
Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease



Abstract
:1. Introduction
2. Familial and Sporadic Forms of PD Epigenetic Aspects
2.1. Genetic Factors
2.2. Environmental Factors
2.3. Epigenetic Aspects in Development of PD
3. Organophosphates and Other Environmental Toxicants as a Cause of Sporadic PD
4. Mitochondrial Dysfunction in PD
5. α-Synuclein in Health and Parkinson’s Disease
5.1. Role of αSyn in DA Synaptic Transmission in the Normal State
5.2. Prion-like Effect of αSyn
5.3. αSyn Inhibits Formation of the TOM Complex
5.4. αSyn Participates in Calcium Transfer between ER and Mitochondria
5.5. αSyn and Mitochondrial Voltage-Dependent Anion Channel (VDAC)
5.6. αSyn and Control of Mitochondrial Bioenergetics
6. Physiological Features of Neurons and Vulnerability of Nervous Systems
6.1. Nigrostriatal Neurons
6.2. Enteric Nervous System and Enteroendocrine Cells
7. Gastrointestinal (GI) Tract in Pathogenesis of PD
7.1. Gut Microbiota May Influence αSyn Aggregation
7.2. αSyn Accumulates in the GI Tract
8. Endothelial Cells (ECs) and Blood Vessel Damage in Pathogenesis of PD
8.1. αSyn, ECs and Vascular Factors in PD
8.2. Neurovascular Unit, Angiogenesis and the BBB Dysfunction in PD
9. Phagocytic Cells and Glymphatic Transport in PD
10. Treatment and Optimization of the Condition of Patients with PD
10.1. Traditional Therapy of PD
10.2. Non-Traditional Pharmaceuticals for Prevention and Treatment PD
10.3. Targeting Autophagy in PD
10.4. Care for Vascular Endothelium in PD
10.5. Use of Nutraceuticals in PD
10.6. Targeting Gut Microflora
10.7. Physiotherapy and Exercise in PD
10.8. Experimental Stem Cell Therapy
10.9. Immunotherapies
10.10. Epigenetic Drugs
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACOX | acyl-CoA oxidase |
| AD | Alzheimer disease |
| ALS | amyotrophic lateral sclerosis |
| AP | action potential |
| AP-1 | activator protein 1 |
| ALP | autophagy-lysosome or autolysosome pathway |
| AQP4 | aquaporin 4 |
| αSyn | α-synuclein |
| BA | bile acids |
| BAM | border-associated macrophages |
| BBB | blood-brain barrier |
| BG-PVS | perivascular space of the basal ganglia |
| c-ABL | Abl kinase |
| CAA | cerebral amyloid angiopathy |
| CNS | central nervous system |
| COMT | catechol O-methyltransferase |
| CPF | chlorpyrifos |
| DA | dopamine |
| DAO | D-amino-acid oxidase |
| DAMP | damage-associated molecular patterns |
| DATs | dopamine transporters |
| DBS | deep brain stimulation |
| DDH1 or AKR1C1 | dihydrodiol dehydrogenase |
| DDO | D-aspartate oxidase |
| DLB | dementia with Lewy bodies |
| DMT2 | type 2 diabetes |
| DNMTs | DNA methyltransferases |
| DMV | dorsal motor nucleus of the vagus |
| DOPAL | 3,4-dihydroxyphenylacetaldehyde |
| DSCs | disease-specific single-nucleotide changes |
| DTI-ALPS | diffusion tensor image analysis along the perivascular space |
| EECs | enteroendocrine cells |
| EET | epoxyeicosatrienoic acids |
| EC | endothelial cells |
| ENS | enteric nervous system |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| EVs | erythrocyte vesicles |
| FAO | fatty acid β-oxidation |
| FHC | ferritin heavy chain |
| FMT | fecal microbial flora transplantation |
| FOS | fructo-oligosaccharides |
| GABA | γ-aminobutyric acid |
| GBA1 | lysosomal glucocerebrosidase |
| GCase 1 | β-glucocerebrosidase 1 |
| GI | gastrointestinal tract |
| GOS | galacto-oligosaccharides |
| GPx | glutathione peroxidase |
| GST-Pi | glutathione-S-transferase Pi |
| GWAS | genome-wide association study |
| HAO | L-α-hydroxyacid oxidase |
| HDAC | histone deacetylase |
| 5-HT | 5-hydroxytryptamine, serotonin |
| ICAM-1 | Inter-Cellular Adhesion Molecule 1 |
| ICDH | isocitrate dehydrogenase |
| IL-1β | interleukin-1β |
| iLBD | incidental LB disease |
| IMM | inner mitochondrial membrane |
| IMS | intermediate syndrome |
| InsP3R | inositol 1,4,5-trisphosphate receptors |
| Kir6 | ATP-sensitive K+ channels |
| LB | Lewy bodies |
| LC | locus coeruleus |
| LRRK2 | leucine-rich repeat kinase 2 |
| LPS | lipopolysaccharide |
| LRRK2 | leucine-rich repeat kinase 2 |
| LTCCs | L-type calcium channels |
| LUHMES | Lund human mesencephalic cells |
| MAM | mitochondria-associated membranes |
| MAO | monoamine oxidase |
| MAO-B | monoamine oxidase B |
| MCh | mitochondria |
| MCP-1 | monocyte chemotactic protein-1 |
| miRNA | microRNA |
| MMP-9 | matrix metalloproteinase-9 |
| mPTP | mitochondrial permeability transition pore |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MT3 | metallothionein-3 |
| NAC | non-amyloid-β component |
| NAM | niacinamide |
| NC | nucleus caudatus, caudate nucleus |
| ncRNA | non-coding RNA |
| NO | nitric oxide |
| Nogo-B | Neurite outgrowth inhibitor-B |
| NOX | NADPH oxidases |
| 3-NPA | 3-nitropropionic acid |
| nt | nucleotides |
| NVU | neurovascular unit |
| OGDH | oxoglutarate dehydrogenase |
| 6-OHDA | 6-hydroxydopamine |
| OMM | outer mitochondrial membrane |
| OPs | organophosphates |
| OPIDP | organophosphate-induced delayed neuropathy |
| OSC | organosulfur compounds |
| PAI-1 | plasminogen activator inhibitor-1 |
| PAOX | polyamine oxidase |
| PD | Parkinson’s disease |
| PDD | Parkinson’s disease dementia |
| PDH | pyruvate dehydrogenase |
| PFFs | pre-formed fibrils |
| PINK1 | phosphatase and tensin homologue (PTEN)-induced kinase 1 |
| PIPOX | L-pipecolic acid oxidase |
| PKA | protein kinase A |
| PMCA | plasma membrane Ca2+-ATPase |
| PNS | peripheral nervous system |
| PON-1 | paraoxonase-1 |
| PP2A | protein phosphatase 2A |
| PrP | prion proteins |
| PVM | perivascular macrophages |
| ROS | reactive oxygen species |
| SCFA | short-chain fatty acids |
| sHsps | small heat shock proteins |
| SN | substantia nigra |
| SNpc | substantia nigra pars compacta |
| SOD | superoxide dismutase |
| TJ | tight junction |
| TM | thrombomodulin |
| TNF-α | tumor necrosis factor alpha |
| TOM | translocase of the outer membrane |
| TOM-CC | TOM core complex |
| tPA | tissue plasminogen activator |
| TUDCA | tauro ursodesoxy cholic acid |
| UDCA | ursodesoxy cholic acid |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VDAC | voltage-dependent anion channel |
| VEGFR | vascular endothelial growth factor receptor |
| VMAT2 | vesicular monoamine transporter 2 |
| VPS35 | vacuolar protein sorting ortholog 35, involved in autophagy |
| VTA | ventral tegmental area |
| vWF | von Willebrand factor |
| WPB | Weibel-Palade bodies |
| XO | xanthine oxidase |
| ZO-1 | zonula occludens-1 |
References
- Schnabel, J. Secrets of the shaking palsy. Nature 2010, 466, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Obeso, I.; Casabona, E.; Rodríguez-Rojas, R.; Bringas, M.L.; Macías, R.; Pavón, N.; Obeso, J.A.; Jahanshahi, M. Unilateral subthalamotomy in Parkinson’s disease: Cognitive, psychiatric and neuroimaging changes. Cortex 2017, 94, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Braak, E.; Braak, H. Silver staining method for demonstrating Lewy bodies in Parkinson’s disease and argyrophilic oligodendrocytes in multiple system atrophy. J. Neurosci. Methods 1999, 87, 111–115. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ. 1998, 5, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Pediaditakis, I.; Kodella, K.R.; Manatakis, D.V.; Le, C.Y.; Hinojosa, C.D.; Tien-Street, W.; Manolakos, E.S.; Vekrellis, K.; Hamilton, G.A.; Ewart, L.; et al. Modeling alpha-synuclein pathology in a human brain-chip to assess blood-brain barrier disruption. Nat. Commun. 2021, 12, 5907. [Google Scholar] [CrossRef]
- Hang, L.; Thundyil, J.; Lim, K.L. Mitochondrial dysfunction and Parkinson disease: A Parkin-AMPK alliance in neuroprotection. Ann. N. Y. Acad. Sci. 2015, 1350, 37–47. [Google Scholar] [CrossRef]
- Goiran, T.; Eldeeb, M.A.; Zorca, C.E.; Fon, E.A. Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease. Cells 2022, 11, 2097. [Google Scholar] [CrossRef]
- Lim, L.; Jackson-Lewis, V.; Wong, L.C.; Shui, G.H.; Goh, A.X.; Kesavapany, S.; Jenner, A.M.; Fivaz, M.; Przedborski, S.; Wenk, M.R. Lanosterol induces mitochondrial uncoupling and protects dopaminergic neurons from cell death in a model for Parkinson’s disease. Cell Death Differ. 2012, 19, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Requejo-Aguilar, R.; Bolaños, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 2016, 100, 123–137. [Google Scholar] [CrossRef]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Trojanowski, J.Q.; Lee, V.M. α-Synuclein pathology in Parkinson’s disease and related a-synucleinopathies. Neurosci. Lett. 2019, 709, 134316. [Google Scholar] [CrossRef] [PubMed]
- Berge, N.V.D.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, N.; Uemura, M.; Luk, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med. 2020, 26, 936–952. [Google Scholar] [CrossRef]
- Mou, Y.; Du, Y.; Zhou, L.; Yue, J.; Hu, X.; Liu, Y.; Chen, S.; Lin, X.; Zhang, G.; Xiao, H.; et al. Gut Microbiota Interact With the Brain Through Systemic Chronic Inflammation: Implications on Neuroinflammation, Neurodegeneration, and Aging. Front. Immunol. 2022, 13, 796288. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Martens, Y.A.; Meneses, A.; Ryu, D.H.; Lu, W.; Raulin, A.C.; Li, F.; Zhao, J.; Chen, Y.; Jin, Y.; et al. LRP1 is a neuronal receptor for α-synuclein uptake and spread. Mol. Neurodegener. 2022, 17, 57. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.Q.; Jia, C.; Lim, Y.J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118. [Google Scholar] [CrossRef]
- Lanore, A.; Lesage, S.; Mariani, L.L.; Menon, P.J.; Ravassard, P.; Cheval, H.; Corti, O.; Brice, A.; Corvol, J.C. Does the Expression and Epigenetics of Genes Involved in Monogenic Forms of Parkinson’s Disease Influence Sporadic Forms? Genes 2022, 13, 479. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Brodin, L.; Shupliakov, O. Retromer in Synaptic Function and Pathology. Front. Synaptic Neurosci. 2018, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.K.; Farrer, M.J. Genetics and genomics of Parkinson’s disease. Genome Med. 2014, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. 2022; epub ahead of print. [Google Scholar] [CrossRef]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson’s Disease: A Multisystem Disorder. Neurosci. Bull. 2022; epub ahead of print. [Google Scholar] [CrossRef]
