
An Ultra-Rare Manifestation of an X-Linked Recessive Disorder: Duchenne Muscular Dystrophy in a Female Patient

 , , ,
, , ,
Abstract
1. Introduction
1.1. Duchenne Muscular Dystrophy
1.2. DMD Carrier Females
1.3. Reciprocal Translocations
1.4. X Chromosome Inactivation
1.5. Translocations in Females with DMD Phenotype—Previously Reported Cases in the Literature
2. Results
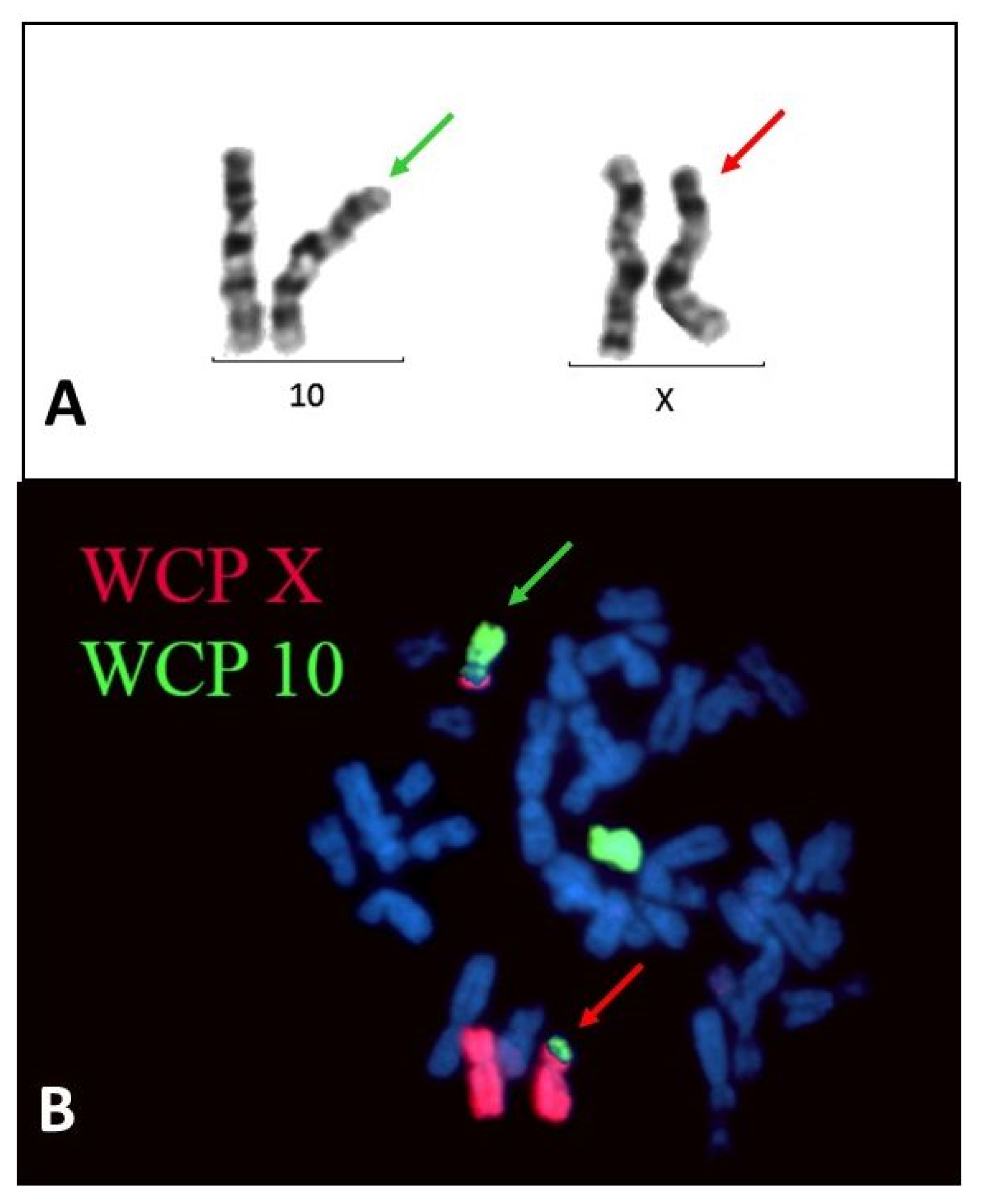
2.1. Case Presentation
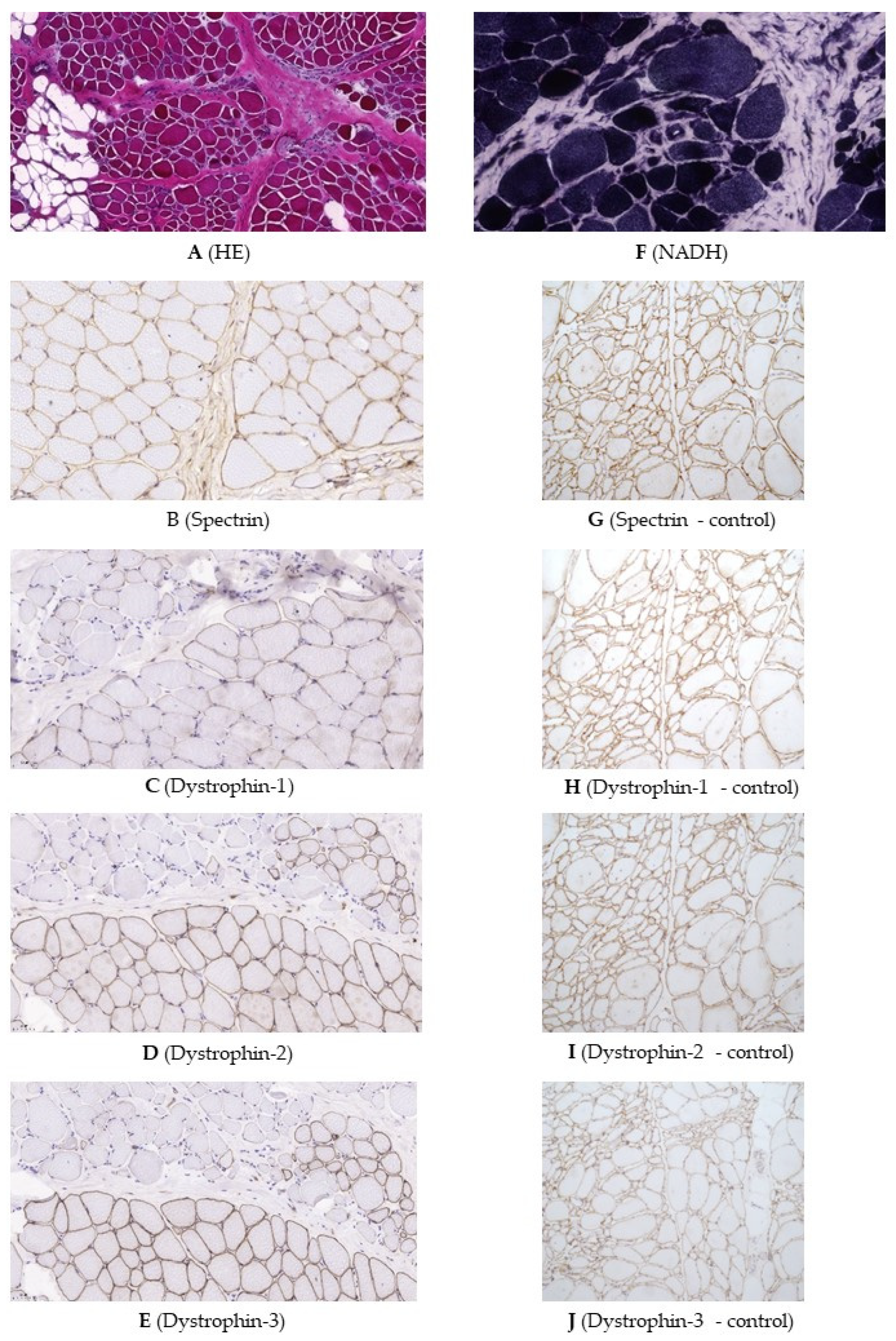
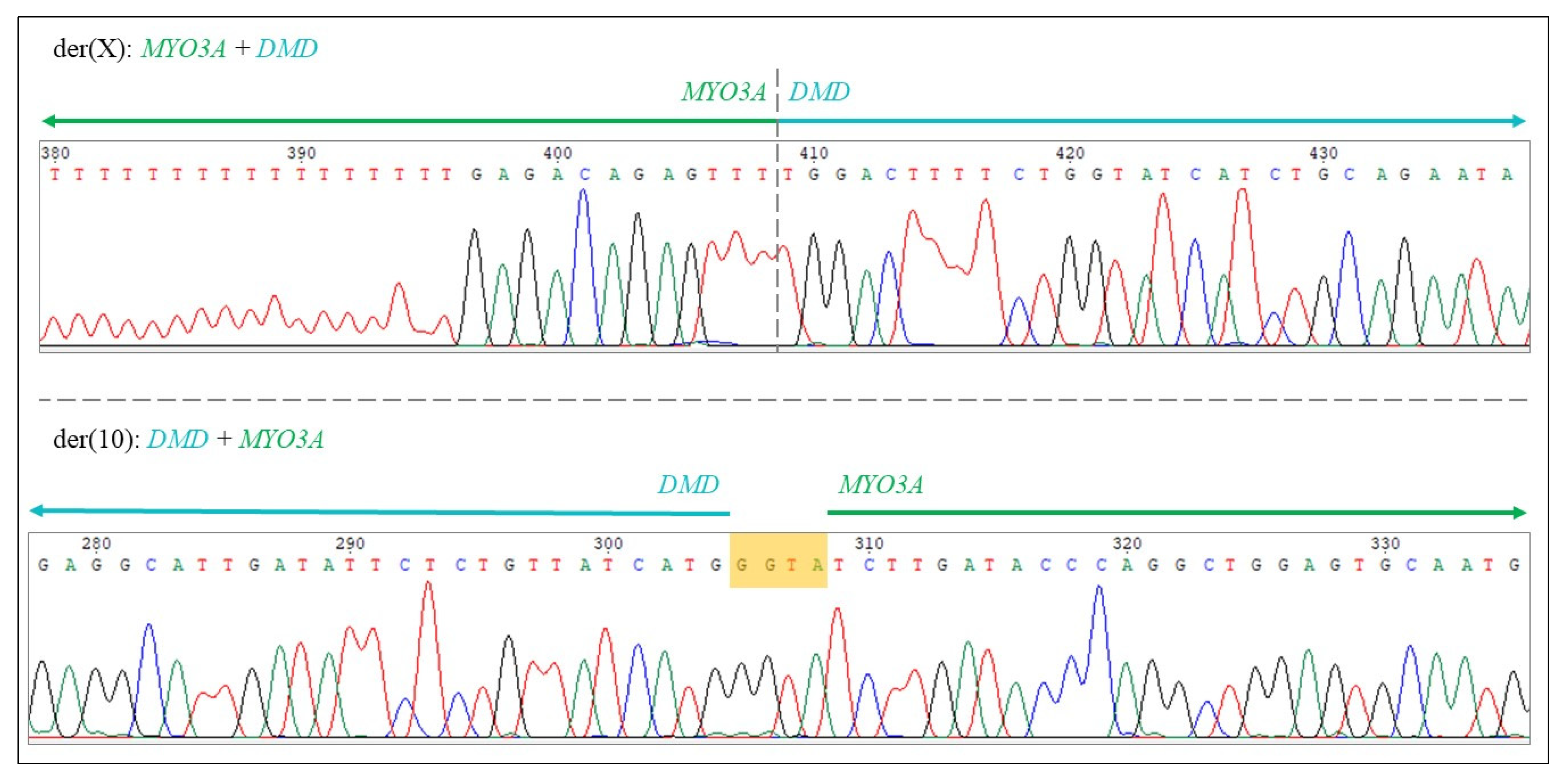
2.2. Genetic Testing and Muscle Biopsy
3. Discussion
4. Methods and Materials
4.1. Methods
4.1.1. Molecular Genetic Testing
4.1.2. Muscle Histology and Dystrophin Immunohistochemistry
4.1.3. Bioinformatical Analysis, Determination of Translocation Breakpoints
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szabo, S.M.; Salhany, R.M.; Deighton, A.; Harwood, M.; Mah, J.; Gooch, K.L. The Clinical Course of Duchenne Muscular Dystrophy in the Corticosteroid Treatment Era: A Systematic Literature Review. Orphanet J. Rare Dis. 2021, 16, 237. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Kornberg, A.J. Duchenne Muscular Dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The Importance of Genetic Diagnosis for Duchenne Muscular Dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An Explanation for the Phenotypic Differences between Patients Bearing Partial Deletions of the DMD Locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Dowling, J.J.; Weihl, C.C.; Spencer, M.J. Molecular and Cellular Basis of Genetically Inherited Skeletal Muscle Disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 713–732. [Google Scholar] [CrossRef]
- Rando, T.A. The Dystrophin-Glycoprotein Complex, Cellular Signaling, and the Regulation of Cell Survival in the Muscular Dystrophies. Muscle Nerve 2001, 24, 1575–1594. [Google Scholar] [CrossRef]
