


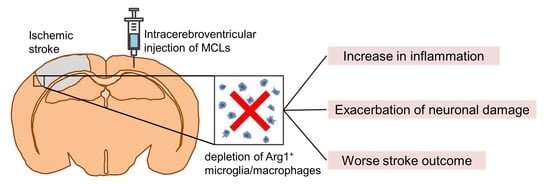
Depletion of Arg1-Positive Microglia/Macrophages Exacerbates Cerebral Ischemic Damage by Facilitating the Inflammatory Response

Abstract
:
1. Introduction
2. Results
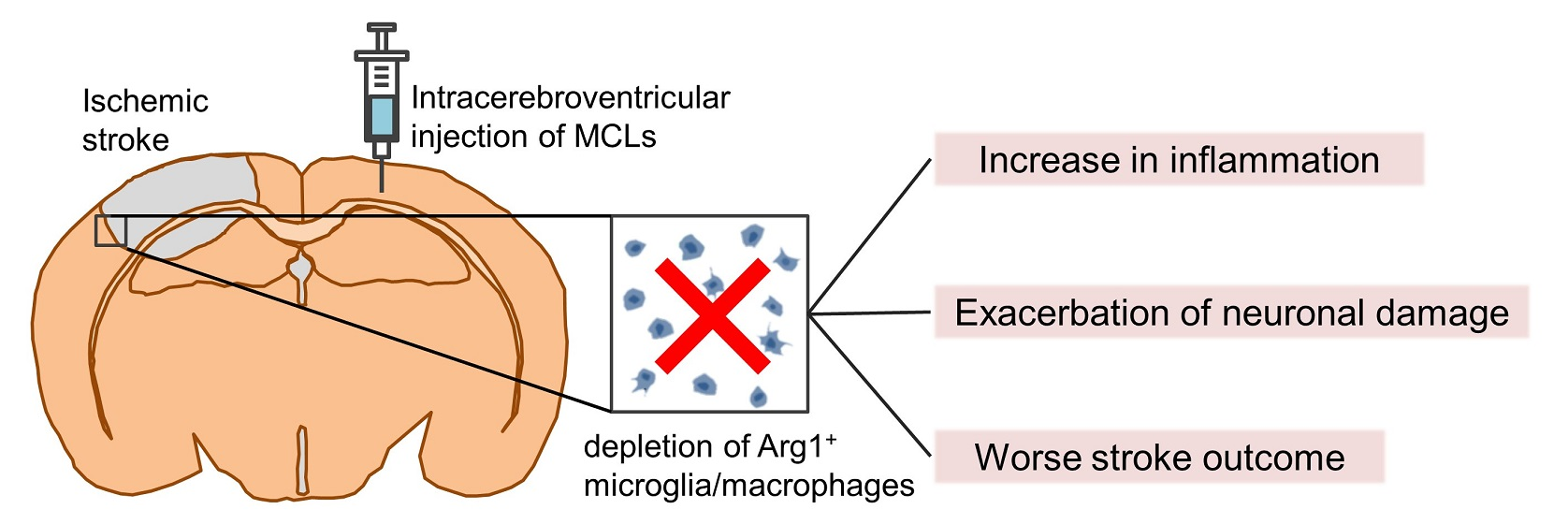
2.1. Dynamics of Arg1+ Cells after Acute Ischemic Stroke
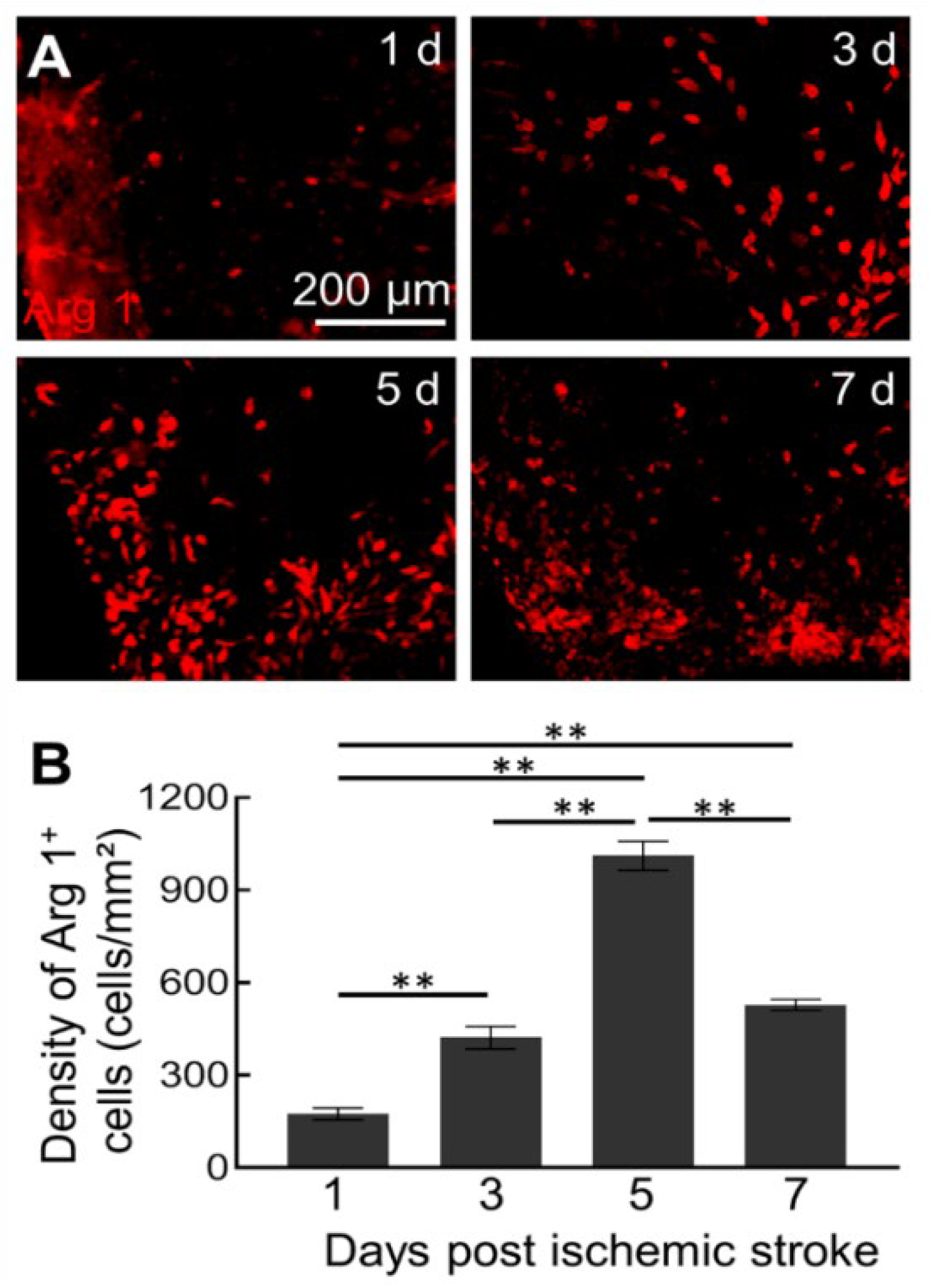
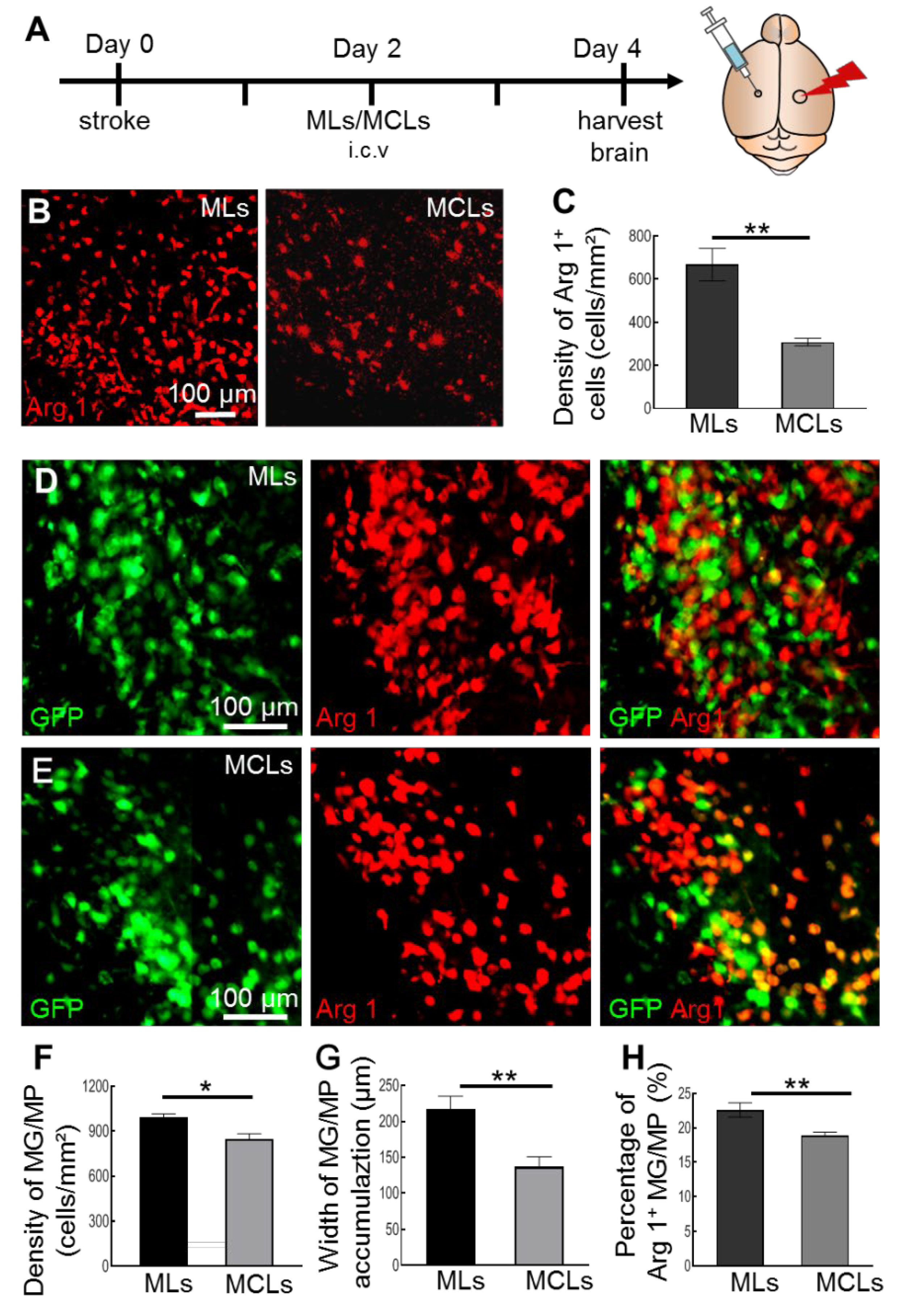
2.2. MCLs Selectively Depletes Arg1+ Microglia/Macrophages in the Brain
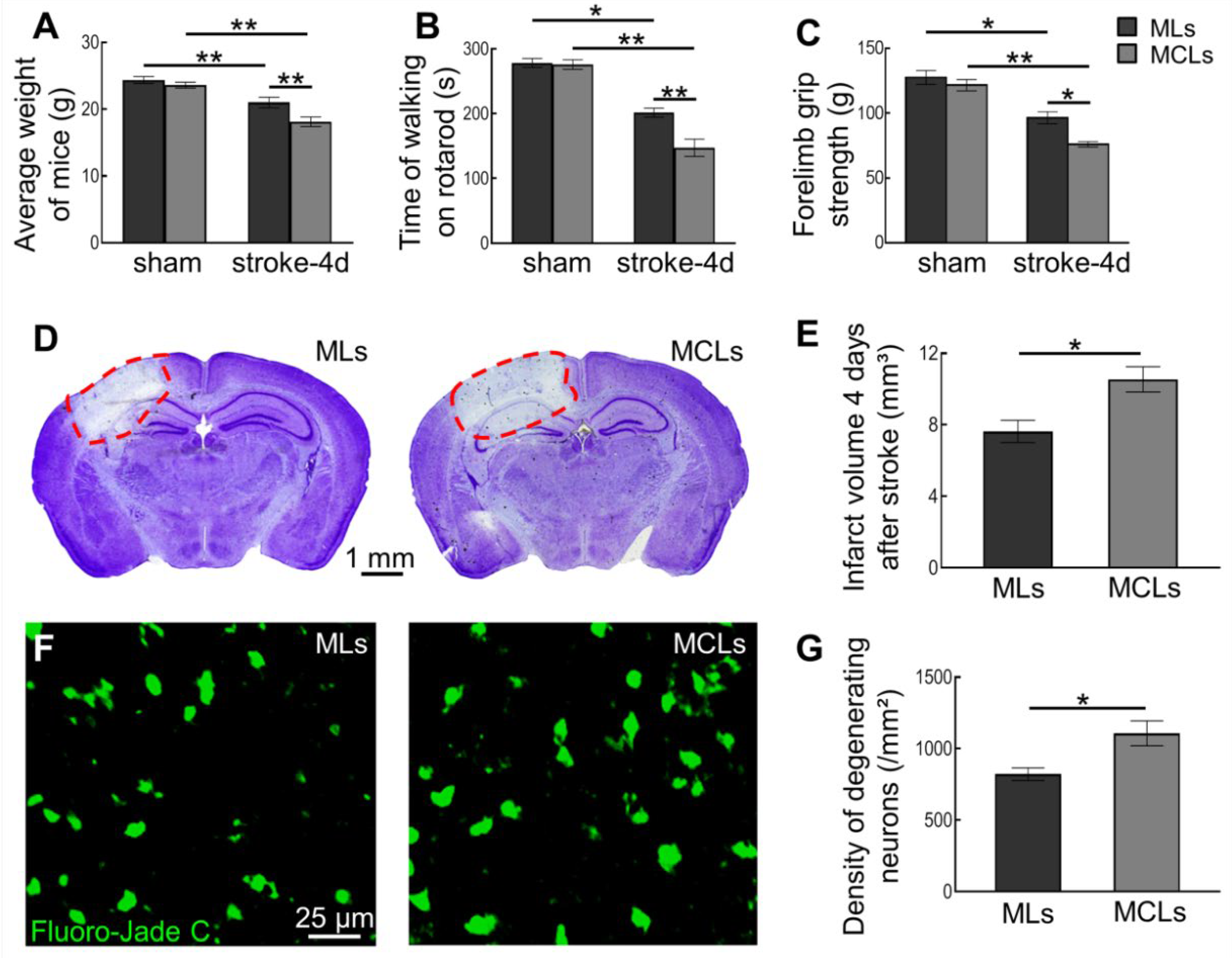
2.3. Depleting Arg1+ Microglia/Macrophages Exacerbates Ischemic Injury
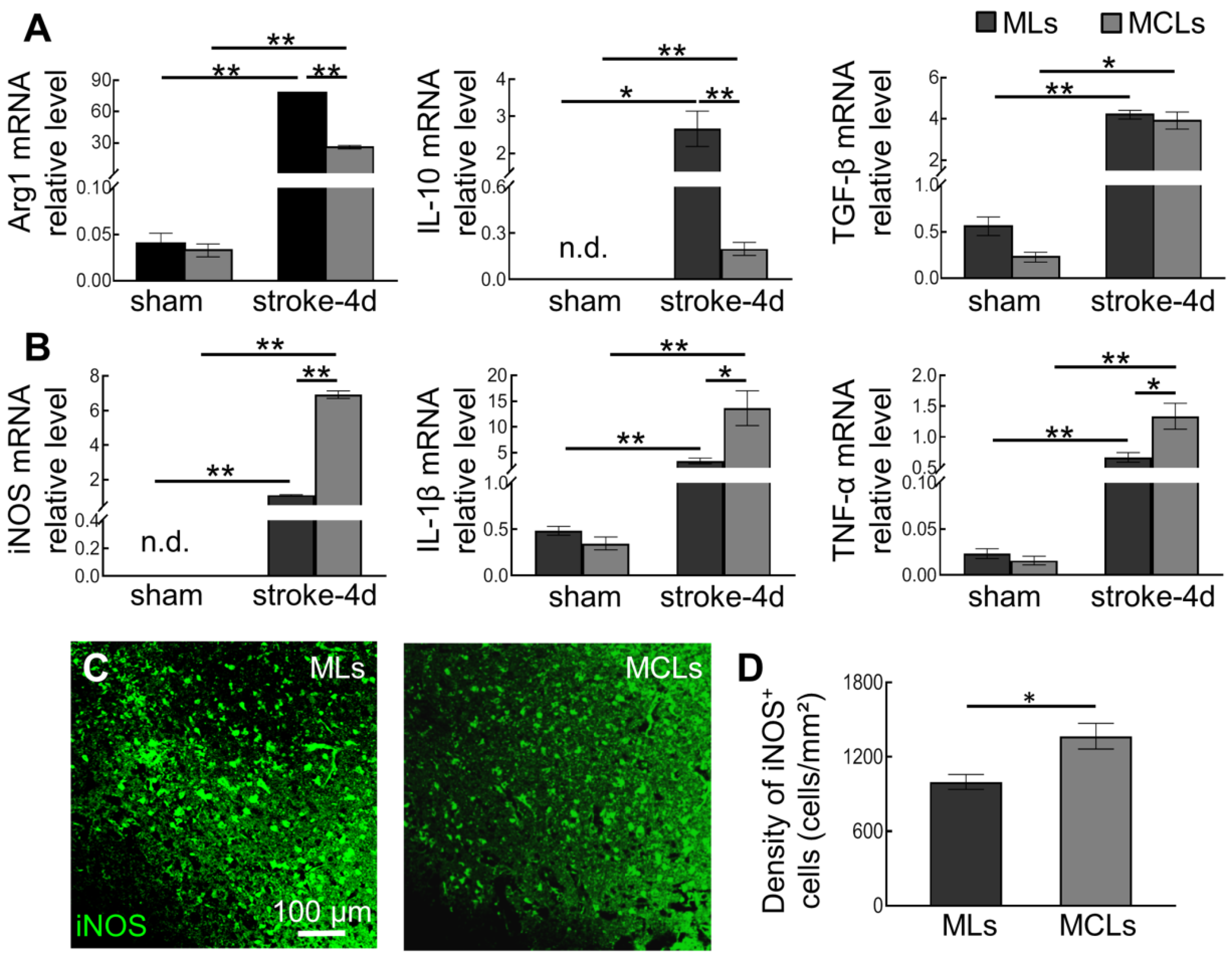
2.4. Depleting Arg1+ Microglia/Macrophages Promotes Inflammation in Ischemic Tissue
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Photothrombotic Ischemic Stroke Model
4.3. Selective Depletion of Arg1+ Microglia/Macrophages after Ischemia
4.4. Infarct Volumetry
4.5. Histochemical Stainings
4.6. Behavioral Tests
4.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Li, T.; Liesz, A. Immunity in Stroke: The Next Frontier. Thromb. Haemost. 2022, 122, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Tian, D.C.; Li, Z.G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.D. Global brain inflammation in stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sippel, T.R.; Shimizu, T.; Strnad, F.; Traystman, R.J.; Herson, P.S.; Waziri, A. Arginase I release from activated neutrophils induces peripheral immunosuppression in a murine model of stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1657–1663. [Google Scholar] [CrossRef] [Green Version]
