
Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Structure of Arteriviruses and the Function of Their Membrane Proteins
1.2. Primary Structure and Modifications of Gp5 and M
1.3. Immune Evasion and Persistence of PRRSV
1.4. Alphafold2: An Artificial-Intelligence-Based Method to Predict Protein Structures De Novo
2. Results
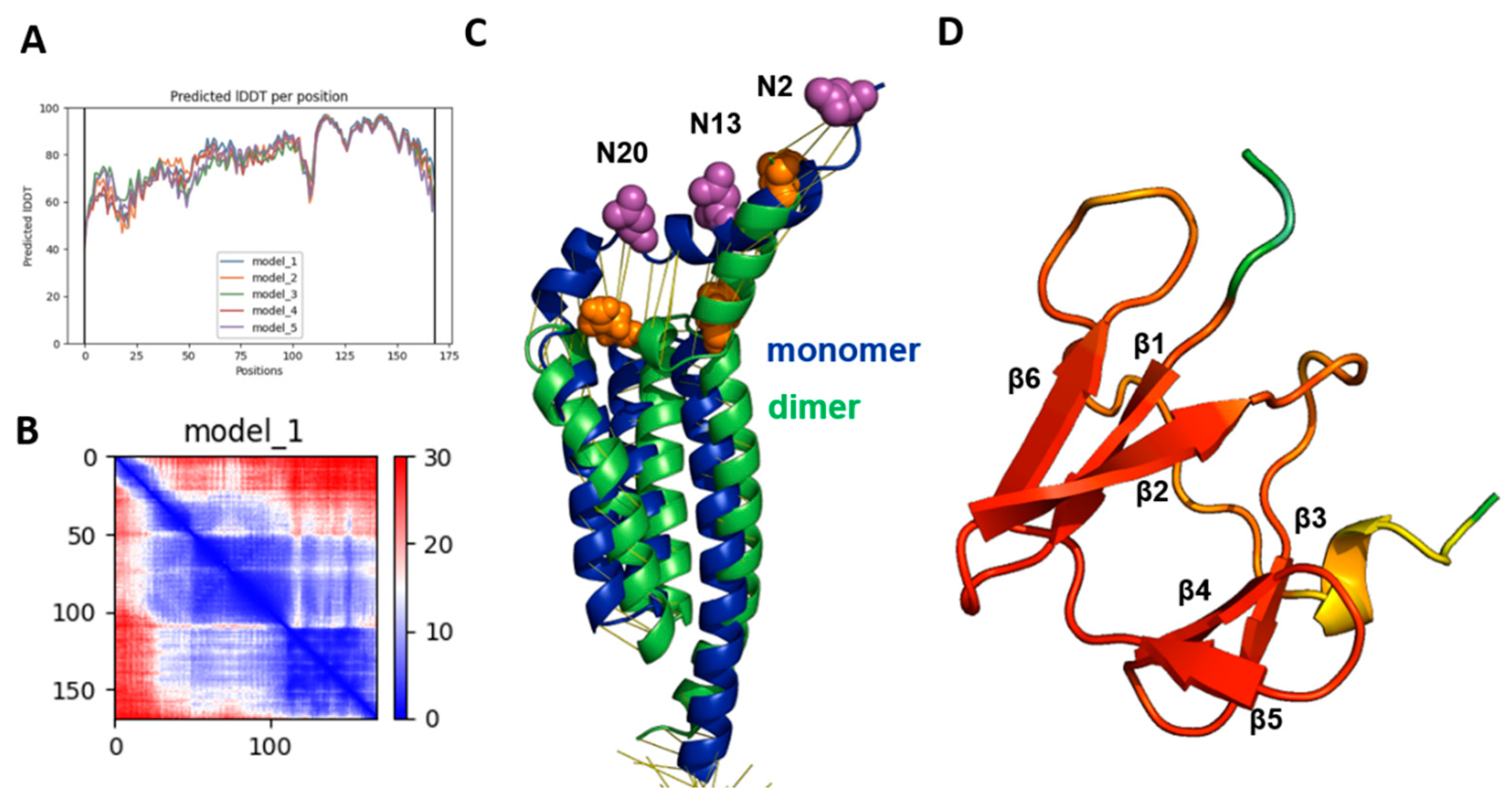
2.1. Evaluation of the Quality of the Gp5/M Dimer Model
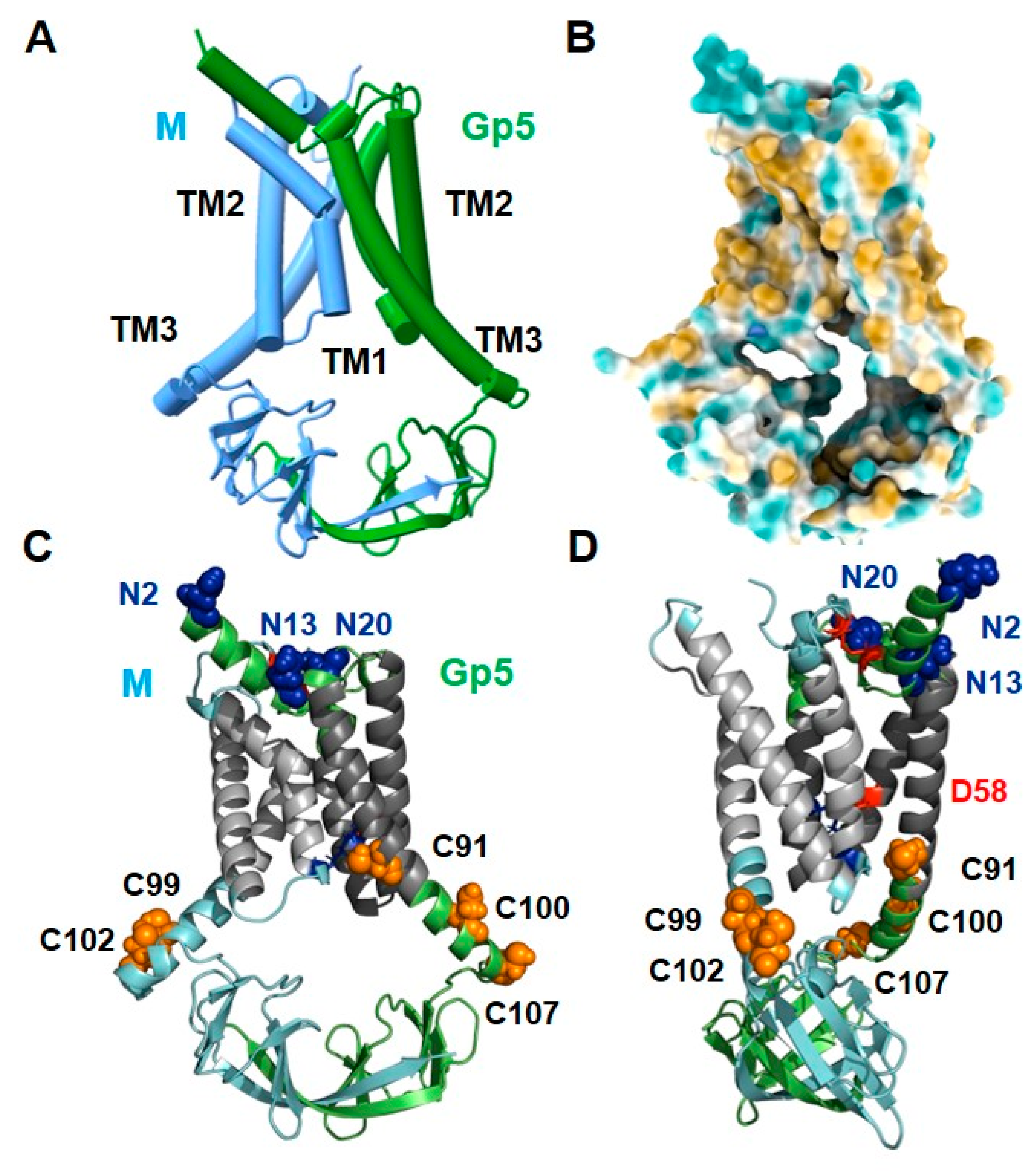
2.2. The Structure of Gp5/M of the PRRSV-2 Reference Strain VR 2332
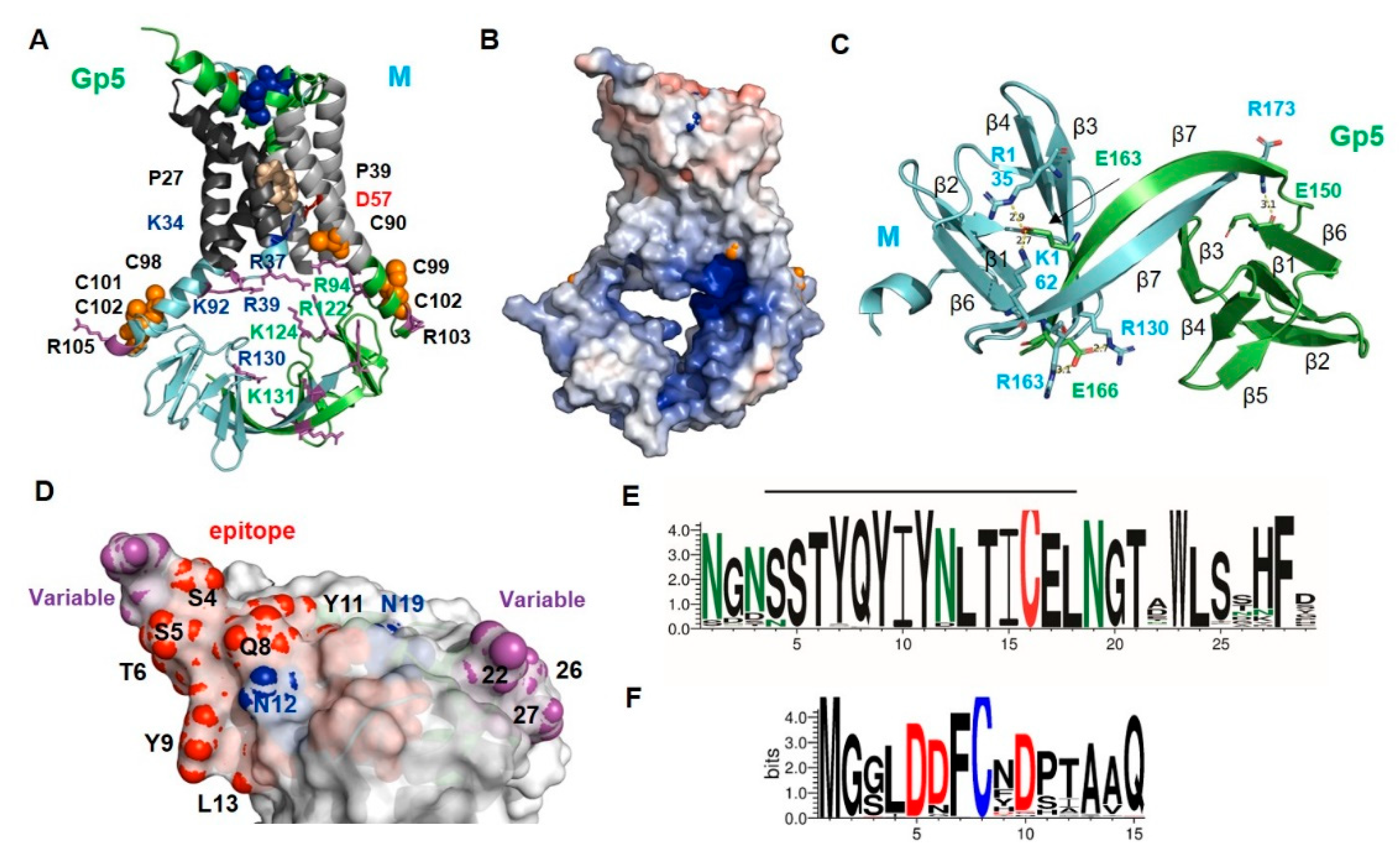
2.3. Structure and Diversity of the Ectodomain of Gp5/M of VR 2332 and Its Epitopes
2.4. Structure of the Longer Ectodomain of Gp5/M of VR 2332 Containing the Decoy Epitope
2.5. Predicted Signal Peptide Cleavage Sites in Gp5 Proteins of PRRSV-2 Strains
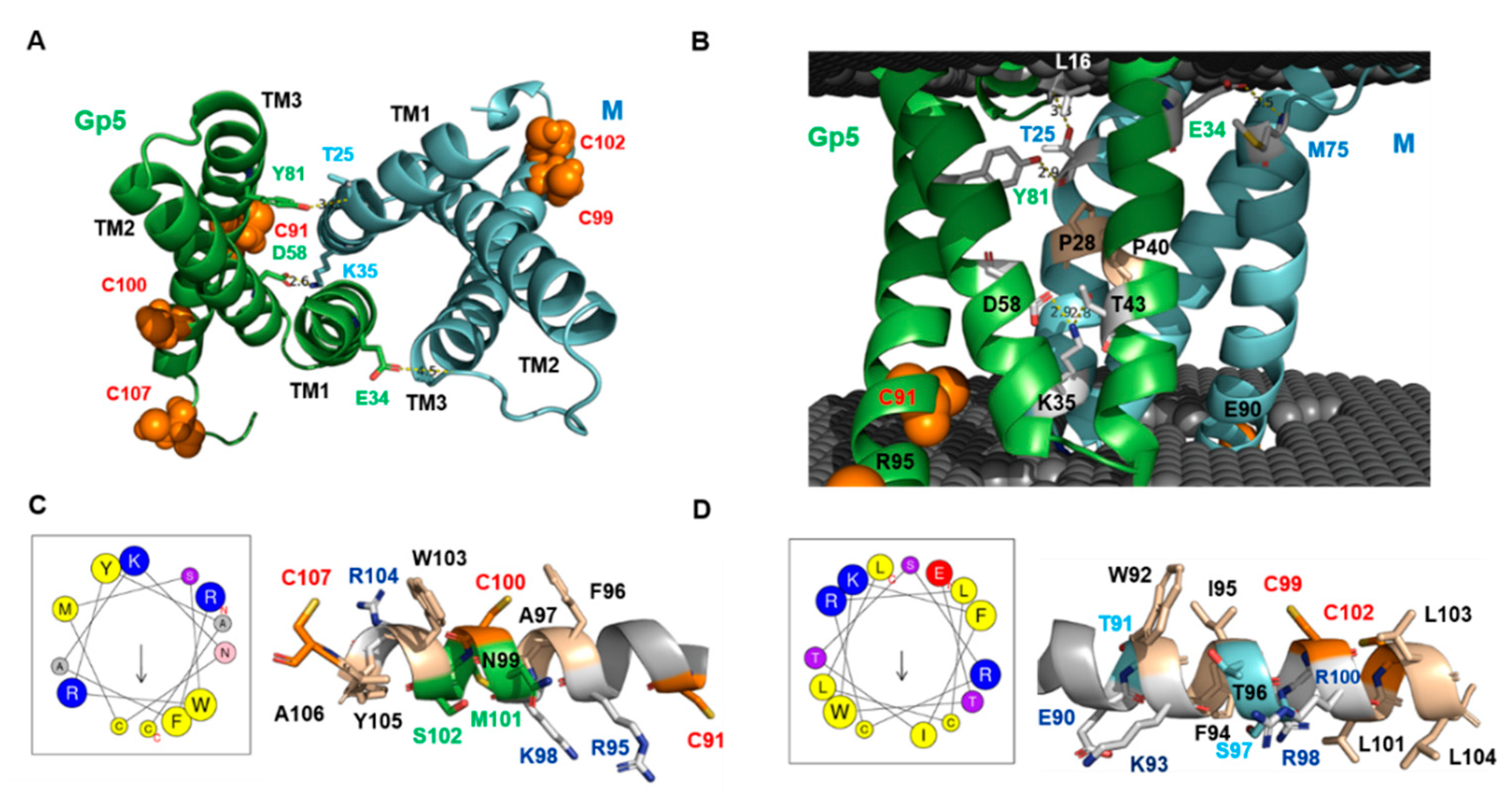
2.6. Peculiar Features of the Transmembrane Region of Gp5/M of VR 2332
2.7. The Basic Endodomain of Gp5/M of VR 2332
2.8. Model of the Gp5/M Dimer of the PRRSV-1 Reference Strain Lelystad
2.9. Model of Monomeric Gp5 Suggests Conformational Changes Occurring upon Dimerization
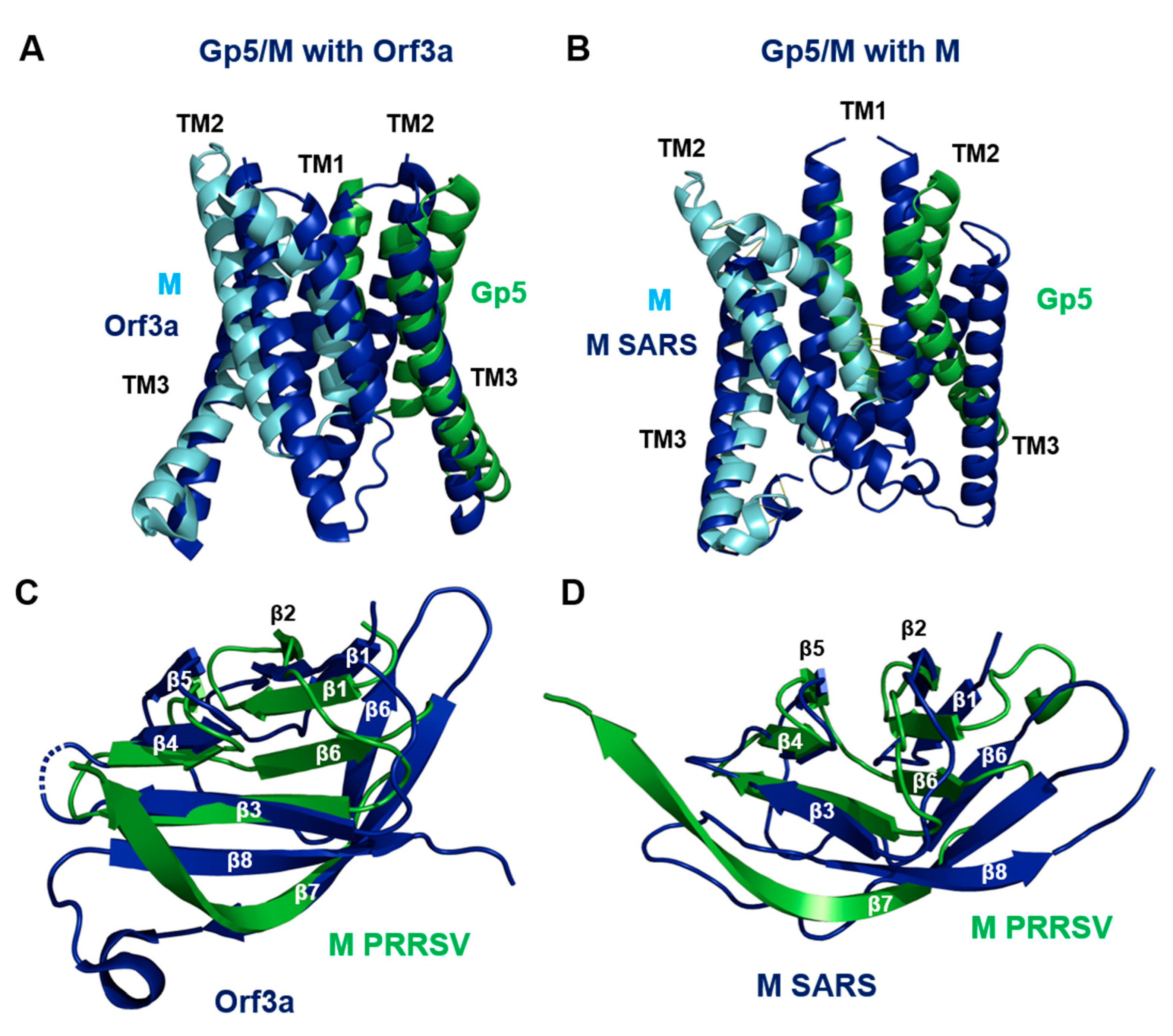
