
The Gene Expression Landscape of Prostate Cancer BM Reveals Close Interaction with the Bone Microenvironment

 , ,
, , 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
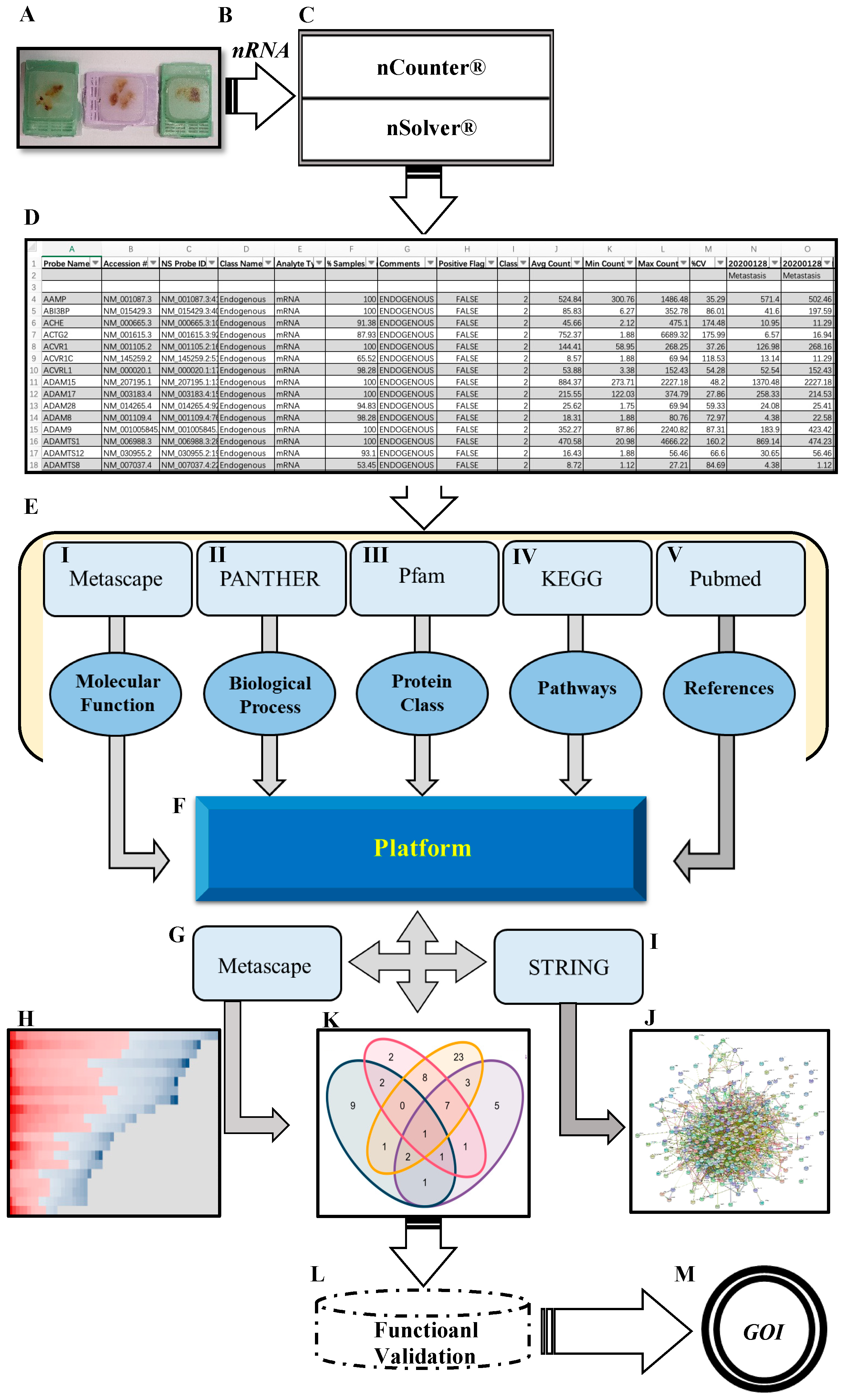
2. Results
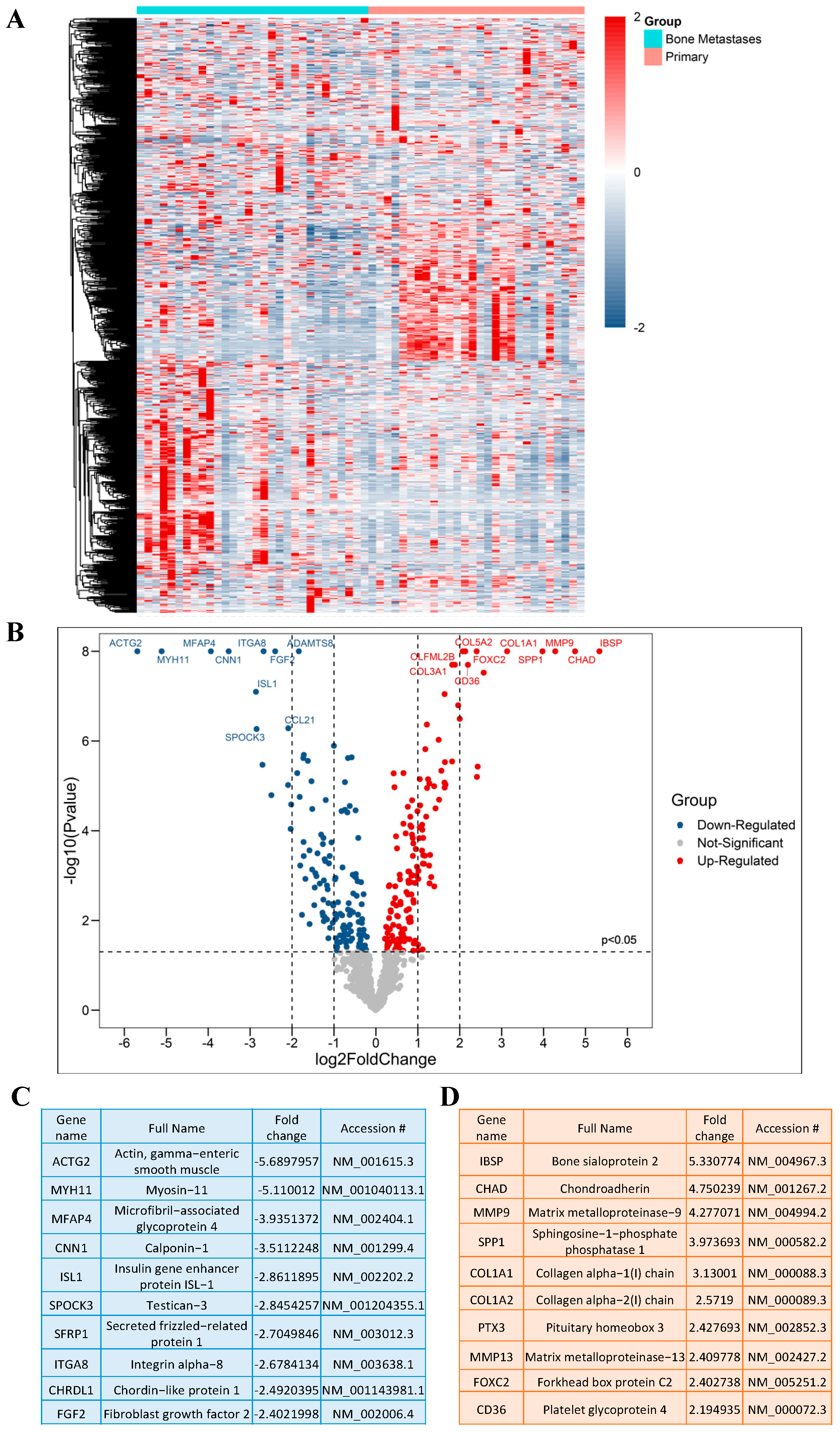
2.1. Differentially Expressed Genes (DEGs) in PCa BM Compared to Primary Tumors
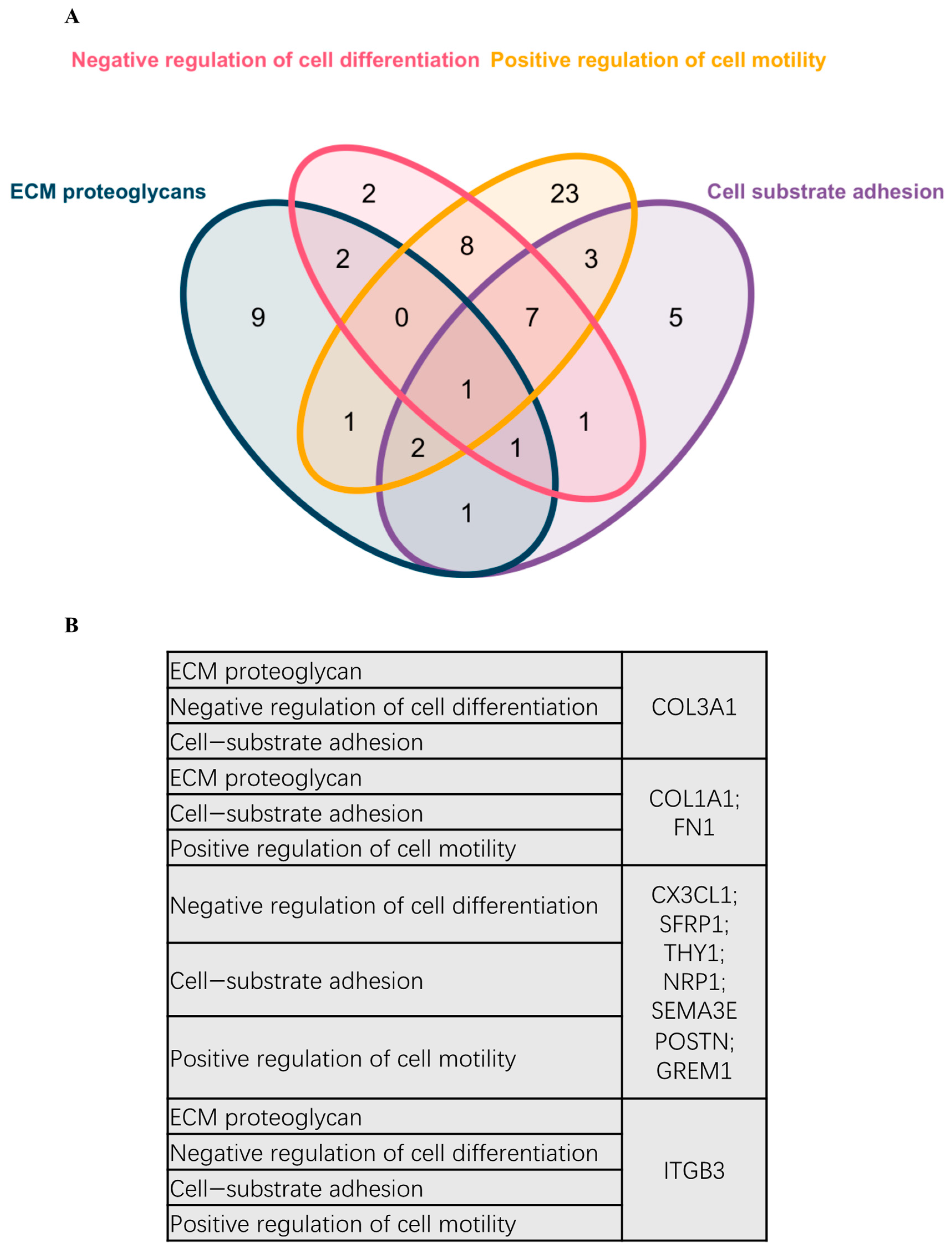
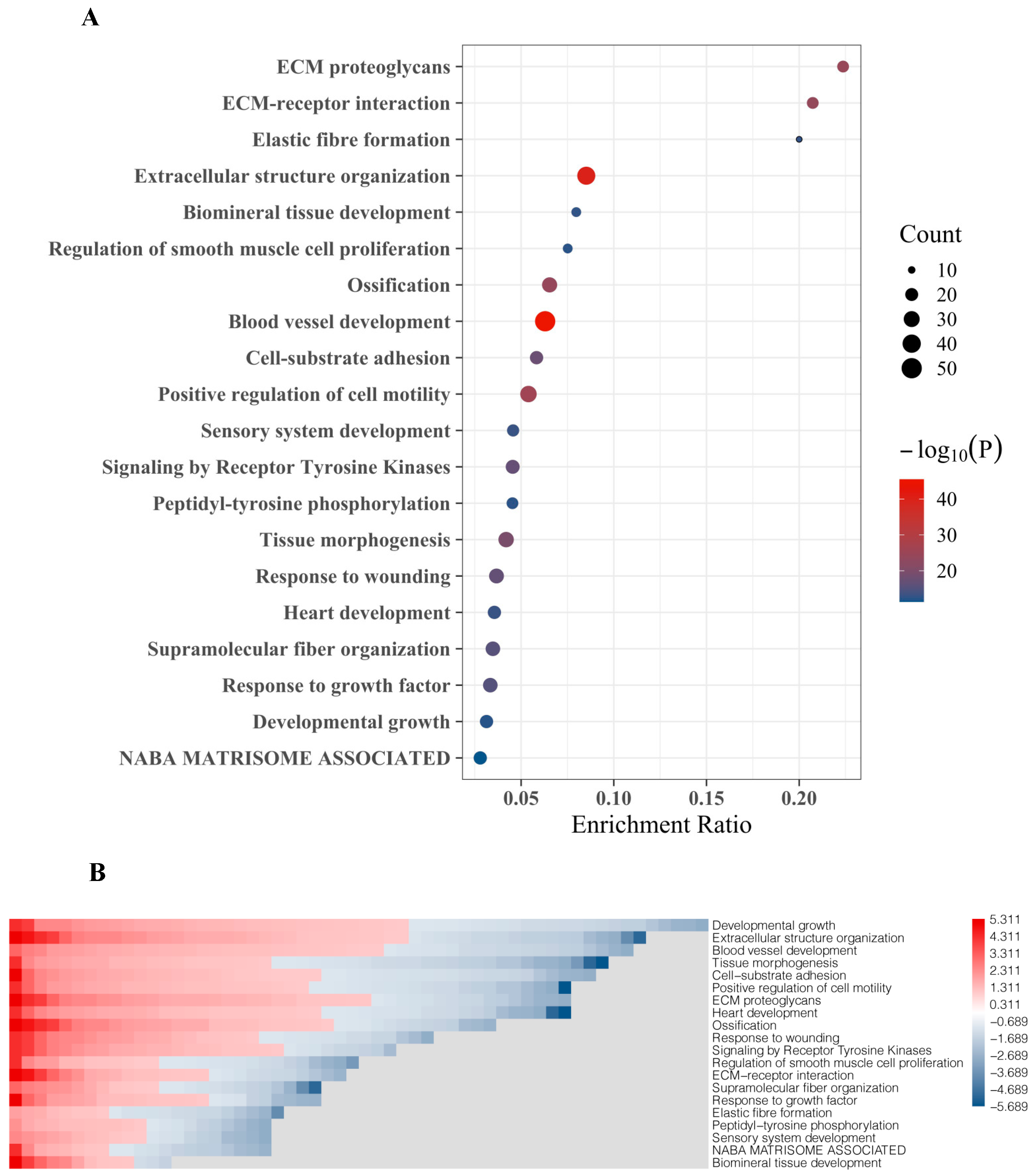
2.2. Functional Assignment of Top DEGs
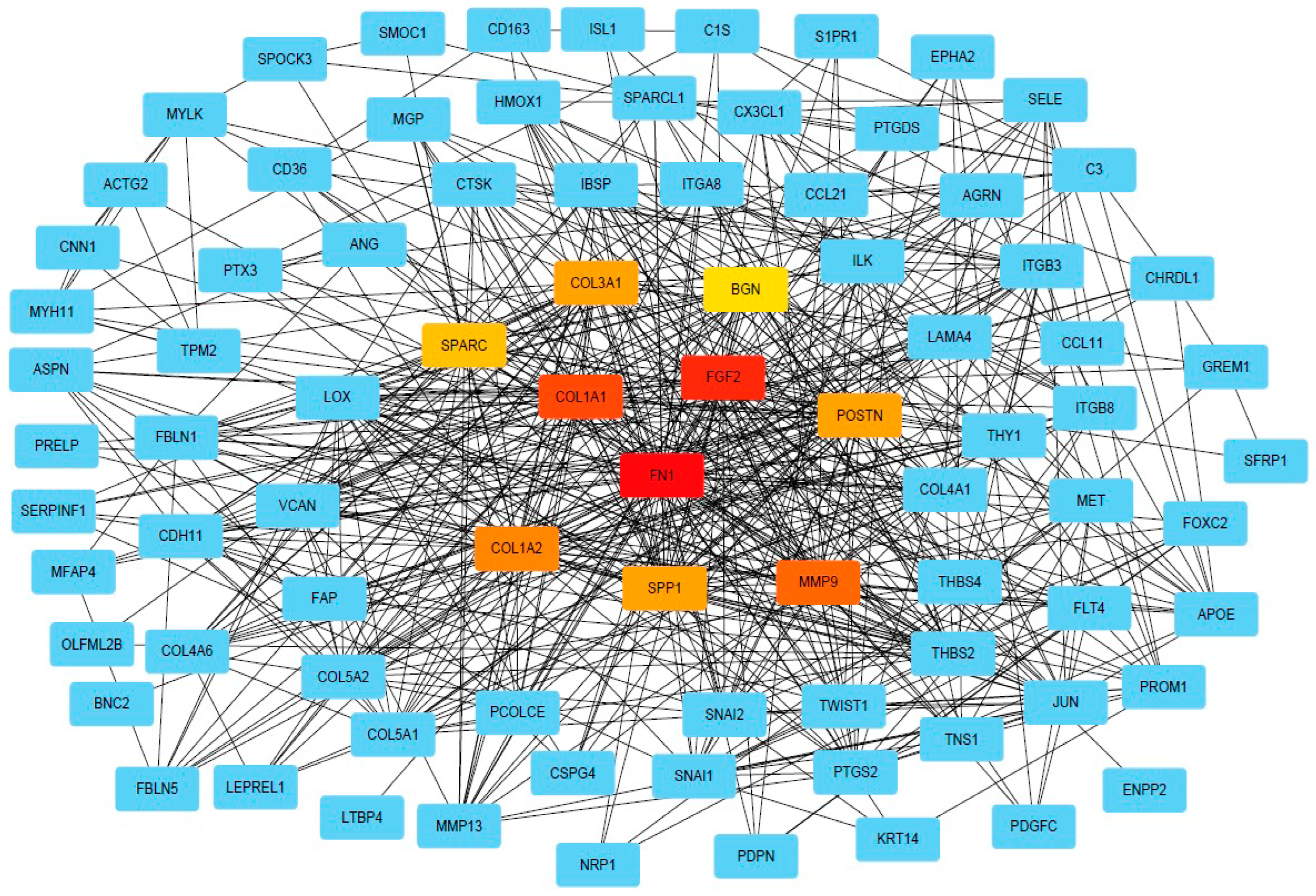
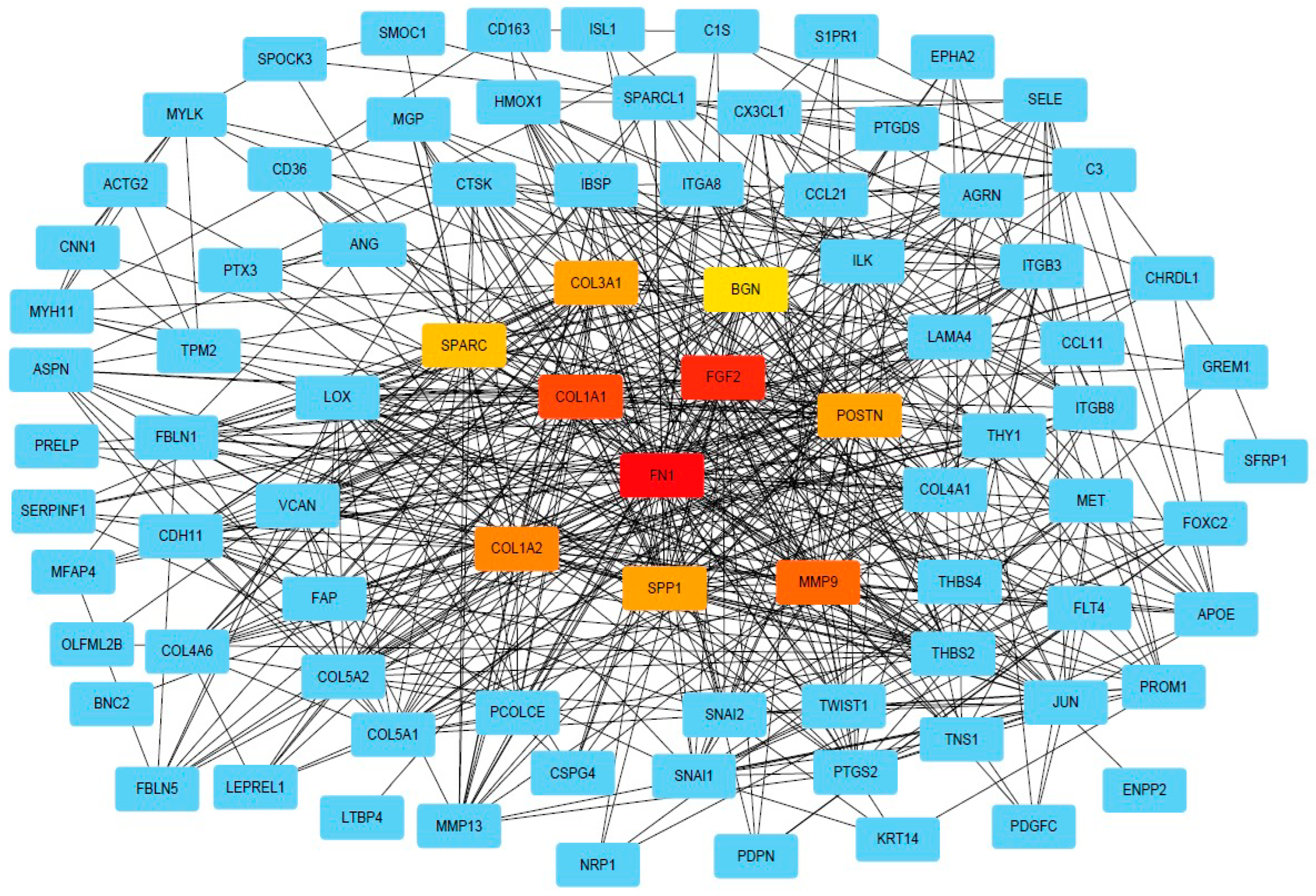
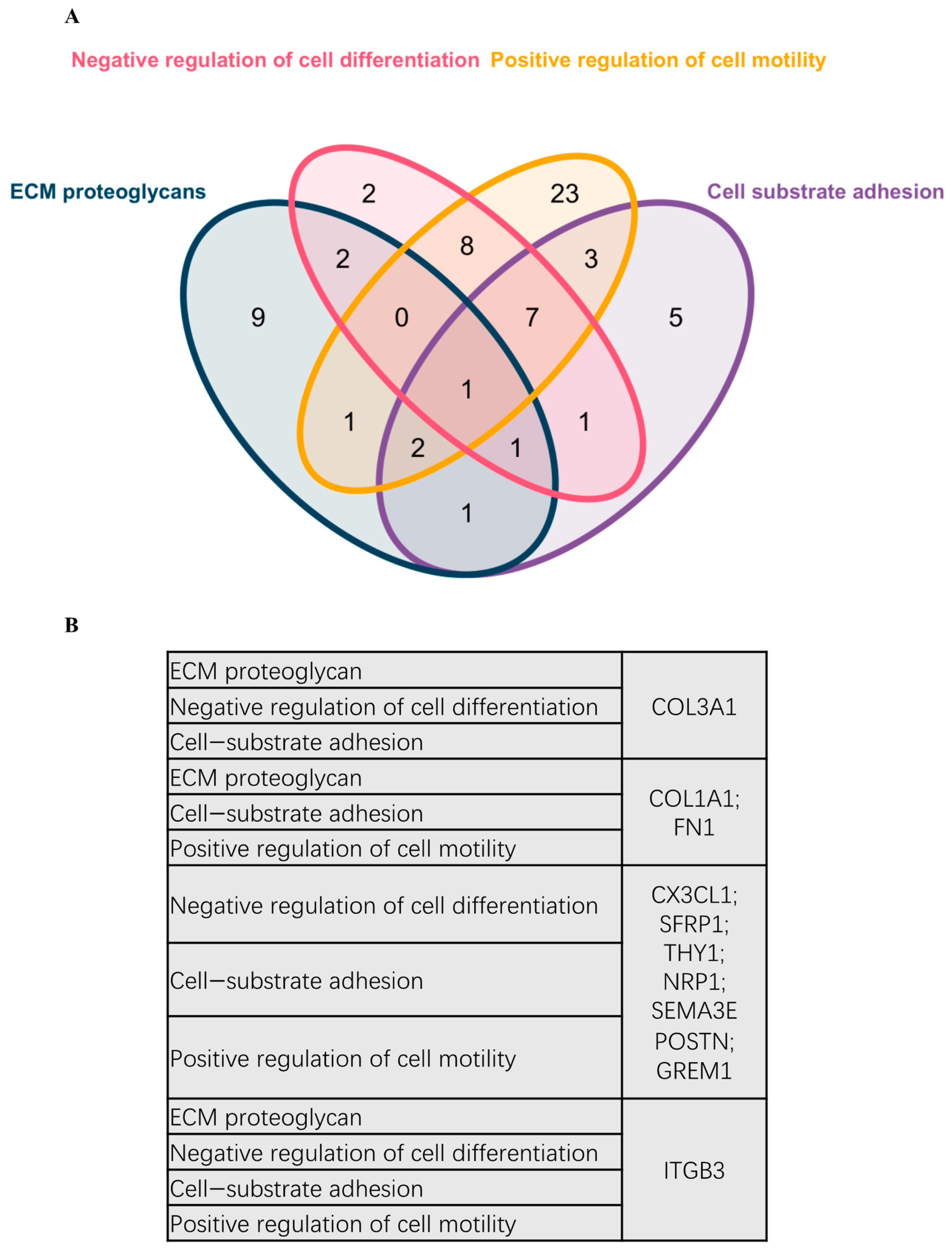
2.3. PPI and Common Functional Assignments of Top DEGs
3. Discussion
4. Materials and Methods
4.1. FFPE Specimens and Cohort Description
4.2. RNA Extraction from Decalcified FFPE Specimens
4.3. mRNA Analysis (Digital Analysis for Transcriptomes)
4.4. DEGs Analysis
4.5. Gene Annotation (GA) and Biological Themes Enrichment Analysis
4.6. Construction of Protein–Protein Interaction (PPI) Network and Hub Proteins Determination
4.7. Data Platform Creation (MCNP Build Up)