- Chanyachukul, T.; Yoovathaworn, K.; Thongsaard, W.; Chongthammakun, S.; Navasumrit, P.; Satayavivad, J. Attenuation of paraquat-induced motor behavior and neurochemical disturbances by L-valine in vivo. Toxicol. Lett. 2004, 150, 259–269. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltazar, M.T.; Dinis-Oliveira, R.J.; de Lourdes Bastos, M.; Tsatsakis, A.M.; Duarte, J.A.; Carvalho, F. Pesticides exposure as etiological factors of Parkinson’s disease and other neurodegenerative diseases—A mechanistic approach. Toxicol. Lett. 2014, 230, 85–103. [Google Scholar] [CrossRef]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Martino, R.; Candundo, H.; Lieshout, P.V.; Shin, S.; Crispo, J.A.G.; Barakat-Haddad, C. Onset and progression factors in Parkinson’s disease: A systematic review. Neurotoxicology 2017, 61, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Yankee, E.L. A review on Parkinson’s disease treatment. Neuroimmunol. Neuroinflamm. 2021, 8, 222–244. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Vilhelmova, H.M.; Hartmanova, I.; Pink, W.K. Chronic parkinsonism in humans due to product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; Sone, N.; Saitoh, T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J. Neurochem. 1987, 48, 1787–1793. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: The dual hit theory revisited. Ann. N.Y. Acad. Sci. 2009, 1170, 615–622. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; van Wezel, R.J.; Kraneveld, A.D. Exploring Braak’s Hypothesis of Parkinson’s Disease. Front Neurol. 2017, 8, 37. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.C.; Li, W.J.; Han, Q.; Chen, C.; Lu, J.M.; Zheng, J.Y.; Xue, S.R. Identification of Parkinson’s disease-related pathways and potential risk factors. J. Int. Med. Res. 2020, 48, 300060520957197. [Google Scholar] [CrossRef]
- Shin, M.-S.; Kim, T.-W.; Lee, J.-M.; Ji, E.-S.; Lim, B.-V. Treadmill Exercise Alleviates Nigrostriatal Dopaminergic Loss of Neurons and Fibers in Rotenone-Induced Parkinson Rats. J. Exerc. Rehabil. 2017, 13, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta 2015, 1852, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Iglesias, O.; Naidoo, V.; Cacabelos, N.; Cacabelos, R. Epigenetic Biomarkers as Diagnostic Tools for Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 23, 13. [Google Scholar] [CrossRef]
- Murthy, M.; Cheng, Y.Y.; Holton, J.L.; Bettencourt, C. Neurodegenerative movement disorders: An epigenetics perspective and promise for the future. Neuropathol. Appl. Neurobiol. 2021, 47, 897–909. [Google Scholar] [CrossRef]
- Rathore, A.S.; Birla, H.; Singh, S.S.; Zahra, W.; Dilnashin, H.; Singh, R.; Keshri, P.K.; Singh, S.P. Epigenetic Modulation in Parkinson’s Disease and Potential Treatment Therapies. Neurochem. Res. 2021, 46, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.; Lorenzo-Betancor, O.; Ross, O.A. Epigenetic regulation in Parkinson’s disease. Acta Neuropathol. 2016, 132, 515–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kia, D.A.; Zhang, D.; Guelfi, S.; Manzoni, C.; Hubbard, L.; Reynolds, R.H.; Botía, J.; Ryten, M.; Ferrari, R.; Lewis, P.A.; et al. United Kingdom Brain Expression Consortium (UKBEC) and the International Parkinson’s Disease Genomics Consortium (IPDGC). Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study, Expression, and Epigenetic Data Sets. JAMA Neurol. 2021, 78, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Schaake, S.; Grütz, K.; Sievert, H.; Reyes, C.J.; König, I.R.; Laabs, B.H.; Jamora, R.D.; Rosales, R.L.; Diesta, C.C.E.; et al. DNA Methylation as a Potential Molecular Mechanism in X-linked Dystonia-Parkinsonism. Mov. Disord. 2020, 35, 2220–2229. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Kobor, M.S. DNA methylation as a mediator of genetic and environmental influences on Parkinson’s disease susceptibility: Impacts of alpha-Synuclein, physical activity, and pesticide exposure on the epigenome. Front. Genet. 2022, 13, 971298. [Google Scholar] [CrossRef]
- Mohd Murshid, N.; Aminullah Lubis, F.; Makpol, S. Epigenetic Changes and Its Intervention in Age-Related Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2022, 42, 577–595. [Google Scholar] [CrossRef]
- Bakhit, Y.; Schmitt, I.; Hamed, A.; Ibrahim, E.A.A.; Mohamed, I.N.; El-Sadig, S.M.; Elseed, M.A.; Alebeed, M.A.; Shaheen, M.T.; Ibrahim, M.O.; et al. Methylation of alpha-synuclein in a Sudanese cohort. Parkinsonism Relat. Disord. 2022, 101, 6–8. [Google Scholar] [CrossRef]
- Gordevicius, J.; Li, P.; Marshall, L.L.; Killinger, B.A.; Lang, S.; Ensink, E.; Kuhn, N.C.; Cui, W.; Maroof, N.; Lauria, R.; et al. Epigenetic inactivation of the autophagy-lysosomal system in appendix in Parkinson’s disease. Nat. Commun. 2021, 12, 5134. [Google Scholar] [CrossRef]
- Go, R.C.P.; Corley, M.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.H.; Maunakea, A.K.; He, Q.; Tiirikainen, M.I. Genome-wide epigenetic analyses in Japanese immigrant plantation workers with Parkinson’s disease and exposure to organochlorines reveal possible involvement of glial genes and pathways involved in neurotoxicity. BMC Neurosci. 2020, 21, 31. [Google Scholar] [CrossRef]
- Nasamran, C.A.; Sachan, A.N.S.; Mott, J.; Kuras, Y.I.; Scherzer, C.R.; Study, H.B.; Ricciardelli, E.; Jepsen, K.; Edland, S.D.; Fisch, K.M.; et al. Differential blood DNA methylation across Lewy body dementias. Alzheimers Dement. 2021, 13, e12156. [Google Scholar] [CrossRef]
- Zhang, H.Q.; Wang, J.Y.; Li, Z.F.; Cui, L.; Huang, S.S.; Zhu, L.B.; Sun, Y.; Yang, R.; Fan, H.H.; Zhang, X.; et al. DNA Methyltransferase 1 Is Dysregulated in Parkinson’s Disease via Mediation of miR-17. Mol. Neurobiol. 2021, 58, 2620–2633. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.C.; Binder, A.M.; Horvath, S.; Kusters, C.; Yan, Q.; Rosario, I.D.; Yu, Y.; Bronstein, J.; Ritz, B. Accelerated hematopoietic mitotic aging measured by DNA methylation, blood cell lineage, and Parkinson’s disease. BMC Genom. 2021, 22, 696. [Google Scholar] [CrossRef] [PubMed]
- Vishweswaraiah, S.; Akyol, S.; Yilmaz, A.; Ugur, Z.; Gordevičius, J.; Oh, K.J.; Brundin, P.; Radhakrishna, U.; Labrie, V.; Graham, S.F. Methylated Cytochrome P450 and the Solute Carrier Family of Genes Correlate with Perturbations in Bile Acid Metabolism in Parkinson’s Disease. Front. Neurosci. 2022, 16, 804261. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Ensink, E.; Li, P.; Gordevičius, J.; Marshall, L.L.; George, S.; Pospisilik, J.A.; Aho, V.T.E.; Houser, M.C.; Pereira, P.A.B.; et al. Bacterial Butyrate in Parkinson’s Disease Is Linked to Epigenetic Changes and Depressive Symptoms. Mov. Disord. 2022, 37, 1644–1653. [Google Scholar] [CrossRef]
- Toker, L.; Tran, G.T.; Sundaresan, J.; Tysnes, O.B.; Alves, G.; Haugarvoll, K.; Nido, G.S.; Dölle, C.; Tzoulis, C. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol. Neurodegener. 2021, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Ranganayaki, S.; Govindaraj, P.; Gayathri, N.; Srinivas Bharath, M.M. Exposure to the neurotoxin 3-nitropropionic acid in neuronal cells induces unique histone acetylation pattern: Implications for neurodegeneration. Neurochem. Int. 2020, 140, 104846. [Google Scholar] [CrossRef] [PubMed]
- Günaydın, C.; Çelik, Z.B.; Bilge, S.S. CIITA expression is regulated by histone deacetylase enzymes and has a role in α-synuclein pre-formed fibril-induced antigen presentation in murine microglial cell line. Immunopharmacol. Immunotoxicol. 2022, 44, 447–455. [Google Scholar] [CrossRef]
- Blount, G.S.; Coursey, L.; Kocerha, J. MicroRNA Networks in Cognition and Dementia. Cells 2022, 11, 1882. [Google Scholar] [CrossRef]
- Zhou, S.; Yu, X.; Wang, M.; Meng, Y.; Song, D.; Yang, H.; Wang, D.; Bi, J.; Xu, S. Long Non-coding RNAs in Pathogenesis of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2021, 9, 719247. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. In MicroRNA Profiling; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1509, pp. 1–10. [Google Scholar] [CrossRef]
- Li, S.; Lei, Z.; Sun, T. The role of microRNAs in neurodegenerative diseases: A review. Cell Biol. Toxicol. 2022, 20, 1–31. [Google Scholar] [CrossRef]
- Selvakumar, S.C.; Preethi, K.A.; Tusubira, D.; Sekar, D. MicroRNAs in the epigenetic regulation of disease progression in Parkinson’s disease. Front. Cell. Neurosci. 2022, 16, 995997. [Google Scholar] [CrossRef] [PubMed]
- Marrs, T.C.; Maynard, R.L. Neurotranmission systems as targets for toxicants: A review. Cell Biol. Toxicol. 2013, 29, 381–396. [Google Scholar] [CrossRef]
- Müller-Vahl, K.R.; Kolbe, H.; Dengler, R. Transient severe parkinsonism after acute organophosphate poisoning. J. Neurol. Neurosurg. Psychiatry 1999, 66, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Hashim, H.Z.; Wan Musa, W.R.; Ngiu, C.S.; Wan Yahya, W.N.; Tan, H.J.; Ibrahim, N. Parkinsonism complicating acute organophosphate insecticide poisoning. Ann. Acad. Med. Singap. 2011, 40, 150–151. [Google Scholar] [PubMed]
- Pezzoli, G.; Cereda, E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 2013, 80, 2035–2041. [Google Scholar] [CrossRef]
- Narayan, S.; Liew, Z.; Paul, K.; Lee, P.C.; Sinsheimer, J.S.; Bronstein, J.M.; Ritz, B. Household organophosphorus pesticide use and Parkinson’s disease. Int. J. Epidemiol. 2013, 42, 1476–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Cockburn, M.; Ly, T.T.; Bronstein, J.M.; Ritz, B. The association between ambient exposure to organophosphates and Parkinson’s disease risk. Occup. Environ. Med. 2014, 71, 275–281. [Google Scholar] [CrossRef]
- Norkaew, S.; Lertmaharit, S.; Wilaiwan, W.; Siriwong, W.; Pérez, H.M.; Robson, M.G. An association between organophosphate pesticides exposure and Parkinsonism amongst people in an agricultural area in Ubon Ratchathani Province, Thailand. Rocz. Panstw. Zakl. Hig. 2015, 66, 21–26. [Google Scholar]
- Chuang, C.S.; Su, H.L.; Lin, C.L.; Kao, C.H. Risk of Parkinson disease after organophosphate or carbamate poisoning. Acta Neurol. Scand. 2017, 136, 129–137. [Google Scholar] [CrossRef]
- Davis, K.L.; Yesavage, J.A.; Berger, P.A. Single case study. Possible organophosphate-induced parkinsonism. J. Nerv. Ment. Dis. 1978, 166, 222–225. [Google Scholar] [CrossRef]
- Bhatt, M.H.; Elias, M.A.; Mankodi, A.K. Acute and reversible parkinsonism due to organophosphate pesticide intoxication: Five cases. Neurology 1999, 52, 1467–1471. [Google Scholar] [CrossRef]
- Das, K.; Ghosh, M.; Nag, C.; Nandy, S.P.; Banerjee, M.; Datta, M.; Devi, G.; Chaterjee, G. Role of familial, environmental and occupational factors in the development of Parkinson’s disease. Neurodegener. Dis. 2011, 8, 345–351. [Google Scholar] [CrossRef]
- Kanthasamy, A.; Jin, H.; Charli, A.; Vellareddy, A.; Kanthasamy, A. Environmental neurotoxicant-induced dopaminergic neurodegeneration: A potential link to impaired neuroinflammatory mechanisms. Pharmacol. Ther. 2019, 197, 61–82. [Google Scholar] [CrossRef]