- Wein, N.; Alfano, L.; Flanigan, K.M. Genetics and Emerging Treatments for Duchenne and Becker Muscular Dystrophy. Pediatr. Clin. North Am. 2015, 62, 723–742. [Google Scholar] [CrossRef]
- Le Rumeur, E. Dystrophin and the Two Related Genetic Diseases, Duchenne and Becker Muscular Dystrophies. Bosn. J. Basic Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef]
- Hoogerwaard, E.M.; Bakker, E.; Ippel, P.F.; Oosterwijk, J.C.; Majoor-Krakauer, D.F.; Leschot, N.J.; Van Essen, A.J.; Brunner, H.G.; van der Wouw, P.A.; Wilde, A.A.; et al. Signs and Symptoms of Duchenne Muscular Dystrophy and Becker Muscular Dystrophy among Carriers in The Netherlands: A Cohort Study. Lancet 1999, 353, 2116–2119. [Google Scholar] [CrossRef]
- Bourke, J.; Turner, C.; Bradlow, W.; Chikermane, A.; Coats, C.; Fenton, M.; Ilina, M.; Johnson, A.; Kapetanakis, S.; Kuhwald, L.; et al. Cardiac Care of Children with Dystrophinopathy and Females Carrying DMD-Gene Variations. Open Heart 2022, 9, e001977. [Google Scholar] [CrossRef] [PubMed]
- Gruber, D.; Lloyd-Puryear, M.; Armstrong, N.; Scavina, M.; Tavakoli, N.P.; Brower, A.M.; Caggana, M.; Chung, W.K. Newborn Screening for Duchenne Muscular Dystrophy-Early Detection and Diagnostic Algorithm for Female Carriers of Duchenne Muscular Dystrophy. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 197–205. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Ellard, S.; Cleaver, R. Emery’s Elements of Medical Genetics and Genomics; Elsevier: Amsterdam, The Netherlands, 2022; ISBN 978-0-7020-7966-5. [Google Scholar]
- Matthews, P.M.; Karpati, G. Pattern of X-Chromosome Inactivation as a Key Determinant of the Clinicopathologic Phenotype of Duchenne Muscular Dystrophy Carriers. Neurology 1996, 46, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.N.; Choi, Y.-C. Female Carriers of Duchenne Muscular Dystrophy. J. Genet. Med. 2013, 10, 94–98. [Google Scholar] [CrossRef]