4.8. Liquid Biopsy (Circulating Tumor Cells (CTC) Detection and mRNA Isolation)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Bernard, B.; Burnett, C.; Sweeney, C.J.; Rider, J.R.; Sridhar, S.S. Impact of age at diagnosis of de novo metastatic prostate cancer on survival. Cancer 2020, 126, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Schopfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Furesi, G.; Rauner, M.; Hofbauer, L.C. Emerging Players in Prostate Cancer-Bone Niche Communication. Trends Cancer 2021, 7, 112–121. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Chi, K.N.; Wyatt, A.W. Clinical implications of genomic alterations in metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 310–322. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Sailer, V.; Schiffman, M.H.; Kossai, M.; Cyrta, J.; Beg, S.; Sullivan, B.; Pua, B.B.; Lee, K.S.; Talenfeld, A.D.; Nanus, D.M.; et al. Bone biopsy protocol for advanced prostate cancer in the era of precision medicine. Cancer 2017, 124, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Saraji, A.; Offermann, A.; Stegmann-Frehse, J.; Hempel, K.; Kang, D.; Krupar, R.; Watermann, C.; Jonigk, D.; Kühnel, M.P.; Kirfel, J.; et al. Cracking it—Successful mRNA extraction for digital gene expression analysis from decalcified, formalin-fixed and paraffin-embedded bone tissue. PLoS ONE 2021, 16, e0257416. [Google Scholar] [CrossRef]