- Paul, K.C.; Sinsheimer, J.S.; Cockburn, M.; Bronstein, J.M.; Bordelon, Y.; Ritz, B. Organophosphate pesticides and PON1 L55M in Parkinson’s disease progression. Environ. Int. 2017, 107, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Quezada, M.T.; Lucero, B.A.; Iglesias, V.P.; Muñoz, M.P.; Cornejo, C.A.; Achu, E.; Baumert, B.; Hanchey, A.; Concha, C.; Brito, A.M.; et al. Chronic exposure to organophosphate (OP) pesticides and neuropsychological functioning in farm workers: A review. Int. J. Occup. Environ. Health 2016, 22, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Fernandez, C.; Flores, P.; Sánchez-Santed, F. A Systematic Review on the Influences of Neurotoxicological Xenobiotic Compounds on Inhibitory Control. Front. Behav. Neurosci. 2019, 13, 139. [Google Scholar] [CrossRef] [Green Version]
- Jokanović, M. Neurotoxic effects of organophosphorus pesticides and possible association with neurodegenerative diseases in man: A review. Toxicology 2018, 410, 125–131. [Google Scholar] [CrossRef]
- Voorhees, J.R.; Rohlman, D.S.; Lein, P.J.; Pieper, A.A. Neurotoxicity in Preclinical Models of Occupational Exposure to Organophosphorus Compounds. Front. Neurosci. 2017, 10, 590. [Google Scholar] [CrossRef]
- Trojsi, F.; Monsurrò, M.R.; Tedeschi, G. Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: State of the art and research perspectives. J. Mol. Sci. 2013, 14, 15286–15311. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Santed, F.; Colomina, M.T.; Herrero Hernández, E. Organophosphate pesticide exposure and neurodegeneration. Cortex 2016, 74, 417–426. [Google Scholar] [CrossRef]
- Costa, L.G. Organophosphorus Compounds at 80: Some Old and New Issues. Toxicol. Sci. 2018, 162, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Kori, R.K.; Singh, M.K.; Jain, A.K.; Yadav, R.S. Neurochemical and Behavioral Dysfunctions in Pesticide Exposed Farm Workers: A Clinical Outcome. Indian J. Clin. Biochem. 2018, 33, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, J.; Ferré, M.; Chevrollier, A.; Codron, P.; Verny, C.; Homedan, C.; Lenaers, G.; Procaccio, V.; May-Panloup, P.; Reynier, P. Neurotoxicity of Insecticides. Curr. Med. Chem. 2017, 24, 2988–3001. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Kandimalla, R.J.L.; Sharma, D.R.; Kaushal, A.; Ruban, A.; Sunkaria, A.; Vallamkondu, J.; Chiarugi, A.; Reddy, P.H.; Gill, K.D. Cell cycle activation in p21 dependent pathway: An alternative mechanism of organophosphate induced dopaminergic neurodegeneration. Biochim. Biophys. Acta 2017, 1863, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Naughton, S.X.; Terry, A.V., Jr. Neurotoxicity in acute and repeated organophosphate exposure. Toxicology 2018, 408, 101–112. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Mehrpour, O.; Forouzanfar, F.; Roshanravan, B.; Samarghandian, S. Oxidative stress and mitochondrial dysfunction in organophosphate pesticide-induced neurotoxicity and its amelioration: A review. Environ. Sci. Pollut. Res. Int. 2020, 27, 24799–24814. [Google Scholar] [CrossRef] [PubMed]
- Karimani, A.; Ramezani, N.; Afkhami Goli, A.; Nazem Shirazi, M.H.; Nourani, H.; Jafari, A.M. Subchronic neurotoxicity of diazinon in albino mice: Impact of oxidative stress, AChE activity, and gene expression disturbances in the cerebral cortex and hippocampus on mood, spatial learning, and memory function. Toxicol. Rep. 2021, 8, 1280–1288. [Google Scholar] [CrossRef]
- Zhao, M.W.; Yang, P.; Zhao, L.L. Chlorpyrifos activates cell pyroptosis and increases susceptibility on oxidative stress-induced toxicity by miR-181/SIRT1/PGC-1α/Nrf2 signaling pathway in human neuroblastoma SH-SY5Y cells: Implication for association between chlorpyrifos and Parkinson’s disease. Environ. Toxicol. 2019, 34, 699–707. [Google Scholar] [CrossRef]
- Anderson, F.L.; von Herrmann, K.M.; Young, A.L.; Havrda, M.C. Bbc3 Loss Enhances Survival and Protein Clearance in Neurons Exposed to the Organophosphate Pesticide Chlorpyrifos. Toxicol. Sci. 2021, 183, 378–392. [Google Scholar] [CrossRef]
- Parashar, A.; Udayabanu, M. Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 38, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Bian, X.; Mahbub, R.; Lu, K. Sex-specific effects of organophosphate diazinon on the gut microbiome and its metabolic functions. Environ. Health Perspect. 2017, 125, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Bian, X.; Chi, L.; Tu, P.; Ru, H.; Lu, K. Organophosphate diazinon altered quorum sensing, cell motility, stress response, and carbohydrate metabolism of gut microbiome. Toxicol. Sci. 2017, 157, 354–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanaway, I.B.; Wallace, J.C.; Shojaie, A.; Griffith, W.C.; Hong, S.; Wilder, C.S.; Green, F.H.; Tsai, J.; Knight, M.; Workman, T.; et al. Human oral buccal microbiomes are associated with farmworker status and azynphos-methyl agricultural pesticide exposure. Appl. Environ. Microbiol. 2017, 83, e02149-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Wang, C.; Li, H.S. Effect of organophosphate pesticides poisoning on cognitive impairment. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2021, 39, 313–316. (In Chinese) [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Higgins, D.S.; Eller, R.V. Quantitative autoradiography of dihydrorotenone binding to complex I of the electron transport chain. J. Neurochem. 1992, 59, 746–749. [Google Scholar] [CrossRef]
- Herb, M.; Gluschko, A.; Schramm, M. Reactive Oxygen Species: Not Omnipresent but Important in Many Locations. Front. Cell Dev. Biol. 2021, 9, 716406. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herb, M.; Gluschko, A.; Wiegmann, K.; Farid, A.; Wolf, A.; Utermöhlen, O.; Krut, O.; Krönke, M.; Schramm, M. Mitochondrial reactive oxygen species enable proinflammatory signaling through disulfide linkage of NEMO. Sci. Signal. 2019, 12, eaar5926. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.Y.; Leem, E.; Lee, J.M.; Kim, S.R. Control of Reactive Oxygen Species for the Prevention of Parkinson’s Disease: The Possible Application of Flavonoids. Antioxidants 2020, 9, 583. [Google Scholar] [CrossRef]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.S.; Burns, M.R.; Brundin, P.; Wesson, D.W. Linking α-synuclein-induced synaptopathy and neural network dysfunction in early Parkinson’s disease. Brain Commun. 2022, 4, fcac165. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-Terminus of the Intrinsically Disordered Protein α-Synuclein Triggers Membrane Binding and Helix Folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, E.A.; Mazzulli, J.R.; Giasson, B.I. Characterization of hydrophobic residue requirements for alpha-synuclein fibrillization. Biochemistry 2009, 48, 9427–9436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilie, I.M.; Caflisch, A. Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.; Dettmer, U.; Luth, E.; Kim, N.; Newman, A.; Bartels, T. Defining the native state of α-synuclein. Neurodegener. Dis. 2014, 13, 114–117. [Google Scholar] [CrossRef]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fountaine, T.M.; Wade-Martins, R. RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 2007, 85, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.L.; Hasadsri, L.; Woods, W.S.; George, J.M. Dynamic transport and localization of alpha-synuclein in primary hippocampal neurons. Mol. Neurodegener. 2010, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Rasmussen, J.E.; Pedersen, S.L.; Jensen, K.J.; Langen, R.; Stamou, D. Membrane curvature sensing by amphipathic helices: A single liposome study using α-synuclein and annexin B12. J. Biol. Chem. 2011, 49, 42603–42614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, U.; Lee, J.C. Membrane Interactions of α-Synuclein Probed by Neutrons and Photons. Acc. Chem. Res. 2021, 54, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [Green Version]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.C.; Yeh, T.H.; Lai, S.C.; Wu-Chou, Y.H.; Chen, C.H.; Mochly-Rosen, D.; Huang, Y.C.; Chen, Y.J.; Chen, C.L.; Chang, Y.M.; et al. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp. Neurol. 2015, 263, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Monoranu, C.; Strobel, S.; Riederer, P. Lewy Bodies: A Spectator or Salient Killer? CNS Neurol. Disord. Drug Targets 2015, 14, 947–955. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Han, L.; Jiang, C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Howell, N.; Andrews, R.M.; Turnbull, D.M. Clinical mitochondrial genetics. J. Med. Genet. 1999, 36, 425–436. [Google Scholar] [PubMed]
- Van Wilpe, S.; Ryan, M.T.; Hill, K.; Maarse, A.C.; Meisinger, C.; Brix, J.; Dekker, P.J.; Moczko, M.; Wagner, R.; Meijer, M.; et al. Tom22 is a multifunctional organizer of the mitochondrial preprotein translocase. Nature 1999, 401, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Lind, M.; Frazier, A.E.; Dudek, J.; Meisinger, C.; Geissler, A.; Sickmann, A.; Meyer, H.E.; Truscott, K.N.; Guiard, B.; et al. Mitochondrial presequence translocase: Switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 2005, 120, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [Green Version]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective Neuronal Vulnerability in Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [Green Version]
- De Miranda, B.R.; Rocha, E.M.; Castro, S.L.; Greenamyre, J.T. Protection from α-Synuclein induced dopaminergic neurodegeneration by overexpression of the mitochondrial import receptor TOM20. NPJ Parkinson’s Dis. 2020, 6, 38. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Cuadros, T.; Parent, A.; Romero-Gimenez, J.; Vila, M.; Perier, C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2018, 9, 1122. [Google Scholar] [CrossRef] [Green Version]
- Needs, H.I.; Protasoni, M.; Henley, J.M.; Prudent, J.; Collinson, I.; Pereira, G.C. Interplay between Mitochondrial Protein Import and Respiratory Complexes Assembly in Neuronal Health and Degeneration. Life 2021, 11, 432. [Google Scholar] [CrossRef]
- Simmen, T.; Lynes, E.M.; Gesson, K.; Thomas, G. Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2010, 1798, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellerich, F.N.; Gizatullina, Z.; Gainutdinov, T.; Muth, K.; Seppet, E.; Orynbayeva, Z.; Vielhaber, S. The control of brain mitochondrial energization by cytosolic calcium: The mitochondrial gas pedal. IUBMB Life 2013, 65, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Suaga, P.; Bravo-San Pedro, J.M.; González-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 2018, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- De Pinto, V. Renaissance of VDAC: New Insights on a Protein Family at the Interface between Mitochondria and Cytosol. Biomolecules 2021, 11, 107. [Google Scholar] [CrossRef]
- Hodge, T.; Colombini, M. Regulation of Metabolite Flux through Voltage-Gating of VDAC Channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef]