- Amos-Landgraf, J.M.; Cottle, A.; Plenge, R.M.; Friez, M.; Schwartz, C.E.; Longshore, J.; Willard, H.F. X Chromosome–Inactivation Patterns of 1,005 Phenotypically Unaffected Females. Am. J. Hum. Genet. 2006, 79, 493–499. [Google Scholar] [CrossRef]
- Boyd, Y.; Buckle, V.; Holt, S.; Munro, E.; Hunter, D.; Craig, I. Muscular Dystrophy in Girls with X;Autosome Translocations. J. Med. Genet. 1986, 23, 484–490. [Google Scholar] [CrossRef]
- Greenstein, R.M.; Reardon, M.P.; Chan, T.S.; Middleton, A.B.; Mulivor, R.A.; Greene, A.E.; Coriell, L.L. An (X;11) Translocation in a Girl with Duchenne Muscular Dystrophy. Cytogenet. Cell Genet. 1980, 27, 268. [Google Scholar] [CrossRef]
- Greenstein, R.M.; Reardon, M.P.; Chan, T.S. An X/Autosome Translocation in a Girl with Duchenne Muscular Dystrophy (Dmd): Evidence for Dmd Gene Localization. Pediatr. Res. 1977, 11, 457. [Google Scholar] [CrossRef]
- Lindenbaum, R.H.; Clarke, G.; Patel, C.; Moncrieff, M.; Hughes, J.T. Muscular Dystrophy in an X; 1 Translocation Female Suggests That Duchenne Locus Is on X Chromosome Short Arm. J. Med. Genet. 1979, 16, 389–392. [Google Scholar] [CrossRef][Green Version]
- Jacobs, P.A.; Hunt, P.A.; Mayer, M.; Bart, R.D. Duchenne Muscular Dystrophy (DMD) in a Female with an X/Autosomal Translocation: Further Evidence That the DMD Locus Is at Xp21. Am. J. Hum. Genet. 1981, 33, 513–518. [Google Scholar]
- Zatz, M.; Vianna-Morgante, A.M.; Campos, P.; Diament, A.J. Translocation (X;6) in a Female with Duchenne Muscular Dystrophy: Implications for the Localisation of the DMD Locus. J. Med. Genet. 1981, 18, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, B.S.; Zackai, E.H.; Tucker, S.H. Further Evidence for Xp21 Location of Duchenne Muscular Dystrophy (DMD) Locus: X;9 Translocation in a Female with DMD. J. Med. Genet. 1983, 20, 461–463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bjerglund Nielsen, L.; Nielsen, I.M. Turner’s Syndrome and Duchenne Muscular Dystrophy in a Girl with an X.; Autosome Translocation. Ann. Genet. 1984, 27, 173–177. [Google Scholar]