2.10. Gp5/M Has a Similar Structure as the Orf3a and M Proteins of SARS-CoV-2
2.11. Orf3a Is Palmitoylated at a Cluster of Cysteine Residues near the C-Terminal Part of TM3
3. Discussion
3.1. Role of the Gp5/M Ectodomain for Neutralization of PRRSV
3.2. Heterogeneity in Signal Peptide Cleavage Sites of Gp5 of PRRSV
3.3. The Function of the Transmembrane Region in Virus Assembly and Budding
3.4. A putative Role of the Endodomain in Genome Recruitment and as a Protein Binding Site
3.5. Gp5 and M Are Members of a Protein Superfamily That Includes M and Orf3a-like Proteins from Coronaviruses
4. Materials and Methods
4.1. Predictions of Protein Structures by Alphafold2
4.2. Assessment of the Quality of the Model of the Gp5/M Dimer
4.3. Visualization and Analysis of Predicted Gp5/M Structures
4.4. Calculation of the Position of Gp5/M within a Virtual Lipid Bilayer
4.5. Sequence Conservation Analysis
4.6. Prediction of Signal Peptide Cleavage Sites in Gp5 of PRRSV-1 and PRRSV-2
4.7. Predicting the Amphiphilic Properties of the C-Terminus of Transmembrane Helix 3
4.8. Docking of a Peptide from the Endodomain of Gp5/M to the Viral N-Protein
4.9. Analysis of Orf3a and Gp5/M for the Presence of Cavities, Tunnels and Pores