- Petrone, A.B.; O’Connell, G.C.; Regier, M.D.; Chantler, P.D.; Simpkins, J.W.; Barr, T.L. The Role of Arginase 1 in Post-Stroke Immunosuppression and Ischemic Stroke Severity. Transl. Stroke Res. 2016, 7, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Quirié, A.; Demougeot, C.; Bertrand, N.; Mossiat, C.; Garnier, P.; Marie, C.; Prigent-Tessier, A. Effect of stroke on arginase expression and localization in the rat brain. Eur. J. Neurosci. 2013, 37, 1193–1202. [Google Scholar] [CrossRef]
- Hamzei Taj, S.; Kho, W.; Riou, A.; Wiedermann, D.; Hoehn, M. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials 2016, 91, 151–165. [Google Scholar] [CrossRef]
- Hansen, R.B.; Laursen, C.C.H.; Nawaz, N.; Madsen, J.S.; Nielsen, H.H.; Kruuse, C.; Møller, A.; Degn, M.; Lambertsen, K.L. Leukocyte TNFR1 and TNFR2 Expression Contributes to the Peripheral Immune Response in Cases with Ischemic Stroke. Cells 2021, 10, 861. [Google Scholar] [CrossRef]
- Iyer, R.K.; Yoo, P.K.; Kern, R.M.; Rozengurt, N.; Tsoa, R.; O’Brien, W.E.; Yu, H.; Grody, W.W.; Cederbaum, S.D. Mouse model for human arginase deficiency. Mol. Cell. Biol. 2002, 22, 4491–4498. [Google Scholar] [CrossRef]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Meininger, C.J.; Hawker, J.R., Jr.; Haynes, T.E.; Kepka-Lenhart, D.; Mistry, S.K.; Morris, S.M., Jr.; Wu, G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am. J. Physiol.-Endocrinol. Metab. 2001, 280, E75–E82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, T.W.; Zhang, C.; Wang, W.; Chang, C.I.; Thengchaisri, N.; Kuo, L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: Counteracting role of arginase. FASEB J. 2003, 17, 2328–2330. [Google Scholar] [CrossRef] [PubMed]