- Verellen-Dumoulin, C.; Freund, M.; De Meyer, R.; Laterre, C.; Frédéric, J.; Thompson, M.W.; Markovic, V.D.; Worton, R.G. Expression of an X-Linked Muscular Dystrophy in a Female Due to Translocation Involving Xp21 and Non-Random Inactivation of the Normal X Chromosome. Hum. Genet. 1984, 67, 115–119. [Google Scholar] [CrossRef]
- Kimura, S.; Mitsuda, T.; Misugi, N.; Saito, F.; Tonomura, A.; Sugita, H. Clinical Features in a Girl with Duchenne Muscular Dystrophy with an X-Autosome Translocation; (X;4)(P21;Q26). Brain Dev. 1986, 8, 619–623. [Google Scholar] [CrossRef]
- Saito, F.; Tonomura, A.; Kimura, S.; Misugi, N.; Sugita, H. High-Resolution Banding Study of an X/4 Translocation in a Female with Duchenne Muscular Dystrophy. Hum. Genet. 1985, 71, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.J.; Smith, A.; MacLeod, P.M.; Masotti, R.; Duncan, A.M. Xp21/Autosome Translocations. Case Report and Risk for Duchenne Muscular Dystrophy. Clin. Genet. 1986, 29, 516–522. [Google Scholar] [CrossRef] [PubMed]
- van Bakel, I.; Holt, S.; Craig, I.; Boyd, Y. Sequence Analysis of the Breakpoint Regions of an X;5 Translocation in a Female with Duchenne Muscular Dystrophy. Am. J. Hum. Genet. 1995, 57, 329–336. [Google Scholar]
- Nevin, N.C.; Hughes, A.E.; Calwell, M.; Lim, J.H. Duchenne Muscular Dystrophy in a Female with a Translocation Involving Xp21. J. Med. Genet. 1986, 23, 171–173. [Google Scholar] [CrossRef][Green Version]
- Ribeiro, M.C.M.; Melaragno, M.I.; Schmidt, B.; Brunoni, D.; Gabbai, A.A.; Hackel, C.; Reynolds, J.F. Duchenne Muscular Dystrophy in a Girl with an (X;15) Translocation. Am. J. Med. Genet. 1986, 25, 231–236. [Google Scholar] [CrossRef]
- Bodrug, S.E.; Roberson, J.R.; Weiss, L.; Ray, P.N.; Worton, R.G.; Van Dyke, D.L. Prenatal Identification of a Girl with a t(X;4)(P21;Q35) Translocation: Molecular Characterisation, Paternal Origin, and Association with Muscular Dystrophy. J. Med. Genet. 1990, 27, 426–432. [Google Scholar] [CrossRef]