4.10. Mutagenesis, Expression and Acylation Analysis of the Orf3a Protein of SARS-CoV-2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chand, R.J.; Trible, B.R.; Rowland, R.R. Pathogenesis of Porcine Reproductive and Respiratory Syndrome Virus. Curr. Opin. Virol. 2012, 2, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Lauck, M.; Bailey, A.L.; Shchetinin, A.M.; Vishnevskaya, T.V.; Bao, Y.; Ng, T.F.; LeBreton, M.; Schneider, B.S.; Gillis, A.; et al. Reorganization and Expansion of the Nidoviral Family Arteriviridae. Arch. Virol. 2016, 161, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, K.; Yu, X.; Zhao, T.; Feng, Y.; Cao, Z.; Wang, C.; Hu, Y.; Chen, X.; Hu, D.; Tian, X.; et al. Emergence of Fatal Prrsv Variants: Unparalleled Outbreaks of Atypical Prrs in China and Molecular Dissection of the Unique Hallmark. PLoS ONE 2007, 2, e526. [Google Scholar] [CrossRef] [PubMed]
- Karniychuk, U.U.; Geldhof, M.; Vanhee, M.; van Doorsselaere, J.; Saveleva, T.A.; Nauwynck, H.J. Pathogenesis and Antigenic Characterization of a New East European Subtype 3 Porcine Reproductive and Respiratory Syndrome Virus Isolate. BMC Vet. Res. 2010, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasuriya, U.B.; Go, Y.Y.; MacLachlan, J.N. Equine Arteritis Virus. Vet. Microbiol. 