- Thysell, E.; Vidman, L.; Ylitalo, E.B.; Jernberg, E.; Crnalic, S.; Iglesias-Gato, D.; Flores-Morales, A.; Stattin, P.; Egevad, L.; Widmark, A.; et al. Gene expression profiles define molecular subtypes of prostate cancer bone metastases with different outcomes and morphology traceable back to the primary tumor. Mol. Oncol. 2019, 13, 1763–1777. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Bozec, A.; Rauner, M.; Jakob, F.; Perner, S.; Pantel, K. Novel approaches to target the microenvironment of bone metastasis. Nat. Rev. Clin. Oncol. 2021, 18, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Boguslawska, J.; Kedzierska, H.; Poplawski, P.; Rybicka, B.; Tanski, Z.; Piekielko-Witkowska, A. Expression of Genes Involved in Cellular Adhesion and Extracellular Matrix Remodeling Correlates with Poor Survival of Patients with Renal Cancer. J. Urol. 2016, 195, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.Q.; Li, Z.H.; Wen, S.W.; Zhang, Y.F.; Li, Y.; Cheng, J.G.; Wang, G.Y. Identification of Commonly Dysregulated Genes in Non-small-cell Lung Cancer by Integrated Analysis of Microarray Data and qRT-PCR Validation. Lung 2015, 193, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, Y.; Li, A. Identification of COL1A1 and COL1A2 as candidate prognostic factors in gastric cancer. World J. Surg. Oncol. 