- Robinson, D.O.; Boyd, Y.; Cockburn, D.; Collinson, M.N.; Craig, I.; Jacobs, P.A. The Parental Origin of de Novo X-Autosome Translocations in Females with Duchenne Muscular Dystrophy Revealed by M27 Beta Methylation Analysis. Genet. Res. 1990, 56, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, J.P.; Francke, U. Common Sequence Motifs at the Rearrangement Sites of a Constitutional X/Autosome Translocation and Associated Deletion. Am. J. Hum. Genet. 1992, 50, 725–741. [Google Scholar]
- Hausmanowa-Petrusewicz, I.; Zaremba, J.; Fidziańska, A.; Zimowski, J.; Bisko, M.; Badurska, B.; Fidziańska, E.; Lusakowska, A.; Borkowska, J. Interrelationship between Gene, Its Product and Phenotype in Duchenne and Becker Muscular Dystrophy. Acta Neurobiol. Exp. 1993, 53, 297–303. [Google Scholar] [PubMed]
- Wenger, S.L.; Steele, M.W.; Hoffman, E.P.; Barmada, M.A.; Wessel, H.B. X Inactivation and Dystrophin Studies in a t(X;12) Female: Evidence for Biochemical Normalization in Duchenne Muscular Dystrophy Carriers. Am. J. Med. Genet. 1992, 43, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Seemann, N.; Selby, K.; McAdam, L.; Biggar, D.; Kolski, H.; Goobie, S.; Yoon, G.; Campbell, C. Canadian Pediatric Neuromuscular Group Symptomatic Dystrophinopathies in Female Children. Neuromuscul. Disord. 2011, 21, 172–177. [Google Scholar] [CrossRef]
- Brioschi, S.; Gualandi, F.; Scotton, C.; Armaroli, A.; Bovolenta, M.; Falzarano, M.S.; Sabatelli, P.; Selvatici, R.; D’Amico, A.; Pane, M.; et al. Genetic Characterization in Symptomatic Female DMD Carriers: Lack of Relationship between X-Inactivation, Transcriptional DMD Allele Balancing and Phenotype. BMC Med. Genet. 2012, 13, 73. [Google Scholar] [CrossRef]
- Viggiano, E.; Picillo, E.; Politano, L. DMD Phenotype in Girls with a de Novo Balanced X;3 Autosome Translocation: A Case Report. J. Genet. Syndr. Gene Ther. 2013, 4, 1000132. [Google Scholar] [CrossRef]