2013, 167, 93–122. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Lauck, M.; Sibley, S.D.; Friedrich, T.C.; Kuhn, J.H.; Freimer, N.B.; Jasinska, A.J.; Phillips-Conroy, J.E.; Jolly, C.J.; Marx, P.A.; et al. Zoonotic Potential of Simian Arteriviruses. J. Virol. 2016, 90, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Dokland, T. The Structural Biology of Prrsv. Virus Res. 2010, 154, 86–97. [Google Scholar] [CrossRef]
- Veit, M.; Matczuk, A.K.; Sinhadri, B.C.; Krause, E.; Thaa, B. Membrane Proteins of Arterivirus Particles: Structure, Topology, Processing and Function. Virus Res 2014, 194, 16–36. [Google Scholar] [CrossRef]
- Snijder, E.J.; Kikkert, M.; Fang, Y. Arterivirus Molecular Biology and Pathogenesis. J. Gen. Virol. 2013, 94 Pt 10, 2141–2163. [Google Scholar] [CrossRef]
- Wieringa, R.; de Vries, A.A.; van der Meulen, J.; Godeke, G.J.; Onderwater, J.J.; van Tol, H.; Koerten, H.K.; Mommaas, A.M.; Snijder, E.J.; Rottier, P.J. Structural Protein Requirements in Equine Arteritis Virus Assembly. J. Virol. 2004, 78, 13019–13027. [Google Scholar] [CrossRef]