- Colombini, M. The published 3D structure of the VDAC channel: Native or not? Trends Biochem. Sci. 2009, 34, 382–389. [Google Scholar] [CrossRef]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Bezrukov, S.M.; Hoogerheide, D.P. Regulation of Mitochondrial Respiration by VDAC Is Enhanced by Membrane-Bound Inhibitors with Disordered Polyanionic C-Terminal Domains. Int. J. Mol. Sci. 2021, 22, 7358. [Google Scholar] [CrossRef] [PubMed]
- Rovini, A.; Gurnev, P.A.; Beilina, A.; Queralt-Martín, M.; Rosencrans, W.; Cookson, M.R.; Bezrukov, S.M.; Rostovtseva, T.K. Molecular mechanism of olesoxime-mediated neuroprotection through targeting α-synuclein interaction with mitochondrial VDAC. Cell. Mol. Life Sci. 2020, 77, 3611–3626. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, C.; Cai, Q.; Lu, Q.; Duan, C.; Zhu, Y.; Yang, H. Voltage-dependent anion channel involved in the α-synuclein-induced dopaminergic neuron toxicity in rats. Acta Biochim. Biophys. Sin. 2013, 45, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Rosencrans, W.M.; Aguilella, V.M.; Rostovtseva, T.K.; Bezrukov, S.M. α-Synuclein Emerges as a Potent Regulator of VDAC-Facilitated Calcium Transport. Cell Calcium 2021, 95, 102355. [Google Scholar] [CrossRef] [PubMed]
- Head, S.A.; Shi, W.; Zhao, L.; Gorshkov, K.; Pasunooti, K.; Chen, Y.; Deng, Z.; Li, R.J.; Shim, J.S.; Tan, W.; et al. Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7276–E7285. [Google Scholar] [CrossRef] [Green Version]
- Tewari, D.; Majumder, D.; Vallabhaneni, S.; Bera, A.K. Aspirin induces cell death by directly modulating mitochondrial voltage-dependent anion channel (VDAC). Sci. Rep. 2017, 7, 45184. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barceló-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell. Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [Green Version]
- Oyarzabal, A.; Marin-Valencia, I. Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 2019, 42, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Vergara, R.C.; Jaramillo-Riveri, S.; Luarte, A.; Moenne-Loccoz, C.; Fuentes, R.; Couve, A.; Maldonado, P.E. The energy homeostasis principle: Neuronal energy regulation drives local network dynamics generating behavior. Front. Comput. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Paxinos, G.; Watson, C.; Halliday, G.M. The substantia nigra and ventral tegmental dopaminergic neurons from development to degeneration. J. Chem. Neuroanat. 2016, 76 Pt B, 98–107. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.Y.; Lee, S.B.; Kim, H.J.; Kim, H.T.; Yang, H.O.; Jang, W. Autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity in an in vitro model of Parkinson’s disease. Neurosci. Lett. 2017, 642, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Schumacker, P.T. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 2013, 288, 10736–10741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foehring, R.C.; Zhang, X.F.; Lee, J.C.; Callaway, J.C. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef] [Green Version]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Mori, F.; Tanji, K.; Orimo, S.; Takahashi, H. Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain. Acta Neuropathol. 2010, 120, 1–12. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Mabbott, N.A. How do PrP(Sc) Prions spread between host species, and within hosts? Pathogens 2017, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Engelender, S.; Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 2017, 40, 4–14. [Google Scholar] [CrossRef]
- Chandra, R.; Hiniker, A.; Kuo, Y.M.; Nussbaum, R.L.; Liddle, R.A. alpha-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2017, 2, e92295. [Google Scholar] [CrossRef] [Green Version]
- Liddle, R.A. Parkinson’s disease from the gut. Brain Res. 2018, 1693 Pt B, 201–206. [Google Scholar] [CrossRef]
- Rodrigues, P.V.; de Godoy, J.V.P.; Bosque, B.P.; Amorim Neto, D.P.; Tostes, K.; Palameta, S.; Garcia-Rosa, S.; Tonoli, C.C.C.; de Carvalho, H.F.; de Castro Fonseca, M. Transcellular propagation of fibrillar α-synuclein from enteroendocrine to neuronal cells requires cell-to-cell contact and is Rab35-dependent. Sci. Rep. 2022, 12, 4168. [Google Scholar] [CrossRef]
- Sun, M.F.; Shen, Y.Q. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s Disease. Ageing Res. Rev. 2018, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Empadinhas, N. The microbiome-mitochondria dance in prodromal Parkinson’s disease. Front. Physiol. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Mahana, D.; Trent, C.M.; Kurtz, Z.D.; Bokulich, N.A.; Battaglia, T.; Chung, J.; Müller, C.L.; Li, H.; Bonneau, R.A.; Blaser, M.J. Antibiotic perturbation of the murine gut microbiome anhnces the adiposity, insulin resistance, and liver disease associated with high-fat diet. Genome Med. 2016, 27, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felice, V.D.; O’Mahony, S.M. The microbiome and disorders of the central nervous system. Pharmacol. Biochem. Behav. 2017, 160, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Cui, L.; Gao, J.; Zhu, M.; Zhang, Y.; Zhang, H.L. Gut Microbial Metabolites in Parkinson’s Disease: Implications of Mitochondrial Dysfunction in the Pathogenesis and Treatment. Mol. Neurobiol. 2021, 58, 3745–3758. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The critical modulators regulating gut-brain axis. J. Cell. Physiol. 2017, 232, 2359–2372. [Google Scholar] [CrossRef] [Green Version]
- Vendrik, K.E.W.; Ooijevaar, R.E.; de Jong, P.R.C.; Laman, J.D.; van Oosten, B.W.; van Hilten, J.J.; Ducarmon, Q.R.; Keller, J.J.; Kuijper, E.J.; Contarino, M.F. Fecal microbiota transplantation in neurological disorders. Front. Cell. Infect. Microbiol. 2020, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef]
- Lohmann, S.; Bernis, M.E.; Tachu, B.J.; Ziemski, A.; Grigoletto, J.; Tamgüney, G. Oral and intravenous transmission of α-synuclein fibrils to mice. Acta Neuropathol. 2019, 138, 515–533. [Google Scholar] [CrossRef]
- Malek, N.; Lawton, M.A.; Swallow, D.M.A.; Grosset, K.A.; Marrinan, S.L.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Hardy, J.; Morris, H.R.; et al. Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson’s disease. Mov. Disord. 2016, 87, 1518–1526. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Seara, M.A.; Mengual, E.; Vidorreta, M.; Aznarez-Sanado, M.; Loayza, F.R.; Villagra, F.; Irigoyen, J.; Pastor, M.A. Cortical hypoperfusion in Parkinson’s disease assessed using arterial spin labeled perfusion MRI. Neuroimage 2012, 59, 2743–2750. [Google Scholar] [CrossRef]
- Ouellette, J.; Lacoste, B. From Neurodevelopmental to Neurodegenerative Disorders: The Vascular Continuum. Front. Aging Neurosci. 2021, 13, 749026. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Pavlovic, D.; Waldvogel, H.; Dragunow, M.; Synek, B.; Turner, C.; Faull, R.; Guan, J. String vessel formation is increased in the brain of Parkinson disease. J. Parkinson’s Dis. 2015, 5, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; De Jong, G.I.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Luiten, P.G.M. Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 2000, 100, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.N.; Berthiaume, A.-A.; Faino, A.V.; McDowell, K.P.; Bhat, N.R.; Hartmann, D.A.; Shih, A.Y. Mild pericyte deficiency is associated with aberrant brain microvascular flow in aged PDGFRβ+/− mice. J. Cereb. Blood Flow Metab. 2020, 40, 2387–2400. [Google Scholar] [CrossRef]
- Hartmann, D.A.; Berthiaume, A.A.; Grant, R.I.; Harrill, S.A.; Koski, T.; Tieu, T.; McDowell, K.P.; Faino, A.V.; Kelly, A.L.; Shih, A.Y. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 2021, 24, 633–645. [Google Scholar] [CrossRef]
- Andreone, B.J.; Lacoste, B.; Gu, C. Neuronal and vascular interactions. Annu. Rev. Neurosci. 2015, 38, 25–46. [Google Scholar] [CrossRef] [Green Version]
- Dabertrand, F.; Harraz, O.F.; Masayo, K.; Longden, T.A.; Rosehart, A.C.; Hill-Eubanks, D.; Joutel, A.; Nelson, M.T. PIP 2 corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2025998118. [Google Scholar] [CrossRef]
- McConnell, H.L.; Li, Z.; Woltjer, R.L.; Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem. Int. 2019, 128, 70–84. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell. Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef] [Green Version]
- Goncharov, N.V.; Popova, P.I.; Golovkin, A.S.; Zalutskaya, N.M.; Palchikova, E.I.; Zanin, K.V.; Avdonin, P.V. Vascular endothelial dysfunction is a pathogenetic factor in the development of neurodegenerative diseases and cognitive impairment. Bekhterev Rev. Psychiatry Med. Psychol. 2020, 3, 11–26. (In Russian) [Google Scholar] [CrossRef]
- Barker, R.A.; Ratcliffe, E.; McLaughlin, M.; Richards, A.; Dunnett, S.B. A role for complement in the rejection of porcine ventral mesencephalic xenografts in a rat model of Parkinson’s disease. J. Neurosci. 2000, 20, 3415–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-synuclein induces blood-brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef]
- Chen, R.; Lee, C.; Lin, X.; Zhao, C.; Li, X. Novel function of VEGF-B as an antioxidant and therapeutic implications. Pharmacol. Res. 2019, 143, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Song, R.; Yang, Z.; Shan, Q.; Chen, W. 6-Hydroxydopamine induces brain vascular endothelial inflammation. IUBMB Life 2017, 69, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auriel, E.; Regev, K.; Korczyn, A.D. Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb. Clin. Neurol. 2014, 119, 577–584. [Google Scholar] [CrossRef]
- Xue, C.; Li, X.; Ba, L.; Zhang, M.; Yang, Y.; Gao, Y.; Sun, Z.; Han, Q.; Zhao, R.C. MSC-Derived Exosomes can Enhance the Angiogenesis of Human Brain MECs and Show Therapeutic Potential in a Mouse Model of Parkinson’s Disease. Aging Dis. 2021, 12, 1211–1222. [Google Scholar] [CrossRef]
- Chen, T.; Hou, R.; Li, C.; Wu, C.; Xu, S. MPTP/MPP+ suppresses activation of protein C in Parkinson’s disease. J. Alzheimers Dis. 2015, 43, 133–142. [Google Scholar] [CrossRef]
- Kursun, O.; Karatas, H.; Bariskaner, H.; Ozturk, S. Arachidonic Acid Metabolites in Neurologic Disorders. CNS Neurol. Disord. Drug Targets 2022, 21, 150–159. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Wang, Q.; Hand, T.; Bilak, M.; McCullough, L.; Andreasson, K. Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 2007, 33, 94–99. [Google Scholar] [CrossRef]
- Koyama, S.; Aizawa, H.; Morita, K.; Fujikane, T.; Sasaki, N.; Kikuchi, K. Plasma von Willebrand factor activities in vascular Parkinsonism: Comparison with Parkinson’s disease. J. Stroke Cerebrovasc. Dis. 2001, 10, 227–230. [Google Scholar] [CrossRef]