- Trippe, H.; Wieczorek, S.; Kötting, J.; Kress, W.; Schara, U. Xp21/A Translocation: A Rarely Considered Genetic Cause for Manifesting Carriers of Duchenne Muscular Dystrophy. Neuropediatrics 2014, 45, 333–335. [Google Scholar] [CrossRef]
- Nozoe, K.T.; Akamine, R.T.; Mazzotti, D.R.; Polesel, D.N.; Grossklauss, L.F.; Tufik, S.; Andersen, M.L.; Moreira, G.A. Phenotypic Contrasts of Duchenne Muscular Dystrophy in Women: Two Case Reports. Sleep Sci. 2016, 9, 129–133. [Google Scholar] [CrossRef]
- Schluth-Bolard, C.; Diguet, F.; Chatron, N.; Rollat-Farnier, P.-A.; Bardel, C.; Afenjar, A.; Amblard, F.; Amiel, J.; Blesson, S.; Callier, P.; et al. Whole Genome Paired-End Sequencing Elucidates Functional and Phenotypic Consequences of Balanced Chromosomal Rearrangement in Patients with Developmental Disorders. J. Med. Genet. 2019, 56, 526–535. [Google Scholar] [CrossRef]
- Toksoy, G.; Durmus, H.; Aghayev, A.; Bagirova, G.; Sevinc Rustemoglu, B.; Basaran, S.; Avci, S.; Karaman, B.; Parman, Y.; Altunoglu, U.; et al. Mutation Spectrum of 260 Dystrophinopathy Patients from Turkey and Important Highlights for Genetic Counseling. Neuromuscul. Disord. 2019, 29, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, F.; Al-Tawari, A.; Alrohaif, H.; Alshuaibi, W.; Mansour, H.; Aartsma-Rus, A.; Mégarbané, A. Case Report: The Genetic Diagnosis of Duchenne Muscular Dystrophy in the Middle East. Front. Pediatr. 2021, 9, 716424. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Walsh, T.; Walsh, V.; Vreugde, S.; Hertzano, R.; Shahin, H.; Haika, S.; Lee, M.K.; Kanaan, M.; King, M.-C.; Avraham, K.B. From Flies’ Eyes to Our Ears: Mutations in a Human Class III Myosin Cause Progressive Nonsyndromic Hearing Loss DFNB30. Proc. Natl. Acad. Sci. USA 2002, 99, 7518–7523. [Google Scholar] [CrossRef]
- Sewry, C.A.; Goebel, H.H. General Pathology of Muscle Disease. In Muscle Disease; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 19–38. ISBN 978-1-118-63546-9. [Google Scholar]