- Wissink, E.H.; Kroese, M.V.; van Wijk, H.A.; Rijsewijk, F.A.; Meulenberg, J.J.; Rottier, P.J. Envelope Protein Requirements for the Assembly of Infectious Virions of Porcine Reproductive and Respiratory Syndrome Virus. J. Virol. 2005, 79, 12495–12506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbe, J.C.; van der Meer, Y.; Spaan, W.J.; Snijder, E.J. Construction of Chimeric Arteriviruses Reveals That the Ectodomain of the Major Glycoprotein Is Not the Main Determinant of Equine Arteritis Virus Tropism in Cell Culture. Virology 2001, 288, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Wei, Z.; Zevenhoven-Dobbe, J.C.; Liu, R.; Tong, G.; Snijder, E.J.; Yuan, S. Arterivirus Minor Envelope Proteins Are a Major Determinant of Viral Tropism in Cell Culture. J. Virol. 2012, 86, 3701–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheije, M.H.; Welting, T.J.; Jansen, H.T.; Rottier, P.J.; Meulenberg, J.J. Chimeric Arteriviruses Generated by Swapping of the M Protein Ectodomain Rule out a Role of This Domain in Viral Targeting. Virology 2002, 303, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Delputte, P.L.; Vanderheijden, N.; Nauwynck, H.J.; Pensaert, M.B. Involvement