2016, 14, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Wen, Y.; Xuan, C.; Chen, Q.; Xiang, Q.; Wang, J.; Liu, Y.; Luo, L.; Zhao, S.; Deng, Y.; et al. Identifying the key genes and microRNAs in prostate cancer bone metastasis by bioinformatics analysis. FEBS Open Bio 2020, 10, 674–688. [Google Scholar] [CrossRef]
- Wu, Y.P.; Ke, Z.B.; Lin, F.; Wen, Y.A.; Chen, S.; Li, X.D.; Chen, S.H.; Sun, X.L.; Huang, J.B.; Zheng, Q.S.; et al. Identification of key genes and pathways in castrate-resistant prostate cancer by integrated bioinformatics analysis. Pathol. Res. Pract. 2020, 216, 153109. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.D.; Carter, K.J.; Jean-Philippe, S.R.; Chang, M.; Mobashery, S.; Thiolloy, S.; Lynch, C.C.; Matrisian, L.M.; Fingleton, B. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008, 68, 6251–6259. [Google Scholar] [CrossRef] [Green Version]
- Yousef, E.M.; Tahir, M.R.; St-Pierre, Y.; Gaboury, L.A. MMP-9 expression varies according to molecular subtypes of breast cancer. BMC Cancer 2014, 14, 609. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Nair, B.B.; Reynolds, J.L.; Sykes, D.E.; Mahajan, S.D.; Chadha, K.C.; Schwartz, S.A. Overexpression of MMP-9 contributes to invasiveness of prostate cancer cell line LNCaP. Immunol. Investig. 