- Koczok, K.; Merő, G.; Szabó, G.P.; Madar, L.; Gombos, É.; Ajzner, É.; Mótyán, J.A.; Hortobágyi, T.; Balogh, I. A Novel Point Mutation Affecting Asn76 of Dystrophin Protein Leads to Dystrophinopathy. Neuromuscul. Disord. 2018, 28, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, Y.; Einhorn, M.; Kamshov, A.; Lev, O.; Trabelsi, A.; Paz-Yaacov, N.; Gross, S.J. Gene-Specific Artificial Intelligence-Based Variant Classification Engine: Results of a Time-Capsule Experiment. Res. Sq. 2019; in press. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural Variant Discovery by Integrated Paired-End and Split-Read Analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Translocation | Additional Information | Reference |
|---|---|---|---|
| 1 | t(X;11) | breakpoints: Xp21 and 11q13 | [18,19] |
| 2 | t(X;1) | [20] | |
| 3 | t(X;5)(p21;q35) | de novo | [21] |
| 4 | t(X;6)(p21;q21) | [22] | |
| 5 | t(X;9)(p21;p22) | [23] | |
| 6 | t(X;3)(p21;ql3) | de novo | [23] |
| 7 | t(X;9) | de novo | [24] |
| 8 | t(X;21)(p21;p12) | [25] | |
| 9 | t(X;4)(p21.1;q26) | de novo | [26,27] |
| 10 | t(X;2)(p21.2;q37) | de novo | [28] |
| 11 | t(X;5)(p21-2;q31-2) | DMD gene disruption in intron flanked by exons 51 and 52 | [29,30] |
| 12 | t(X;15)(p21;q26) | de novo | [31] |
| 13 | t(X;4)(p21;q35) | de novo, prenatal diagnosis | [32] |
| 14 | t(X;9)(p21-2;q21-3) | de novo | [33] |
| 15 | t(X;4)(p21;q31) | [34] | |
| 16 | t(X;22) | breakpoint in Xp21.2 | [35] |
| 17 | t(X;12)(p21.2;q24.33) | de novo | [36] |
| 18 | t(X;7)(p21.2;p15.1) | skewed X-inactivation | [37] |
| 19 | t(X;9)(p21.1;p22.1) | array-CGH: no CNV within the DMD gene | [38] |
| 20 | t(X;3)(p21;p24) | de novo, prenatal diagnosis | [39] |
| 21 | t(X;4)(p21;q31) | de novo, arr(1-22,X)x2 | [40] |
| 22 | t(X;4)(p21;q13) | array-CGH: no CNV within the DMD gene | [41] |
| 23 | t(X;13;15)(p21;q22;q22), t(6;11)(q21;q24) | WGS: DMD gene disruption | [42] |