of the Matrix Protein in Attachment of Porcine Reproductive and Respiratory Syndrome Virus to a Heparinlike Receptor on Porcine Alveolar Macrophages. J. Virol. 2002, 76, 4312–4320. [Google Scholar] [CrossRef] [Green Version]
- Van Breedam, W.; Delputte, P.L.; van Gorp, H.; Misinzo, G.; Vanderheijden, N.; Duan, X.; Nauwynck, H.J. Porcine Reproductive and Respiratory Syndrome Virus Entry into the Porcine Macrophage. J. Gen. Virol. 2010, 91 Pt 7, 1659–1667. [Google Scholar] [CrossRef]
- Van Breedam, W.; van Gorp, H.; Zhang, J.Q.; Crocker, P.R.; Delputte, P.L.; Nauwynck, H.J. The M/Gp(5) Glycoprotein Complex of Porcine Reproductive and Respiratory Syndrome Virus Binds the Sialoadhesin Receptor in a Sialic Acid-Dependent Manner. PLoS Pathog. 2010, 6, e1000730. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, K.M.; Rowland, R.R.; Ewen, C.L.; Trible, B.R.; Kerrigan, M.A.; Cino-Ozuna, A.G.; Samuel, M.S.; Lightner, J.E.; McLaren, D.G.; Mileham, A.J.; et al. Gene-Edited Pigs Are Protected from Porcine Reproductive and Respiratory Syndrome Virus. Nat. Biotechnol. 2016, 34, 20–22. [Google Scholar] [CrossRef]