- Xu, Q.; Lai, Q.; Wang, J.; Zhuang, L.; Cheng, L.; Mo, Y.; Liu, L.; Zhao, Z.; Zhang, Y.; Weng, S.; et al. Association between plasminogen activator inhibitor-1 gene polymorphisms and susceptibility to Parkinson’s disease in Chinese patients. Acta Neurol. Belg. 2021; epub ahead of print. [Google Scholar] [CrossRef]
- Pan, H.; Zhao, Y.; Zhai, Z.; Zheng, J.; Zhou, Y.; Zhai, Q.; Cao, X.; Tian, J.; Zhao, L. Role of plasminogen activator inhibitor-1 in the diagnosis and prognosis of patients with Parkinson’s disease. Exp. Ther. Med. 2018, 15, 5517–5522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.Y.; Paul, G. Human α-synuclein overexpression in a mouse model of Parkinson’s disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Sci. Rep. 2021, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, M.; Choi, Y.K. The Role of a Neurovascular Signaling Pathway Involving Hypoxia-Inducible Factor and Notch in the Function of the Central Nervous System. Biomol. Ther. 2020, 28, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res. 2009, 42, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, Y.; Kurosaki, R.; Watanabe, H.; Michimata, M.; Matsubara, M.; Imai, Y.; Araki, T. Cerebral alterations in a MPTP-mouse model of Parkinson’s disease--an immunocytochemical study. J. Neural Transm. 2003, 110, 1129–1144. [Google Scholar] [CrossRef]
- Zhang, L.; Dawson, V.L.; Dawson, T.M. Role of nitric oxide in Parkinson’s disease. Pharmacol. Ther. 2006, 109, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Nassir, C.M.N.C.M.; Ghazali, M.M.; Hashim, S.; Idris, N.S.; Yuen, L.S.; Hui, W.J.; Norman, H.H.; Gau, C.H.; Jayabalan, N.; Na, Y.; et al. Diets and Cellular-Derived Microparticles: Weighing a Plausible Link With Cerebral Small Vessel Disease. Front. Cardiovasc. Med. 2021, 8, 632131. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Xie, Q.; Jia, J.; Liu, X.; Sun, J.; Deng, Y.; Yi, L. Integrated Analysis and Identification of Novel Biomarkers in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.P.; Hegde, M.; Shetty, S.S.; Rafic, T.; Mutalik, S.; Rao, B.S.S. Targeting receptor-ligand chemistry for drug delivery across blood-brain barrier in brain diseases. Life Sci. 2021, 274, 119326. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Shingo, T.; Kobayashi, K.; Takeuchi, A.; Yano, A.; Muraoka, K.; Matsui, T.; Miyoshi, Y.; Hamada, H.; Date, I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur. J. Neurosci. 2004, 19, 1494–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlin, K.E.; Francardo, V.; Lindgren, H.S.; Sillivan, S.E.; O’Sullivan, S.S.; Luksik, A.S.; Vassoler, F.M.; Lees, A.J.; Konradi, C.; Cenci, M.A. Vascular endothelial growth factor is upregulated by L-dopa in the parkinsonian brain: Implications for the development of dyskinesia. Brain 2011, 134 Pt 8, 2339–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okouchi, M.; Okayama, N.; Alexander, J.S.; Aw, T.Y. NRF2-dependent glutamate-L-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia-induced brain endothelial cell apoptosis. Curr. Neurovasc. Res. 2006, 3, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Tang, B.S.; Guo, J.F. Research advances on neurite outgrowth inhibitor B receptor. J. Cell. Mol. Med. 2020, 24, 7697–7705. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Guo, W.; He, H.; Zhang, H.; Ye, Q.; Zhang, Q.; Liao, J.; Shen, Y.; Wang, J.; Xiao, Y.; et al. Decreased soluble Nogo-B in serum as a promising biomarker for Parkinson’s disease. Front. Neurosci. 2022, 16, 894454. [Google Scholar] [CrossRef]
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity and Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 5121. [Google Scholar] [CrossRef] [PubMed]
- Florey, L. The Endothelial Cell. Br. Med. J. 1966, 2, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef]
- Patel, A.; Toia, G.V.; Colletta, K.; Bradaric, B.D.; Carvey, P.M.; Hendey, B. An angiogenic inhibitor, cyclic RGDfV, attenuates MPTP-induced dopamine neuron toxicity. Exp. Neurol. 2011, 231, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, W.-L.; Bennett, N.; He, X.; Skepper, J.N.; Martynyuk, N.; Wijeyekoon, R.; Moghe, P.V.; Williams-Gray, C.H.; Barker, R.A. α-Synuclein pre-formed fibrils impair tight junction protein expression without affecting cerebral endothelial cell function. Exp. Neurol. 2016, 285, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Goncharov, N.V.; Popova, P.I.; Avdonin, P.P.; Kudryavtsev, I.V.; Serebryakova, M.K.; Korf, E.A.; Avdonin, P.V. Markers of Endothelial Cells in Normal and Pathological Conditions. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2020, 14, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Bradaric, B.D.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural Transm. 2012, 119, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of α-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: Another mechanism for initiation and progression of Parkinson’s disease? Acta Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Bogale, T.A.; Faustini, G.; Longhena, F.; Mitola, S.; Pizzi, M.; Bellucci, A. Alpha-Synuclein in the Regulation of Brain Endothelial and Perivascular Cells: Gaps and Future Perspectives. Front. Immunol. 2021, 12, 611761. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.; Wang, P.; Chan, R.B.; Liu, Z.; Yu, Z.; Liu, X.; Yang, Y.; Zhang, J. Astrocytic VEGFA: An essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia 2022, 70, 337–353. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Cytokines in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 58, 143–151. [Google Scholar]
- Kim, K.S.; Park, J.Y.; Jou, I.; Park, S.M. Regulation of Weibel-Palade body exocytosis by α-synuclein in endothelial cells. J. Biol. Chem. 2010, 285, 21416–21425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, D.; Bauer, B.; Wolthuis, R.M.; Wittinghofer, A.; Cool, R.H.; Bayer, P. Structure determination of the Ras-binding domain of the Ral-specific guanine nucleotide exchange factor Rlf. Biochemistry 1998, 37, 13453–13462. [Google Scholar] [CrossRef] [PubMed]
- Rondaij, M.G.; Bierings, R.; Kragt, A.; Van Mourik, J.A.; Voorberg, J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arteriosc. Thromb. Vasc. Biol. 2006, 26, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Moskalenko, S.; Tong, C.; Rosse, C.; Mirey, G.; Formstecher, E.; Daviet, L.; Camonis, J.; White, M.A. Ral GTPases regulate exocyst assembly through dual subunit interactions. J. Biol. Chem. 2003, 278, 51743–51748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Chen, S.; Luo, Y.; Xia, Y.; Ma, Q.; Yao, Q.; Wu, J. Interaction between ICAM1 in endothelial cells and LFA1 in T cells during the pathogenesis of experimental Parkinson’s disease. Exp. Ther. Med. 2020, 20, 1021–1029. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke induction of α-synuclein mediates ischemic brain damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.Y.; Ji, D.; Tan, C.; Sun, J.; Yao, H. Research progress on the role and regulatory mechanism of pathogenic Th17 cells in neuroinflammation. Yi Chuan Hered. 2022, 44, 289–299. [Google Scholar] [CrossRef]
- Smith, B.C.; Tinkey, R.A.; Shaw, B.C.; Williams, J.L. Targetability of the neurovascular unit in inflammatory diseases of the central nervous system. Immunol. Rev. 2022. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef]
- Pisani, V.; Stefani, A.; Pierantozzi, M.; Natoli, S.; Stanzione, P.; Franciotta, D.; Pisani, A. Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson’s disease. J. Neuroinflamm. 2012, 9, 188. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood–brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Farrall, A.; Armitage, P.A.; Carpenter, T.; Chappell, F.; Doubal, F.; Chowdhury, D.; Cvoro, V.; Dennis, M.S. Changes in background blood-brain barrier integrity between lacunar and cortical ischemic stroke subtypes. Stroke 2008, 39, 1327–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcia, C.; Bautista, V.; Sanchez-Bahillo, A.; Fernandez-Villalba, E.; Faucheux, B.; Poza y Poza, M.; Fernandez Barreiro, A.; Hirsch, E.C.; Herrero, M.T. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J. Neural Transm. 2005, 112, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Rite, I.; Machado, A.; Cano, J.; Venero, J.L. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J. Neurochem. 2007, 101, 1567–1582. [Google Scholar] [CrossRef]
- Chao, Y.-X.; He, B.-P.; Wah Tay, S.S. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson’s disease. J. Neuroimmunol. 2009, 216, 39–50. [Google Scholar] [CrossRef]
- Nguyen, B.; Bix, G.; Yao, Y. Basal lamina changes in neurodegenerative disorders. Mol. Neurodegener. 2021, 16, 81. [Google Scholar] [CrossRef]
- Elabi, O.F.; Cunha, J.P.M.C.M.; Gaceb, A.; Fex, M.; Paul, G. High-fat diet-induced diabetes leads to vascular alterations, pericyte reduction, and perivascular depletion of microglia in a 6-OHDA toxin model of Parkinson disease. J. Neuroinflammation. 2021, 18, 175. [Google Scholar] [CrossRef]
- Sui, Y.-T.; Bullock, K.M.; Erickson, M.A.; Zhang, J.; Banks, W.A. Alpha synuclein is transported into and out of the brain by the blood–brain barrier. Peptides 2014, 62, 197–202. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Breuer, O.; Lawhorn, C.; Miller, T.; Smith, D.M.; Brown, L.L. Functional architecture of the mammalian striatum: Mouse vascular and striosome organization and their anatomic relationships. Neurosci. Lett. 2005, 385, 198–203. [Google Scholar] [CrossRef]
- Chen, B.; Friedman, B.; Cheng, Q.; Tsai, P.; Schim, E.; Kleinfeld, D.; Lyden, P.D. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke 2009, 40, e666–e674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Storkebaum, E.; De Almodóvar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef]
- Wada, K.; Arai, H.; Takahashi, M.; Fukae, J.; Oizumi, H.; Yasuda, T.; Mizuno, Y.; Mochizuki, H. Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport 2006, 17, 705–709. [Google Scholar] [CrossRef]
- Eichmann, A.; Simons, M. VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 2012, 24, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Welser, J.V.; Milner, R. Absence of the αvβ3 integrin dictates the time-course of angiogenesis in the hypoxic central nervous system: Accelerated endothelial proliferation correlates with compensatory increases in α5β1 integrin expression. J. Cereb. Blood Flow Metab. 2010, 30, 1031–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. [Google Scholar] [CrossRef]
- Akiyoshi, R.; Wake, H.; Kato, D.; Horiuchi, H.; Ono, R.; Ikegami, A.; Haruwaka, K.; Omori, T.; Tachibana, Y.; Moorhouse, A.J.; et al. Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro 2018, 5, ENEURO.0088-18.2018. [Google Scholar] [CrossRef]