| 24 | t(X;9)(p.21.1;q12) | DMD gene sequencing: DMD gene disruption | [43] |
| 25 | t(X;1)(p21.3;p22.2) | de novo | [44] |
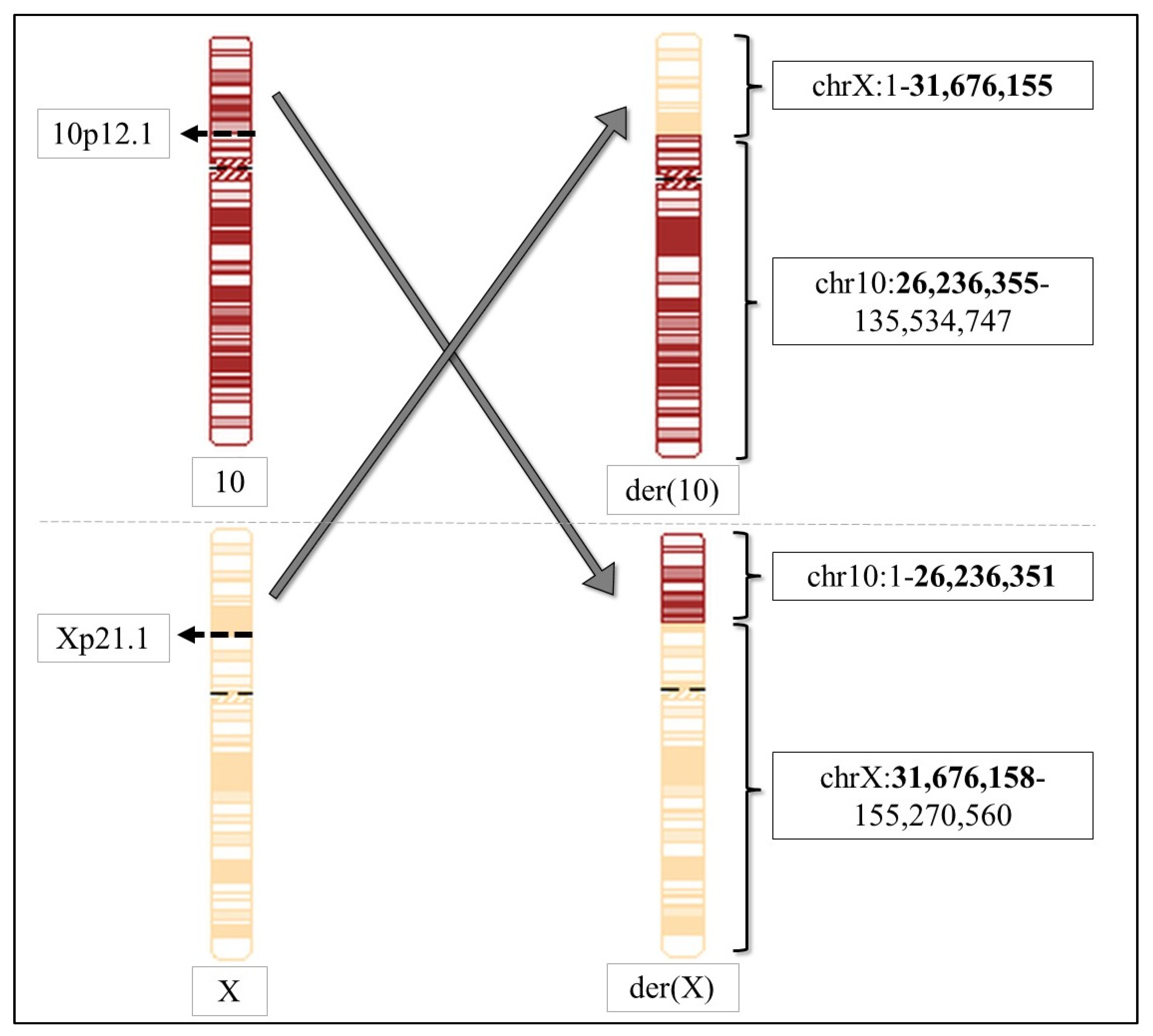
| 26 | t(X;10)(p21.1;p12.1) | array-CGH: two VUS inherited from two parents | this study |
| der (10) | der (X) | |
|---|---|---|
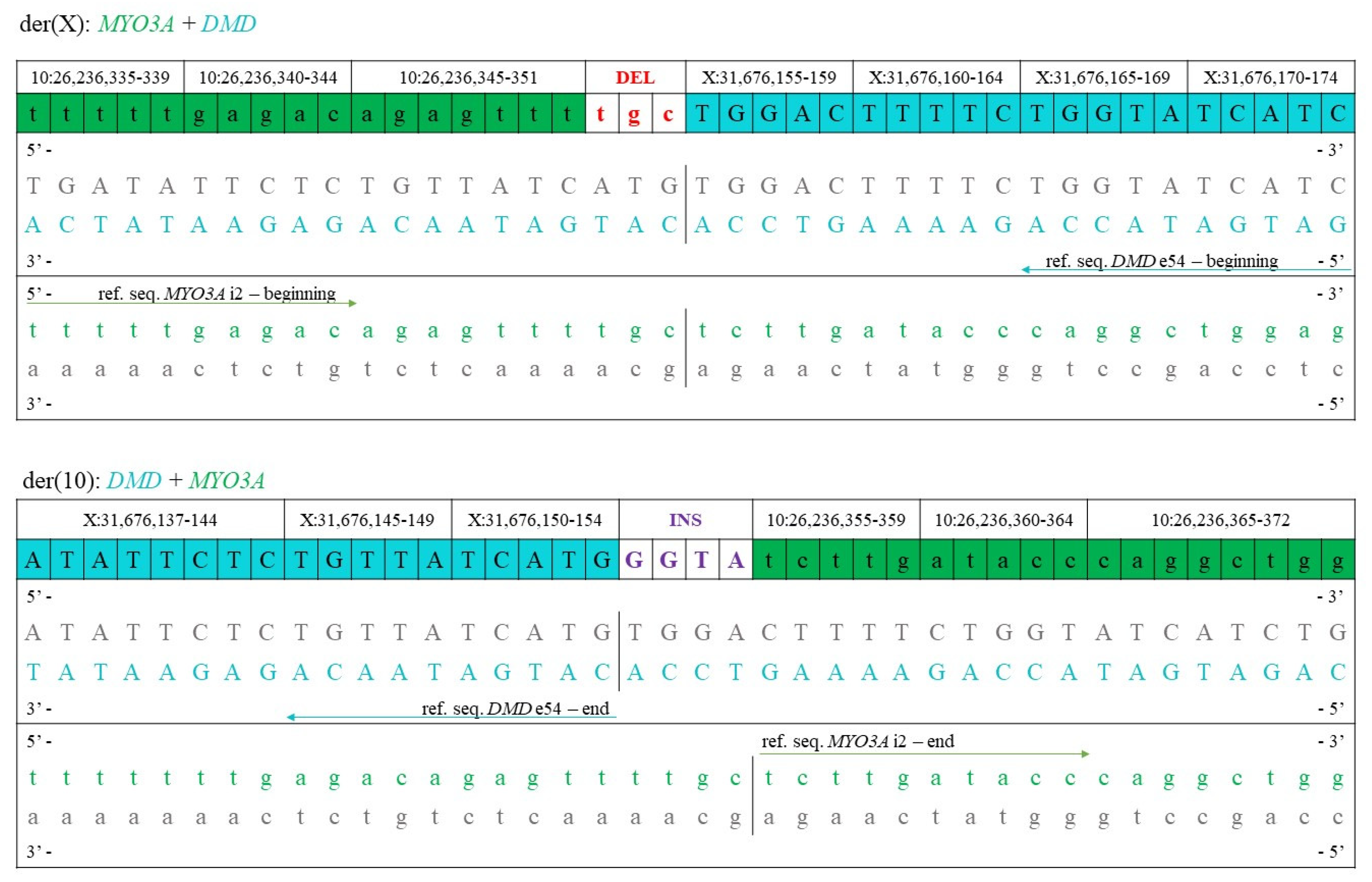
| Breakpoint in chrX | Xp21.1 | Xp21.1 |
| Coordinates of chrX | 1–31,676,155 | 31,676,158–155,270,560 |
| Breakpoint in chr10 | 10p12.1 | 10p12.1 |
| Coordinates of chr10 | 26,236,355–135,534,747 | 1–26,236,351 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szűcs, Z.; Pinti, É.; Haltrich, I.; Szén, O.P.; Nagy, T.; Barta, E.; Méhes, G.; Bidiga, L.; Török, O.; Ujfalusi, A.; et al. An Ultra-Rare Manifestation of an X-Linked Recessive Disorder: Duchenne Muscular Dystrophy in a Female Patient. Int. J. Mol. Sci. 2022, 23, 13076. https://doi.org/10.3390/ijms232113076
Szűcs Z, Pinti É, Haltrich I, Szén OP, Nagy T, Barta E, Méhes G, Bidiga L, Török O, Ujfalusi A, et al. An Ultra-Rare Manifestation of an X-Linked Recessive Disorder: Duchenne Muscular Dystrophy in a Female Patient. International Journal of Molecular Sciences. 2022; 23(21):13076. https://doi.org/10.3390/ijms232113076
Chicago/Turabian StyleSzűcs, Zsuzsanna, Éva Pinti, Irén Haltrich, Orsolya Pálné Szén, Tibor Nagy, Endre Barta, Gábor Méhes, László Bidiga, Olga Török, Anikó Ujfalusi, and et al. 2022. "An Ultra-Rare Manifestation of an X-Linked Recessive Disorder: Duchenne Muscular Dystrophy in a Female Patient" International Journal of Molecular Sciences 23, no. 21: 13076. https://doi.org/10.3390/ijms232113076
APA StyleSzűcs, Z., Pinti, É., Haltrich, I., Szén, O. P., Nagy, T., Barta, E., Méhes, G., Bidiga, L., Török, O., Ujfalusi, A., Koczok, K., & Balogh, I. (2022). An Ultra-Rare Manifestation of an X-Linked Recessive Disorder: Duchenne Muscular Dystrophy in a Female Patient. International Journal of Molecular Sciences, 23(21), 13076. https://doi.org/10.3390/ijms232113076