C.; Devens, F.; Hartschuh, W.; Hofele, C.; Lichter, P.; Joos, S. Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol. Rep. 2006, 15, 1141–1145. [Google Scholar] [CrossRef] [Green Version]
- Lillie, T.H.; Whalley, T.D.; Gomperts, B.D. Modulation of the exocytotic reaction of permeabilised rat mast cells by ATP, other nucleotides and Mg2+. Biochim. Biophys. Acta 1991, 1094, 355–363. [Google Scholar] [CrossRef]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.A.; Lu, F.; Bashllari, D.; Wang, L.; Opipari, A.W.; Castle, V.P. Alternative NHEJ Pathway Components Are Therapeutic Targets in High-Risk Neuroblastoma. Mol. Cancer Res. 2015, 13, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.A.; Chukkapalli, S.; Bashllari, D.; Thomas, T.T.; Van Noord, R.A.; Lawlor, E.R.; Hoenerhoff, M.J.; Opipari, A.W.; Opipari, V.P. Alternative NHEJ pathway proteins as components of MYCN oncogenic activity in human neural crest stem cell differentiation: Implications for neuroblastoma initiation. Cell Death Dis. 2017, 8, 3208. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y. Contribution of Microhomology to Genome Instability: Connection between DNA Repair and Replication Stress. Int. J. Mol. Sci. 2022, 23, 12937. https://doi.org/10.3390/ijms232112937
Jiang Y. Contribution of Microhomology to Genome Instability: Connection between DNA Repair and Replication Stress. International Journal of Molecular Sciences. 2022; 23(21):12937. https://doi.org/10.3390/ijms232112937
Chicago/Turabian StyleJiang, Yuning. 2022. "Contribution of Microhomology to Genome Instability: Connection between DNA Repair and Replication Stress" International Journal of Molecular Sciences 23, no. 21: 12937. https://doi.org/10.3390/ijms232112937
APA StyleJiang, Y. (2022). Contribution of Microhomology to Genome Instability: Connection between DNA Repair and Replication Stress. International Journal of Molecular Sciences, 23(21), 12937. https://doi.org/10.3390/ijms232112937