- Werneburg, S.; Feinberg, P.A.; Johnson, K.M.; Schafer, D.P. A microglia-cytokine axis to modulate synaptic connectivity and function. Curr. Opin. Neurobiol. 2017, 47, 138–145. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welser, J.V.; Li, L.; Milner, R. Microglial activation state exerts a biphasic influence on brain endothelial cell proliferation by regulating the balance of TNF and TGF-β1. J. Neuroinflamm. 2010, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, T.; Kerkhofs, D.; Mizuno, T.; Steinbusch, H.W.M.; Foulquier, S. Vessel-associated immune cells in cerebrovascular diseases: From perivascular macrophages to vessel-associated microglia. Front. Neurosci. 2019, 13, 1291. [Google Scholar] [CrossRef] [Green Version]
- Orr, C.F.; Rowe, D.B.; Halliday, G.M. An inflammatory review of Parkinson’s disease. Prog. Neurobiol. 2002, 68, 325–340. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, T.J.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Dieriks, B.V.; Curtis, M.A. α-synuclein inclusions are abundant in non-neuronal cells in the anterior olfactory nucleus of the Parkinson’s disease olfactory bulb. Sci. Rep. 2020, 10, 6682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Pu, T.; Feng, W.; Lu, M.; Zheng, Y.; Du, R.; Xiao, M.; Hu, G. Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated α-synuclein. Transl. Neurodegener. 2019, 8, 7. [Google Scholar] [CrossRef]
- Cui, H.; Wang, W.; Zheng, X.; Xia, D.; Liu, H.; Qin, C.; Tian, H.; Teng, J. Decreased AQP4 Expression Aggravates ɑ-Synuclein Pathology in Parkinson’s Disease Mice, Possibly via Impaired Glymphatic Clearance. J. Mol. Neurosci. 2021, 71, 2500–2513. [Google Scholar] [CrossRef]
- Chung, S.J.; Yoo, H.S.; Shin, N.Y.; Park, Y.W.; Lee, H.S.; Hong, J.M.; Kim, Y.J.; Lee, S.K.; Lee, P.H.; Sohn, Y.H. Perivascular Spaces in the Basal Ganglia and Long-term Motor Prognosis in Newly Diagnosed Parkinson Disease. Neurology 2021, 96, e2121–e2131. [Google Scholar] [CrossRef]
- Ma, X.; Li, S.; Li, C.; Wang, R.; Chen, M.; Chen, H.; Su, W. Diffusion Tensor Imaging Along the Perivascular Space Index in Different Stages of Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 773951. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Gu, L.Y.; Tian, J.; Dai, S.B.; Chen, Y.; Zheng, R.; Si, X.L.; Jin, C.Y.; Song, Z.; Yan, Y.P.; et al. MRI-visible perivascular spaces are associated with cerebrospinal fluid biomarkers in Parkinson’s disease. Aging 2020, 12, 25805–25818. [Google Scholar] [CrossRef] [PubMed]
- Taoka, T.; Naganawa, S. Imaging for central nervous system (CNS) interstitial fluidopathy: Disorders with impaired interstitial fluid dynamics. Jpn. J. Radiol. 2021, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.L.; Vitek, J.L. Surgical treatment for Parkinson’s disease. Lancet Neurol. 2004, 3, 719–728. [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. 1961. Wien Klin. Wochenschr. 2001, 113, 851–854, (In English and German). [Google Scholar]
- Oertel, W.; Schulz, J.B. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J. Neurochem. 2016, 139 (Suppl. 1), 325–337. [Google Scholar] [CrossRef] [PubMed]
- Le Witt, P.A.; Calne, D.B. Recent advances in the treatment of Parkinson’s disease: The role of bromocriptine. J. Neural Transm. 1981, 51, 175–184. [Google Scholar] [CrossRef]
- Przuntek, H.; Welzel, D.; Gerlach, M.; Blümner, E.; Danielczyk, W.; Kaiser, H.J.; Kraus, P.H.; Letzel, H.; Riederer, P.; Uberla, K. Early institution of bromocriptine in Parkinson’s disease inhibits the emergence of levodopa-associated motor side effects. Long-term results of the PRADO study. J. Neural Transm. 1996, 103, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Honig, H.; Antonini, A.; Martinez-Martin, P.; Forgacs, I.; Faye, G.C.; Fox, T.; Fox, K.; Mancini, F.; Canesi, M.; Odin, P.; et al. Intrajejunal levodopa infusion in Parkinson’s disease: A pilot multicenter study of effects on nonmotor symptoms and quality of life. Mov. Disord. 2009, 24, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Mariani, L.L.; Mangone, G.; Le Febvre de Nailly, D.; Charbonnier-Beaupel, F.; Corvol, J.C. Molecular basis of dopamine replacement therapy and its side effects in Parkinson’s disease. Cell Tissue Res. 2018, 373, 111–135. [Google Scholar] [CrossRef]
- Liu, J.; Lu, Y.; Tang, M.; Shao, F.; Yang, D.; Chen, S.; Xu, Z.; Zhai, L.; Chen, J.; Li, Q.; et al. Fucoxanthin Prevents Long-Term Administration l-DOPA-Induced Neurotoxicity through the ERK/JNK-c-Jun System in 6-OHDA-Lesioned Mice and PC12 Cells. Mar. Drugs 2022, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R. Anticholinergic drugs used in Parkinson’s disease: An overlooked class of drugs from a pharmacokinetic perspective. J. Pharm. Pharm. Sci. 1999, 2, 39–46. [Google Scholar]
- Chung, K.A.; Lobb, B.M.; Nutt, J.G.; Horak, F.B. Effects of a central cholinesterase inhibitor on reducing falls in Parkinson disease. Neurology 2010, 75, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahoor, I.; Shafi, A.; Haq, E. Pharmacological treatment of Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; pp. 129–144. [Google Scholar]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2008, 4, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessoa, R.R.; Moro, A.; Munhoz, R.P.; Teive, H.A.G.; Lees, A.J. Apomorphine in the treatment of Parkinson’s disease: A review. Arq. Neuropsiquiatr. 2018, 76, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Dellapina, E.; Gerdelat-Mas, A.; Ory-Magne, F.; Pourcel, L.; Galitzky, M.; Calvas, F.; Simonetta-Moreau, M.; Thalamas, C.; Payoux, P.; Brefel-Courbon, C. Apomorphine effect on pain threshold in Parkinson’s disease: A clinical and positron emission tomography study. Mov. Disord. 2011, 26, 153–157. [Google Scholar] [CrossRef]
- Jost, W.H. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J. Neural Transm. 2022, 129, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. CMAJ 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshirfar, M.; Baker, P.; Ronquillo, Y. Amantadine Keratopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrino, R.; Schapira, A.H.V. Glucocerebrosidase and Parkinson disease: Molecular, clinical, and therapeutic implications. Neuroscientist 2018, 24, 540–559. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, I.; Gordino, G.; Moreira, S.; Nunes, M.J.; Azevedo, C.; Gama, M.J.; Rodrigues, E.; Rodrigues, C.M.P.; Castro-Caldas, M. Tauroursodeoxycholic acid protects against mitochondrial dysfunction and cell death via mitophagy in human neuroblastoma cells. Mol. Neurobiol. 2017, 54, 6107–6119. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic acid ameliorates apoptotic cascade in the rotenone model of Parkinson’s disease: Modulation of mitochondrial perturbations. Mol. Neurobiol. 2016, 53, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Loh, S.H.Y.; Martins, L.M. Enhancing NAD salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostelnik, A.; Cegan, A.; Pohanka, M. Anti-Parkinson Drug Biperiden Inhibits Enzyme Acetylcholinesterase. BioMed Res. Int. 2017, 2017, 2532764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochfeld, W.E.; Lee, S.; Rubinsztein, D.C. Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol. Sin. 2013, 34, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Iranshahy, M.; Javadi, B.; Sahebkar, A. Protective effects of functional foods against Parkinson’s disease: A narrative review on pharmacology, phytochemistry, and molecular mechanisms. Phytother. Res. 2022, 36, 1952–1989. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z. Chronic caffeine treatment protects against α-synucleinopathy by reestablishing autophagy activity in the mouse striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostini, F.; Masato, A.; Bubacco, L.; Bisaglia, M. Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. Int. J. Mol. Sci. 2021, 23, 398. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Xie, J.; Xiao, J.; Li, D.; Xu, B.; Wang, X. Inhibition of PDE4 by FCPR16 induces AMPK-dependent autophagy and confers neuroprotection in SH-SY5Y cells and neurons exposed to MPP+-induced oxidative insult. Free Radic. Biol. Med. 2019, 135, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Höllerhage, M.; Fussi, N.; Rösler, T.W.; Wurst, W.; Behrends, C.; Höglinger, G.U. Multiple molecular pathways stimulating macroautophagy protect from alpha-synuclein-induced toxicity in human neurons. Neuropharmacology 2019, 149, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Iranpanah, A.; Gravandi, M.M.; Moradi, S.Z.; Ranjbari, M.; Majnooni, M.B.; Echeverría, J.; Qi, Y.; Wang, M.; Liao, P.; et al. Natural products attenuate PI3K/Akt/mTOR signaling pathway: A promising strategy in regulating neurodegeneration. Phytomedicine 2021, 91, 153664. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Song, J.X.; Lu, J.H.; Yuan, Z.W.; Liu, L.F.; Durairajan, S.S.K. Corynoxine, a natural autophagy enhancer, promotes the clearance of alpha-synuclein via Akt/mTOR pathway. J. Neuroimmune Pharmacol. 2014, 9, 380–387. [Google Scholar] [CrossRef]
- Xu, Y.D.; Cui, C.; Sun, M.F.; Zhu, Y.L.; Chu, M.; Shi, Y.W.; Lin, S.L.; Yang, X.S.; Shen, Y.Q. Neuroprotective effects of loganin on MPTP-induced Parkinson’s disease mice: Neurochemistry, glial reaction and autophagy studies. J. Cell Biochem. 2017, 118, 3495–3510. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.B.; Cheng, L.; Mudge, A.W.; Harwood, A.J. A common mechanism of action for three mood-stabilizing drugs. Nature 2002, 417, 292–295. [Google Scholar] [CrossRef]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.V.; Smith, G.P.; Williamson, D.S. Development of LRRK2 inhibitors for the treatment of Parkinson’s disease. Prog. Med. Chem. 2017, 56, 37–80. [Google Scholar] [CrossRef]
- Henderson, J.L.; Kormos, B.L.; Hayward, M.M.; Coffman, K.J.; Jasti, J.; Kurumbail, R.G. Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J. Med. Chem. 2015, 58, 419–432. [Google Scholar] [CrossRef] [PubMed]
- West, A.B. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.H.; Tan, J.Q.; Durairajan, S.S.K.; Liu, L.F.; Zhang, Z.H.; Ma, L. Isorhynchophylline, a natural alkaloid, promotes the degradation of α-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Ma, Z.; Liu, C.; Wang, C.; Deng, Y.; Liu, W. Corynoxine B ameliorates HMGB1-dependent autophagy dysfunction during manganese exposure in SH-SY5Y human neuroblastoma cells. Food Chem. Toxicol. 2019, 124, 336–348. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Semple, B.; Piperi, C.; Othman, I.; Shaikh, M.F. Potential Neuroprotective Effect of the HMGB1 Inhibitor Glycyrrhizin in Neurological Disorders. ACS Chem. Neurosci. 2020, 11, 485–500. [Google Scholar] [CrossRef]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Männistö, P.T.; Maguire-Zeiss, K.A.; Myöhänen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef]