2011, 40, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Enciu, A.M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? Biomed. Res. Int. 2018, 2018, 7801202. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Cai, X.; Long, L.; Xie, L.; Ma, H.; Zhou, Y.; Liu, S.; Zeng, C. CD36 promotes the epithelial-mesenchymal transition and metastasis in cervical cancer by interacting with TGF-β. J. Transl. Med. 2019, 17, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Tao, B.; Fan, Q.; Wang, S.; Chen, L.; Zhang, J.; Hao, Y.; Dong, S.; Wang, Z.; Wang, W.; et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine 2019, 45, 108–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Børretzen, A.; Gravdal, K.; Haukaas, S.A.; Beisland, C.; Akslen, L.A.; Halvorsen, O.J. FOXC2 expression and epithelial-mesenchymal phenotypes are associated with castration resistance, metastasis and survival in prostate cancer. J. Pathol. Clin. Res. 2019, 5, 272–286. [Google Scholar] [CrossRef]
- Khodavirdi, A.C.; Song, Z.; Yang, S.; Zhong, C.; Wang, S.; Wu, H.; Pritchard, C.; Nelson, P.S.; Roy-Burman, P. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006, 66, 883–888. [Google Scholar] [CrossRef] [Green Version]
- Chandran, U.R.; Ma, C.; Dhir, R.; Bisceglia, M.; Lyons-Weiler, M.; Liang, W.; Michalopoulos, G.; Becich, M.; Monzon, F.A. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 2007, 7, 64. [Google Scholar] [CrossRef]
- Falagario, U.G.; Busetto, G.M.; Netti, G.S.; Sanguedolce, F.; Selvaggio, O.; Infante, B.; Ranieri, E.; Stallone, G.; Carrieri, G.; Cormio, L. Prospective Validation of Pentraxin-3 as a Novel Serum Biomarker to Predict the Risk of Prostate Cancer in Patients Scheduled for Prostate Biopsy. Cancers 2021, 13, 1611. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Cormio, L.; Netti, G.S.; Infante, B.; Selvaggio, O.; Fino, G.D.; Ranieri, E.; Bruno, F.; Prattichizzo, C.; Sanguedolce, F.; et al. Pentraxin 3: A novel biomarker for predicting progression from prostatic inflammation to prostate cancer. Cancer Res. 2014, 74, 4230–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, P.; Hao, Y.; Yu, T.; Liu, L.; Song, Y.; Li, Y. Interaction between DNMT3B and MYH11 via hypermethylation regulates gastric cancer progression. BMC Cancer 2021, 21, 914. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cheng, J.; Zhang, F.; Luo, X.; Zhang, Z.; Chen, S. Estrogen receptor α is involved in the regulation of ITGA8 methylation in estrogen receptor-positive breast cancer. Ann. Transl. Med. 2020, 8, 993. [Google Scholar] [CrossRef]
- Lu, X.; Wan, F.; Zhang, H.; Shi, G.; Ye, D. ITGA2B and ITGA8 are predictive of prognosis in clear cell renal cell carcinoma patients. Tumor Biol. 2016, 37, 253–262. [Google Scholar] [CrossRef]
- Wang, L.Y.; Cui, J.J.; Zhu, T.; Shao, W.H.; Zhao, Y.; Wang, S.; Zhang, Y.P.; Wu, J.C.; Zhang, L. Biomarkers identified for prostate cancer patients through genome-scale screening. Oncotarget 2017, 8, 92055–92063. [Google Scholar] [CrossRef]
- Nakada, M.; Yamada, A.; Takino, T.; Miyamori, H.; Takahashi, T.; Yamashita, J.; Sato, H. Suppression of membrane-type 1 matrix metalloproteinase (MMP)-mediated MMP-2 activation and tumor invasion by testican 3 and its splicing variant gene product, N-Tes. Cancer Res. 2001, 61, 8896–8902. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraji, A.; Duan, K.; Watermann, C.; Hempel, K.; Roesch, M.C.; Krupar, R.; Stegmann-Frehse, J.; Jonigk, D.; Kuehnel, M.P.; Klapper, W.; et al. The Gene Expression Landscape of Prostate Cancer BM Reveals Close Interaction with the Bone Microenvironment. Int. J. Mol. Sci. 2022, 23, 13029. https://doi.org/10.3390/ijms232113029