- Wanrooy, B.J.; Wen, S.W.; Wong, C.H. Dynamic roles of neutrophils in post-stroke neuroinflammation. Immunol. Cell Biol. 2021, 99, 924–935. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, G.C.; Treadway, M.B.; Tennant, C.S.; Lucke-Wold, N.; Chantler, P.D.; Barr, T.L. Shifts in Leukocyte Counts Drive the Differential Expression of Transcriptional Stroke Biomarkers in Whole Blood. Transl. Stroke Res. 2019, 10, 26–35. [Google Scholar] [CrossRef]
- Tratsiakovich, Y.; Yang, J.; Gonon, A.T.; Sjöquist, P.O.; Pernow, J. Arginase as a target for treatment of myocardial ischemia-reperfusion injury. Eur. J. Pharmacol. 2013, 720, 121–123. [Google Scholar] [CrossRef]
- Elms, S.C.; Toque, H.A.; Rojas, M.; Xu, Z.; Caldwell, R.W.; Caldwell, R.B. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia 2013, 56, 654–662. [Google Scholar] [CrossRef] [Green Version]
- Moretto, J.; Girard, C.; Demougeot, C. The role of arginase in aging: A systematic review. Exp. Gerontol. 2019, 116, 54–73. [Google Scholar] [CrossRef]
- Schneider, E.; Dy, M. The role of arginase in the immune response. Immunol. Today 1985, 6, 136–140. [Google Scholar] [CrossRef]
- Jian, Z.; Liu, R.; Zhu, X.; Smerin, D.; Zhong, Y.; Gu, L.; Fang, W.; Xiong, X. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front. Immunol. 2019, 10, 2167. [Google Scholar] [CrossRef]