- Zhuang, X.X.; Wang, S.F.; Tan, Y.; Song, J.X.; Zhu, Z.; Wang, Z.Y.; Wu, M.Y.; Cai, C.Z.; Huang, Z.J.; Tan, J.Q.; et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, C.; Liu, J.; Tong, B.C.K.; Zhu, Z.; Malampati, S. A curcumin derivative activates TFEB and protects against parkinsonian neurotoxicity in vitro. Int. J. Mol. Sci. 2020, 21, 1515. [Google Scholar] [CrossRef] [Green Version]
- Khalifeh, M.; Barreto, G.E.; Sahebkar, A. Trehalose as a promising therapeutic candidate for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 1173–1189. [Google Scholar] [CrossRef]
- Ysselstein, D.; Young, T.J.; Nguyen, M.; Padmanabhan, S.; Hirst, W.D.; Dzamko, N.; Krainc, D. Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity. Mov. Disord. 2021, 36, 2719–2730. [Google Scholar] [CrossRef]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H.V. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S. A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.H.; Olanow, C.W. Parkinson’s Disease Modification Through Abl Kinase Inhibition: An Opportunity. Mov. Disord. 2022, 37, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sn, S.; Pandurangi, J.; Murumalla, R.; Dj, V.; Garimella, L.; Acharya, A.; Rai, S.; Paul, A.; Yarreiphang, H.; Pillai, M.S.; et al. Small molecule modulator of aggrephagy regulates neuroinflammation to curb pathogenesis of neurodegeneration. eBioMedicine 2019, 50, 260–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Chen, J.; Wu, X.; Gui, C.; Mao, K.; Zou, F. Role of c-Abl-GSK3β signaling in MPP1-induced autophagy-lysosomal dysfunction. Toxicol. Sci. 2018, 165, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib effects in Parkinson’s disease and dementia with lewy bodies. J. Parkinson’s Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Contu, L.; Hawkes, C.A. A Review of the Impact of Maternal Obesity on the Cognitive Function and Mental Health of the Offspring. Int. J. Mol. Sci. 2017, 18, 1093. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.2; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M.; Berk, M.; Carvalho, A.F.; Puri, B.K. Nutritional ketosis as an intervention to relieve astrogliosis: Possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders. Eur. Psychiatry 2020, 63, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defago, M.D.; Elorriaga, N.; Irazola, V.E.; Rubinstein, A.L. Influence of food patterns on endothelial biomarkers: A systematic review. Clin. Hypertens. 2020, 16, 907–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Disease-modifying treatment of Parkinson’s disease by phytochemicals: Targeting multiple pathogenic factors. J. Neural Transm. 2022, 129, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Virmani, A.; Pinto, L.; Binienda, Z.; Ali, S. Food, nutrigenomics, and neurodegeneration--neuroprotection by what you eat! Mol. Neurobiol. 2013, 48, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.W.; Wang, Y.Q.; Wei, L.C.; Shi, M.; Chan, Y.S. Chinese herbs and herbal extracts for neuroprotection of dopaminergic neurons and potential therapeutic treatment of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2007, 6, 273–281. [Google Scholar] [CrossRef]
- Lee, W.H.; Loo, C.Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its derivatives: Their application in neuropharmacology and neuroscience in the 21st century. Curr. Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, M.A.; Beydoun, H.A.; Fanelli-Kuczmarski, M.T.; Weiss, J.; Hossain, S.; Canas, J.A.; Evans, M.K.; Zonderman, A.B. Association of Serum Antioxidant Vitamins and Carotenoids with Incident Alzheimer Disease and All-Cause Dementia among US Adults. Neurology 2022, 98, e2150–e2162. [Google Scholar] [CrossRef]
- Chao, J.; Yu, M.S.; Ho, Y.S.; Wang, M.; Chang, R.C. Dietary oxyresveratrol prevents Parkinsonian mimetic 6-hydroxydopamine neurotoxicity. Free Radic. Biol. Med. 2008, 45, 1019–1026. [Google Scholar] [CrossRef]
- Katila, N.; Duwa, R.; Bhurtel, S.; Khanal, S.; Maharjan, S.; Jeong, J.H.; Lee, S.; Choi, D.Y.; Yook, S. Enhancement of blood-brain barrier penetration and the neuroprotective effect of resveratrol. J. Control. Release 2022, 346, 1–19. [Google Scholar] [CrossRef]
- Ji, H.F.; Shen, L. The multiple pharmaceutical potential of curcumin in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 369–373. [Google Scholar] [CrossRef]
- Gadad, B.S.; Subramanya, P.K.; Pullabhatla, S.; Shantharam, I.S.; Rao, K.S. Curcumin-glucoside, a novel synthetic derivative of curcumin, inhibits alpha-synuclein oligomer formation: Relevance to Parkinson’s disease. Curr. Pharm. Des. 2012, 18, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.L.; Tapias, V.; Gathagan, R.; Emes, A.; Brandon, T.E.; Smith, A.D. Blueberry juice augments exercise-induced neuroprotection in a Parkinson’s disease model through modulation of GDNF levels. IBRO Neurosci. Rep. 2022, 12, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Belinskaia, D.A.; Ukolov, A.I.; Jenkins, R.O.; Avdonin, P.V. Organosulfur compounds as nutraceuticals. In Nutraceuticals: Efficacy, Safety and Toxicity, 2nd ed.; Gupta, R.C., Lall, R., Srivastava, A., Eds.; Elsevier Inc.: San Diego, CA, USA, 2021; pp. 911–924. ISBN 978-0-12-821038-3. [Google Scholar] [CrossRef]
- Jones, D.P. Redox sensing: Orthogonal control in cell cycle and apoptosis signalling. J. Intern. Med. 2010, 268, 432–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Reprint of: Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2014, 66, 45–57. [Google Scholar] [CrossRef]
- Strandwitz, P.; Kim, K.H.; Terekhova, D.; Liu, J.K.; Sharma, A.; Levering, J.; McDonald, D.; Dietrich, D.; Ramadhar, T.R.; Lekbua, A.; et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 2019, 4, 396–403. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Janik, R.; Thomason, L.A.M.; Stanisz, A.M.; Forsythe, P.; Bienenstock, J.; Stanisz, G.J. Magnetic resonance spectroscopy reveals oral Lactobacillus promotion of increases in brain GABA, N-acetyl aspartate and glutamate. Neuroimage 2016, 125, 988–995. [Google Scholar] [CrossRef]
- Forssten, S.; Ouwehand, A.C. Dose-response recovery of probiotic strains in simulated gastro-intestinal passage. Microorganisms 2020, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Dinan, T.G.; Stanton, C.; Cryan, J.F. Psychobiotics: A novel class of psychotropic. Biol. Psychiatry 2013, 74, 720–726. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Taghizadeh, M.; Daneshvar Kakhaki, R.; Kouchaki, E.; Bahmani, F.; Borzabadi, S.; Oryan, S.; Mafi, A.; Asemi, Z. Clinical and metabolic response to probiotic administration in people with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1031–1035. [Google Scholar] [CrossRef]
- Burokas, A.; Arboleya, S.; Moloney, R.D.; Peterson, V.L.; Murphy, K.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Targeting the microbiota-gut-brain axis: Prebiotics have anxiolytic and antidepressant-like effects and reverse the impact of chronic stress in mice. Biol. Psychiatry 2017, 82, 472–487. [Google Scholar] [CrossRef]
- Chunchai, T.; Thunapong, W.; Yasom, S.; Wanchai, K.; Eaimworawuthikul, S.; Metzler, G.; Lungkaphin, A.; Pongchaidecha, A.; Sirilun, S.; Chaiyasut, C.; et al. Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J. Neuroinflamm. 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barichella, M.; Pacchetti, C.; Bolliri, C.; Cassani, E.; Iorio, L.; Pusani, C.; Pinelli, G.; Privitera, G.; Cesari, I.; Faierman, S.A.; et al. Probiotics and prebiotic fiber for constipation associated with Parkinson disease: An RCT. Neurology 2016, 87, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Zhu, L.; Jing, X.; Liang, Y.; Tao, E. Rifampicin and Parkinson’s disease. Neurol. Sci. 2013, 34, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Reglodi, D.; Renaud, J.; Tamas, A.; Tizabi, Y.; Socías, S.B.; del-Bel, E.; Raisman-Vozari, R. Novel tactics for neuroprotection in Parkinson’s disease: Role of antibiotics, polyphenols and neuropeptides. Prog. Neurobiol. 2017, 155, 120–148. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.-H.; Bellesi, M.; Chen, A.-C.; Lin, C.L.; Li, H.H.; Lin, P.J.; Liao, W.C.; Hung, C.S.; Schwarting, R.K.; Ho, Y.J. A new avenue for treating neuronal diseases: Ceftriaxone, an old antibiotic demonstrating behavioral neuronal effects. Behav. Brain Res. 2019, 364, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertsalmi, T.H.; Pekkonen, E.; Scheperjans, F. Antibiotic exposure and risk of Parkinson’s disease in Finland: A nationwide case-control study. Mov. Disord. 2020, 35, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Khanna, S. Fecal Microbiota Transplantation. JAMA 2017, 318, 102. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.K.; Verma, S.; Jain, V.; Surapaneni, B.K.; Vinayek, R.; Phillips, L.; Nair, P.P. Parkinson’s Disease: The Emerging Role of Gut Dysbiosis, Antibiotics, Probiotics, and Fecal Microbiota Transplantation. J. Neurogastroenterol. Motil. 2019, 25, 363–376. [Google Scholar] [CrossRef]
- Borrione, P.; Tranchita, E.; Sansone, P.; Parisi, A. Effects of physical activity in Parkinson’s disease: A new tool for rehabilitation. World J. Methodol. 2014, 4, 133–143. [Google Scholar] [CrossRef]
- Radder, D.L.M.; Sturkenboom, I.H.; van Nimwegen, M.; Keus, S.H.; Bloem, B.R.; de Vries, N.M. Physical therapy and occupational therapy in Parkinson’s disease. Int. J. Neurosci. 2017, 127, 930–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habets, J.G.V.; Heijmans, M.; Kuijf, M.L.; Janssen, M.L.F.; Temel, Y.; Kubben, P.L. An update on adaptive deep brain stimulation in Parkinson’s disease. Mov. Disord. 2018, 33, 1834–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuter, A.; Lefaucheur, J.P.; Modolo, J. Closed-loop cortical neuromodulation in Parkinson’s disease: An alternative to deep brain stimulation? Clin. Neurophysiol. 2014, 125, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Lauze, M.; Daneault, J.F.; Duval, C. The Effects of Physical Activity in Parkinson’s Disease: A Review. J. Parkinson’s Dis. 2016, 6, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Wong-Yu, I.S.; Mak, M.K. Effects of Exercise on Falls, Balance, and Gait Ability in Parkinson’s Disease: A Meta-analysis. Neurorehabil. Neural Repair 2016, 30, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Tejada, A.; Neisewander, J.; Katsanos, C.S. Regulation of Voluntary Physical Activity Behavior: A Review of Evidence Involving Dopaminergic Pathways in the Brain. Brain Sci. 2022, 12, 333. [Google Scholar] [CrossRef]