Saraji A, Duan K, Watermann C, Hempel K, Roesch MC, Krupar R, Stegmann-Frehse J, Jonigk D, Kuehnel MP, Klapper W, et al. The Gene Expression Landscape of Prostate Cancer BM Reveals Close Interaction with the Bone Microenvironment. International Journal of Molecular Sciences. 2022; 23(21):13029. https://doi.org/10.3390/ijms232113029
Chicago/Turabian StyleSaraji, Alireza, Kang Duan, Christian Watermann, Katharina Hempel, Marie C. Roesch, Rosemarie Krupar, Janine Stegmann-Frehse, Danny Jonigk, Mark Philipp Kuehnel, Wolfram Klapper, and et al. 2022. "The Gene Expression Landscape of Prostate Cancer BM Reveals Close Interaction with the Bone Microenvironment" International Journal of Molecular Sciences 23, no. 21: 13029. https://doi.org/10.3390/ijms232113029
APA StyleSaraji, A., Duan, K., Watermann, C., Hempel, K., Roesch, M. C., Krupar, R., Stegmann-Frehse, J., Jonigk, D., Kuehnel, M. P., Klapper, W., Merseburger, A. S., Kirfel, J., Perner, S., Offermann, A., & Sailer, V. (2022). The Gene Expression Landscape of Prostate Cancer BM Reveals Close Interaction with the Bone Microenvironment. International Journal of Molecular Sciences, 23(21), 13029. https://doi.org/10.3390/ijms232113029