- Kollikowski, A.M.; Schuhmann, M.K.; Nieswandt, B.; Müllges, W.; Stoll, G.; Pham, M. Local Leukocyte Invasion during Hyperacute Human Ischemic Stroke. Ann. Neurol. 2020, 87, 466–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.S.; Marlier, A.; Xu, L.; Doilicho, N.; Linberg, D.; Guo, J.; Cantley, L.G. Arginase-1 Is Required for Macrophage-Mediated Renal Tubule Regeneration. J. Am. Soc. Nephrol. 2022, 33, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Rao, Y.; Huang, Y.; Zhou, T.; Feng, R.; Xiong, S.; Yuan, T.F.; Qin, S.; Lu, Y.; Zhou, X.; et al. Efficient Strategies for Microglia Replacement in the Central Nervous System. Cell Rep. 2020, 33, 108443. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Portier, I.; Rustad, J.L.; Cody, M.J.; de Araujo, C.V.; Hoki, C.; Alexander, M.D.; Grandhi, R.; Dyer, M.R.; Neal, M.D.; et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J. Clin. Investig. 2022, 132, e154225. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Fouda, A.Y.; Xu, Z.; Shosha, E.; Lemtalsi, T.; Chen, J.; Toque, H.A.; Tritz, R.; Cui, X.; Stansfield, B.K.; Huo, Y.; et al. Arginase 1 promotes retinal neurovascular protection from ischemia through suppression of macrophage inflammatory responses. Cell Death Dis. 2018, 9, 1001. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.W.; Li, L.; Huang, Y.Y.; Zhao, C.Q.; Xue, S.J.; Chen, J.; Yang, Z.Z.; Xu, J.F.; Su, X. Vagal-α7nAChR signaling is required for lung anti-inflammatory responses and arginase 1 expression during an influenza infection. Acta Pharmacol. Sin. 2021, 42, 1642–1652. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2018, 301, 120–132. [Google Scholar] [CrossRef]