- Alghadir, A.H.; Gabr, S.A.; Al-Eisa, E.S. Effects of moderate aerobic exercise on cognitive abilities and redox state biomarkers in older adults. Oxid. Med. Cell. Longev. 2016, 2016, 2545168. [Google Scholar] [CrossRef] [Green Version]
- De Vries, N.M.; Sturkenboom, I.H.; Bloem, B.R. Physiotherapy and Occupational Therapy and Mild to Moderate Parkinson Disease. JAMA Neurol. 2016, 73, 893–894. [Google Scholar] [CrossRef]
- Bloem, B.R.; de Vries, N.M.; Ebersbach, G. Nonpharmacological treatments for patients with Parkinson’s disease. Mov. Disord. 2015, 30, 1504–1520. [Google Scholar] [CrossRef]
- LaHue, S.C.; Comella, C.L.; Tanner, C.M. The best medicine? The influence of physical activity and inactivity on Parkinson’s disease. Mov. Disord. 2016, 31, 1444–1454. [Google Scholar] [CrossRef]
- Canning, C.G.; Sherrington, C.; Lord, S.R.; Close, J.C.; Heritier, S.; Heller, G.Z.; Howard, K.; Allen, N.E.; Latt, M.D.; Murray, S.M.; et al. Exercise for falls prevention in Parkinson disease. Neurology 2015, 84, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.L.; Thilarajah, S.; Tan, D. Effectiveness of resistance training on muscle strength and physical function in people with Parkinson’s disease: A systematic review and metaanalysis. Clin. Rehabil. 2016, 30, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Oguh, O.; Eisenstein, A.; Kwasny, M.; Simuni, T. Back to the basics: Regular exercise matters in Parkinson’s disease: Results from the National Parkinson Foundation QII registry study. Park. Relat. Disord. 2014, 20, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.F.; Yang, T.; Yu, S.X.; Huang, H.D.; Jiang, L.L.; Gu, J.W.; Kuang, Y.Q. Aerobic exercise for Parkinson’s disease: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2014, 9, e100503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, F.O.; Santana, V.; Corcos, D.M.; Ugrinowitsch, C.; Silva-Batista, C. Effects of Endurance Training on Motor Signs of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Sports Med. 2022, 52, 1789–1815. [Google Scholar] [CrossRef]
- Wu, C.; Xu, Y.; Guo, H.; Tang, C.; Chen, D.; Zhu, M. Effects of Aerobic Exercise and Mind-Body Exercise in Parkinson’s Disease: A Mixed-Treatment Comparison Analysis. Front. Aging Neurosci. 2021, 13, 739115. [Google Scholar] [CrossRef]
- Di Marco, R.; Pistonesi, F.; Cianci, V.; Biundo, R.; Weis, L.; Tognolo, L.; Baba, A.; Rubega, M.; Gentile, G.; Tedesco, C.; et al. Effect of Intensive Rehabilitation Program in Thermal Water on a Group of People with Parkinson’s Disease: A Retrospective Longitudinal Study. Healthcare 2022, 10, 368. [Google Scholar] [CrossRef]
- Shi, K.; Liu, X.; Qiao, D.; Hou, L. Effects of Treadmill Exercise on Spontaneous Firing Activities of Striatal Neurons in a Rat Model of Parkinson’s Disease. Mot. Control 2017, 21, 58–71. [Google Scholar] [CrossRef]
- Fedotova, E.I.; Dolgacheva, L.P.; Abramov, A.Y.; Berezhnov, A.V. Lactate and Pyruvate Activate Autophagy and Mitophagy that Protect Cells in Toxic Model of Parkinson’s Disease. Mol. Neurobiol. 2021, 59, 177–190. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Heris, R.M.; Shirvaliloo, M.; Abbaspour-Aghdam, S.; Hazrati, A.; Shariati, A.; Youshanlouei, H.R.; Niaragh, F.J.; Valizadeh, H.; Ahmadi, M. The potential use of mesenchymal stem cells and their exosomes in Parkinson’s disease treatment. Stem Cell Res. Ther. 2022, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Whone, A. Monoclonal Antibody Therapy in Parkinson’s Disease—The End? N. Engl. J. Med. 2022, 387, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Butler, Y.R.; Liu, Y.; Kumbhar, R.; Zhao, P.; Gadhave, K.; Wang, N.; Li, Y.; Mao, X.; Wang, W. α-Synuclein fibril-specific nanobody reduces prion-like α-synuclein spreading in mice. Nat. Commun. 2022, 13, 4060. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.S.; Ooi, Y.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Immunotherapies for Parkinson’s Disease: Progression of Clinical Development. CNS Neurol. Disord. Drug Targets 2021, 20, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. SPARK Investigators. Trial of Cinpanemab in Early Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.P.; et al. PASADENA Investigators; Prasinezumab Study Group. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Qi, Z.; Li, J.; Li, M.; Du, X.; Zhang, L.; Wang, S.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. The Essential Role of Epigenetic Modifications in Neurodegenerative Diseases with Dyskinesia. Cell. Mol. Neurobiol. 2021, 42, 2459–2472. [Google Scholar] [CrossRef]
- Kim, J.S.; Lim, H.S.; Moon, B.C.; Ang, M.J.; Kim, S.H.; Moon, C.J.; Seong, B.; Jang, Y.J.; Kim, H.Y.; Kim, C. Epigenetic mechanisms involved in the neuroprotective effect of scorpion extract in a Parkinson’s disease murine model based on multi-omics approach. J. Tradit. Chin. Med. 2021, 41, 390–396. [Google Scholar] [CrossRef]
- Fang, M.; Chen, D.; Yang, C.S. Dietary polyphenols may affect DNA methylation. J. Nutr. 2007, 137 (Suppl. 1), 223S–228S. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Saadat, A. Neurodegeneration and epigenetics: A review. Neurologia 2021, in press. [CrossRef] [PubMed]
- Farrow, S.L.; Cooper, A.A.; O’Sullivan, J.M. Redefining the hypotheses driving Parkinson’s diseases research. NPJ Parkinson’s Dis. 2022, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Desrosiers, C.; Frasnelli, J. Machine Learning for the Diagnosis of Parkinson’s Disease: A Review of Literature. Front. Aging Neurosci. 2021, 13, 184. [Google Scholar] [CrossRef] [PubMed]
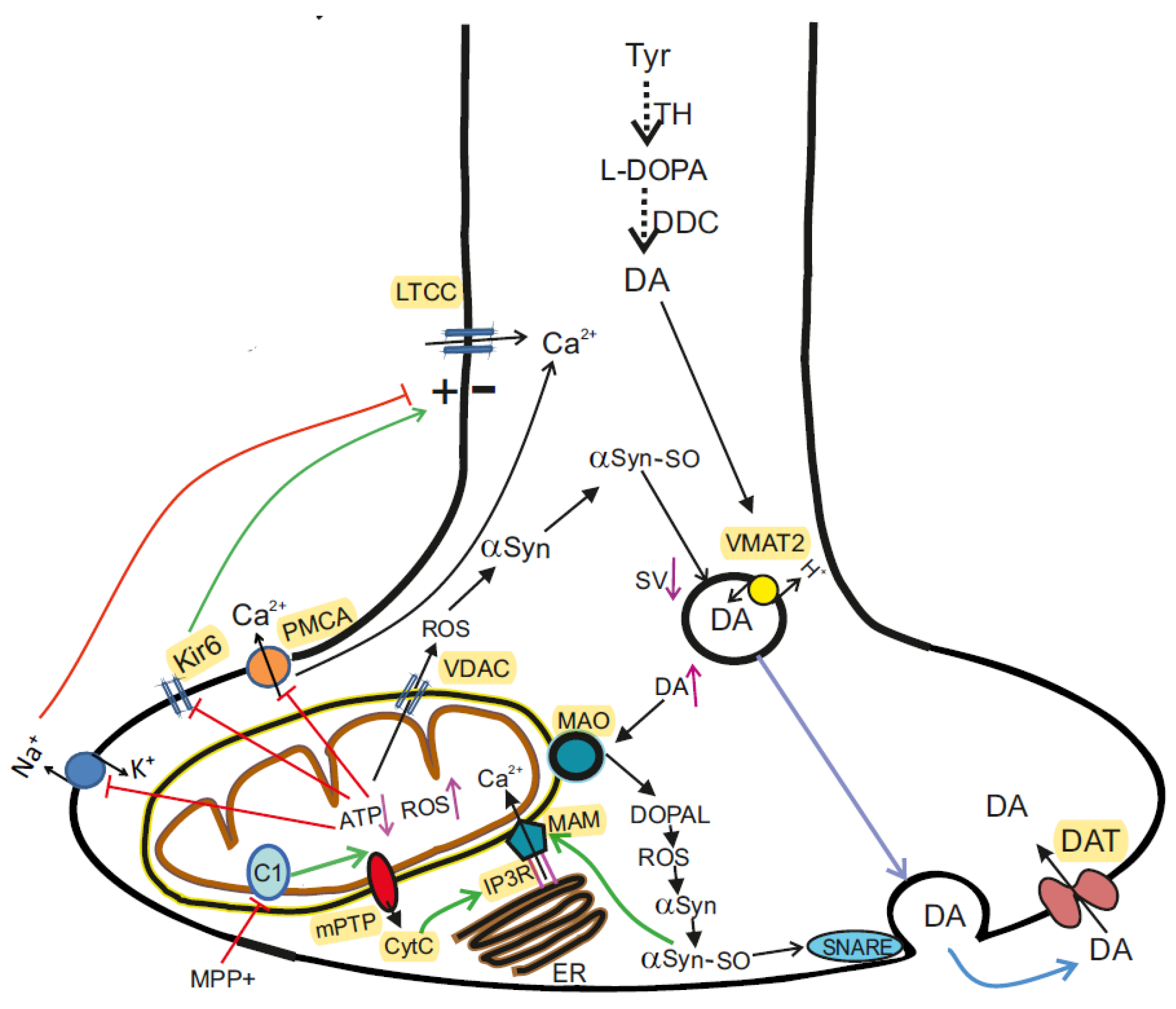



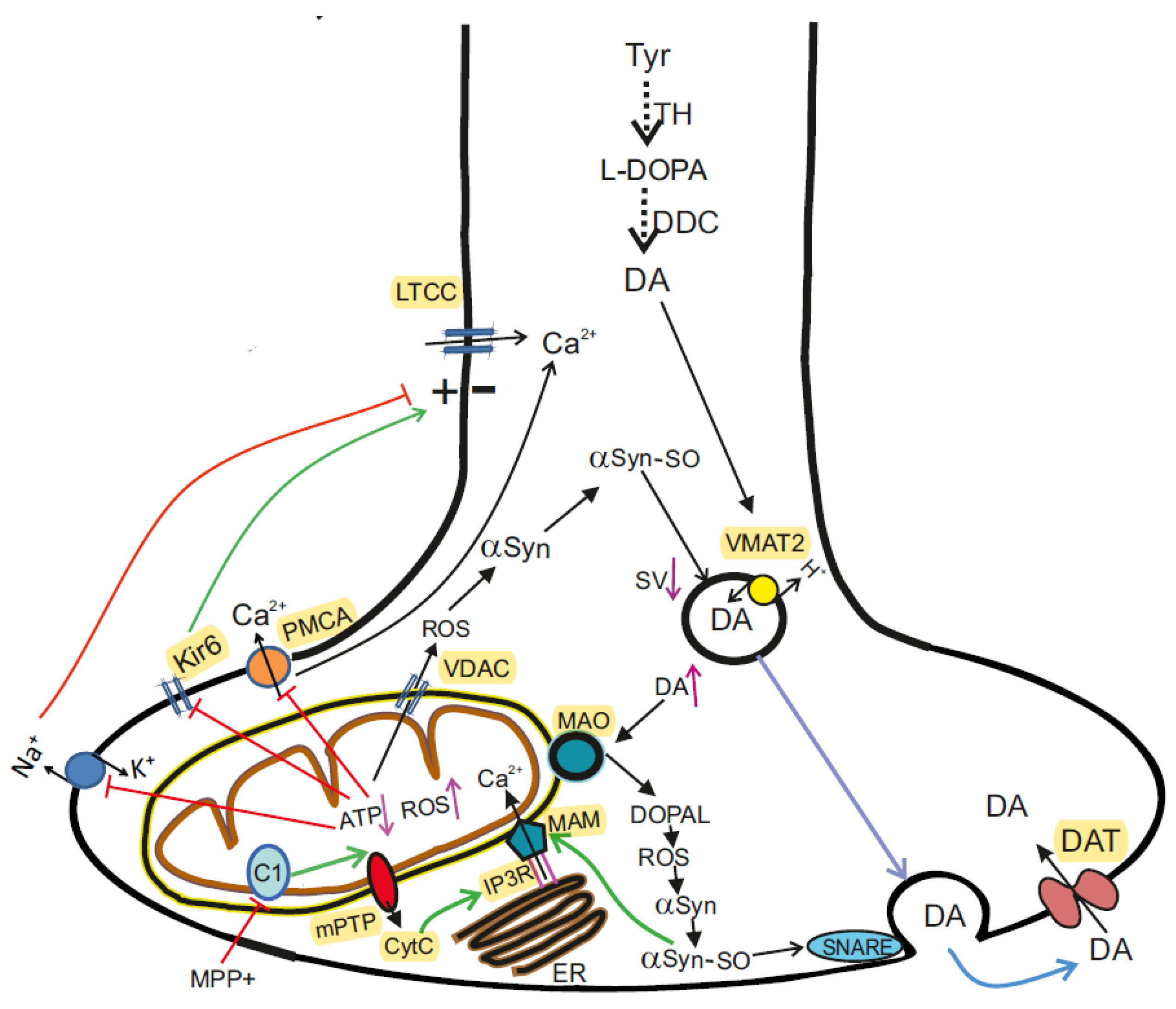{kind=link}
{kind=link}
{kind=link}
| Physiological Functions | Regulatory Factors | Involvement in Pathogenesis of PD |
|---|---|---|
| Inflammation | IL-1β, IL-6, TNF-α, MMP-9, MCP-1, Complement activation, Oxidative stress | [224,225,226,227,228,229] |
| Leukocyte trafficking | ICAM1, VCAM, E-selectin | [196,228,230] |
| Haemostasis and coagulation | Anticoagulant factors: TM, tPA, PGI2 Procoagulant factors: vWF, TXA2, PAI1 | [143,231,232,233,234,235,236] |
| Control of VSMC and pericytes, Vascular tone | Microparticles, Jagged-1/NOTCH-1, miR-126, NO | [228,237,238,239,240,241,242,243] |
| Vascular permeability, Angiogenesis and Metabolism | VEGF/VEGFR, HIF activation, Nogo-B, Glutamate metabolism, Jagged-1/NOTCH-1 | [227,230,238,244,245,246,247,248,249,250] |
| ## | Substances or the Type of Intervention | The Target or Mechanism of Action | References |
|---|---|---|---|
| 1 | pallidotomy, thalamotomy, subthalamotomy | reducing excessive inhibition from the globus pallidus and SN | [36,308] |
| 2 | L-DOPA or levodopa | substitution for DA loss in the striatum | [310] |
| 3 | pergolide, pramipexole dihydrochloride, ropinirole hydrochloride, rotigotine, apomorphine hydrochloride | DA-like agonists that bind to dopaminergic postsynaptic receptors | [319,320] |
| 4 | rasagiline, selegiline, | MAO inhibitors, prolongation of DA and L-DOPA action | [321,322] |
| 5 | entacapone | COMT inhibitor | [36,323] |
| 6 | salbutamol | β2AR agonist, regulators of the αSyn gene | [325] |
| 7 | UDCA, TUDCA | anti-apoptotic agents, upregulates mitophagy and the expression of PINK1 and parkin | [203,327] |
| 8 | flavonoids and carotinoids | reduction of excessive ROS production and oxidative stress | [314,315] |
| 9 | NAM | maintains mitochondrial function by increasing NAD levels in dopaminergic neurons | [203,330] |
| 10 | biperiden | antagonist of muscarinic receptor | [331] |
| 11 | Hsp27 (HSPB1) | binding of αSyn fibrils and inhibition their cytotoxicity | [333] |
| 12 | resveratrol, caffeine, metformin, A769662, GSK621, rosuvastatin, FCPR16, temozolomide | autophagy modulators by activating AMPK | [184,334,335,336,337,338,339] |
| 13 | rapamycin, its analogs CCI-779 and AP23573, corynoxine, loganin, PI-103 | autophagy modulators by activating mTORC1 | [339,340,341,342,343] |
| 14 | sodium valproate, carbamazepine, L-690.330 | autophagy modulators by inhibiting IMPase | [339,344] |
| 15 | LRRK2-IN-1, GNE-7915, PF-06447475, DNL151, DNL201 | autophagy modulators by inhibiting LRRK2 | [345,346,347,348] |
| 16 | isorhynchophylline, corynoxine B, glycyrrhizic acid, KYP-2047 | autophagy modulators by inhibiting Beclin-1 | [349,350,351,352] |
| 17 | ambroxol, isofagomine, NCGC607 | autophagy modulators by inhibiting GCase 1 | [326,356,357,358,359] |
| 18 | fruits and vegetables, nutraceuticals and phytochemicals | antioxidant, anti-inflammatory and anti-apoptotic activities; improvement of the functional state of endothelium and brain blood circulation; modulation of the cell survival genes | [292,334,369,370,371,372,373,374,375,376,377,378] |
| 19 | probiotics, prebiotics, antibiotics, FMT | restore the composition of the gut microbiota, replenish beneficial metabolites, and reduce harmful metabolites | [203,204,383,384,385,386,387,388,389,390] |
| 20 | deep brain stimulation | an alternative to ablative operations in the globus pallidus, subthalamic nucleus and thalamus | [308,399,400] |
| 21 | moderate physical activity | positively affects endothelial cells and cognitive functions, gait parameters and other motor performance | [404,405,408,409,410] |
| 22 | mesenchymal stem cells (MSCs) | differentiate into dopaminergic neurons and produce neurotrophic substances | [417,418] |
| 23 | monoclonal antibodies and nanobodies | targeting the aggregated αSyn | [421,422,423,424] |
| 24 | folate, vitamin B6, vitamin B12, and S-adenosylmethionine, 5-azacytidine, dietary polyphenols, scorpion extract | drugs and substances affecting epigenetic modifications—DNA methylation and histone acetylation | [426,427,428] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolgacheva, L.P.; Zinchenko, V.P.; Goncharov, N.V. Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease. Int. J. Mol. Sci. 2022, 23, 13043. https://doi.org/10.3390/ijms232113043
Dolgacheva LP, Zinchenko VP, Goncharov NV. Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease. International Journal of Molecular Sciences. 2022; 23(21):13043. https://doi.org/10.3390/ijms232113043
Chicago/Turabian StyleDolgacheva, Lyudmila P., Valery P. Zinchenko, and Nikolay V. Goncharov. 2022. "Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease" International Journal of Molecular Sciences 23, no. 21: 13043. https://doi.org/10.3390/ijms232113043
APA StyleDolgacheva, L. P., Zinchenko, V. P., & Goncharov, N. V. (2022). Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease. International Journal of Molecular Sciences, 23(21), 13043. https://doi.org/10.3390/ijms232113043