- Li, T.; Pang, S.; Yu, Y.; Wu, X.; Guo, J.; Zhang, S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain 2013, 136, 3578–3588. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Nolte, K.; Brook, G.; Liebenstund, L.; Weinandy, A.; Höllig, A.; Veldeman, M.; Willuweit, A.; Langen, K.J.; Rossaint, R.; et al. Post-stroke treatment with argon attenuated brain injury, reduced brain inflammation and enhanced M2 microglia/macrophage polarization: A randomized controlled animal study. Crit. Care 2019, 23, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Q.; Ding, D.H.; Wang, X.Y.; Sun, Y.Y.; Wu, J. Lipoxin A4 regulates microglial M1/M2 polarization after cerebral ischemia-reperfusion injury via the Notch signaling pathway. Exp. Neurol. 2021, 339, 113645. [Google Scholar] [CrossRef]
- Li, T.; Zhao, J.; Xie, W.; Yuan, W.; Guo, J.; Pang, S.; Gan, W.B.; Gómez-Nicola, D.; Zhang, S. Specific depletion of resident microglia in the early stage of stroke reduces cerebral ischemic damage. J. Neuroinflammation 2021, 18, 81. [Google Scholar] [CrossRef]
- Cai, W.; Dai, X.; Chen, J.; Zhao, J.; Xu, M.; Zhang, L.; Yang, B.; Zhang, W.; Rocha, M.; Nakao, T.; et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 2019, 4, e131355. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Levard, D.; Buendia, I.; Lanquetin, A.; Glavan, M.; Vivien, D.; Rubio, M. Filling the gaps on stroke research: Focus on inflammation and immunity. Brain Behav. Immun. 2021, 91, 649–667. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Liu, H.; Pearson, L.; Kapoor, M.; Harrison, J.C.; Sammut, I.A.; Jackson, D.M.; Appleton, I. Neuroprotective effects of spermine following hypoxic-ischemic-induced brain damage: A mechanistic study. FASEB J. 2004, 18, 1114–1116. [Google Scholar] [CrossRef]
- Li, T.; Xu, T.; Zhao, J.; Gao, H.; Xie, W. Depletion of iNOS-positive inflammatory cells decelerates neuronal degeneration and alleviates cerebral ischemic damage by suppressing the inflammatory response. Free Radic. Biol. Med. 2022, 181, 209–220. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.H.; Jacobs, A.T.; Morris, S.M., Jr.; Ignarro, L.J. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am. J. Physiol.-Cell Physiol. 2000, 279, C248–C256. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yi, P.; Yuan, D.M.K.; Jie, Z.; Kwota, Z.; Soong, L.; Cong, Y.; Sun, J. IL-33 induces immunosuppressive neutrophils via a type 2 innate lymphoid cell/IL-13/STAT6 axis and protects the liver against injury in LCMV infection-induced viral hepatitis. Cell. Mol. Immunol. 2019, 16, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.; Chen, S.; Chen, S.; Peng, Q.; Jin, H.; Hu, B. The role of leukocytes in acute ischemic stroke-related thrombosis: A notable but neglected topic. Cell. Mol. Life Sci. 2021, 78, 6251–6264. [Google Scholar] [CrossRef]
- Saiki, S.; Sasazawa, Y.; Fujimaki, M.; Kamagata, K.; Kaga, N.; Taka, H.; Li, Y.; Souma, S.; Hatano, T.; Imamichi, Y.; et al. A metabolic profile of polyamines in parkinson disease: A promising biomarker. Ann. Neurol. 2019, 86, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Barakat, W.; Fahmy, A.; Askar, M.; El-Kannishy, S. Effectiveness of arginase inhibitors against experimentally induced stroke. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 603–612. [Google Scholar] [CrossRef]
- Ahmad, A.S.; Shah, Z.A.; Doré, S. Protective Role of Arginase II in Cerebral Ischemia and Excitotoxicity. J. Neurol. Neurosci. 2016, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Seifert, H.A.; Vandenbark, A.A.; Offner, H. Regulatory B cells in experimental stroke. Immunology 2018, 154, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhao, Y.; Gu, Y.; Xu, C. Neuroprotective actions of aminoguanidine involve reduced the activation of calpain and caspase-3 in a rat model of stroke. Neurochem. Int. 2010, 56, 634–641. [Google Scholar] [CrossRef]
- Stewart, V.C.; Heales, S.J. Nitric oxide-induced mitochondrial dysfunction: Implications for neurodegeneration. Free Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, S.; Li, Y.; Sun, Y.; Xiong, X.; Hu, X.; Chen, J.; Qiu, S. Interleukins and Ischemic Stroke. Front. Immunol. 2022, 13, 828447. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Kleinschnitz, C. Regulatory T Cells in Post-stroke Immune Homeostasis. Transl. Stroke Res. 2016, 7, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jia, W.; Xu, W.; Wu, Q.; Wu, J. Downregulation of CD151 restricts VCAM-1 mediated leukocyte infiltration to reduce neurobiological injuries after experimental stroke. J. Neuroinflammation 2021, 18, 118. [Google Scholar] [CrossRef]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [Green Version]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Loihl, A.K.; Asensio, V.; Campbell, I.L.; Murphy, S. Expression of nitric oxide synthase (NOS)-2 following permanent focal ischemia and the role of nitric oxide in infarct generation in male, female and NOS-2 gene-deficient mice. Brain Res. 1999, 830, 155–164. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | 5′-TGAACGGGAAGCTCACTGG-3′ | 5′-TCCACCACCCTGTTGCTGTA-3′ |
| iNOS | 5′-CAAGCACCTTGGAAGAGGAG-3′ | 5′-AAGGCCAAACACAGCATACC-3′ |
| IL-1β | 5′-CCTCGTGCTGTCGGACCCATA-3′ | 5′-CAGGCTTGTGCTCTGCTTGTGA-3′ |
| TNF-α | 5′-GACGTGGAACTGGCAGAAGA-3′ | 5′-ACTGATGAGAGGGAGGCCAT-3′ |
| Arg1 | 5′-TCACCTGAGCTTTGATGTCG-3′ | 5′-CTGAAAGGAGCCCTGTCTTG-3′ |
| IL-10 | 5′-CCAAGCCTTATCGGAAATGA-3′ | 5′-TTTTCACAGGGGAGAAATCG-3′ |
| TGFβ | 5′-GGCGATACCTCAGCAACCG-3′ | 5′-CTAAGGCGAAAGCCCTCAAT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zhao, J.; Gao, H. Depletion of Arg1-Positive Microglia/Macrophages Exacerbates Cerebral Ischemic Damage by Facilitating the Inflammatory Response. Int. J. Mol. Sci. 2022, 23, 13055. https://doi.org/10.3390/ijms232113055
Li T, Zhao J, Gao H. Depletion of Arg1-Positive Microglia/Macrophages Exacerbates Cerebral Ischemic Damage by Facilitating the Inflammatory Response. International Journal of Molecular Sciences. 2022; 23(21):13055. https://doi.org/10.3390/ijms232113055
Chicago/Turabian StyleLi, Ting, Jin Zhao, and Hao Gao. 2022. "Depletion of Arg1-Positive Microglia/Macrophages Exacerbates Cerebral Ischemic Damage by Facilitating the Inflammatory Response" International Journal of Molecular Sciences 23, no. 21: 13055. https://doi.org/10.3390/ijms232113055
APA StyleLi, T., Zhao, J., & Gao, H. (2022). Depletion of Arg1-Positive Microglia/Macrophages Exacerbates Cerebral Ischemic Damage by Facilitating the Inflammatory Response. International Journal of Molecular Sciences, 23(21), 13055. https://doi.org/10.3390/ijms232113055