- Xie, J.; Christiaens, I.; Yang, B.; Breedam, W.V.; Cui, T.; Nauwynck, H.J. Molecular Cloning of Porcine Siglec-3, Siglec-5 and Siglec-10, and Identification of Siglec-10 as an Alternative Receptor for Porcine Reproductive and Respiratory Syndrome Virus (Prrsv). J. Gen. Virol. 2017, 98, 2030–2042. [Google Scholar] [CrossRef]
- Nauwynck, H.J.; Duan, X.; Favoreel, H.W.; van Oostveldt, P.; Pensaert, M.B. Entry of Porcine Reproductive and Respiratory Syndrome Virus into Porcine Alveolar Macrophages Via Receptor-Mediated Endocytosis. J. Gen. Virol. 1999, 80 Pt 2, 297–305. [Google Scholar] [CrossRef]
- Nitschke, M.; Korte, T.; Tielesch, C.; Ter-Avetisyan, G.; Tunnemann, G.; Cardoso, M.C.; Veit, M.; Herrmann, A. Equine Arteritis Virus Is Delivered to an Acidic Compartment of Host Cells Via Clathrin-Dependent Endocytosis. Virology 2008, 377, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, J.G.; Slade, D.E.; Shields, S.L.; Jolie, R.; Mannan, R.M.; Ankenbauer, R.G.; Welch, S.K. Cd163 Expression Confers Susceptibility to Porcine Reproductive and Respiratory Syndrome Viruses. J. Virol. 2007, 81, 7371–7379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.B.; Dinh, P.X.; Ansari, I.H.; de Lima, M.; Osorio, F.A.; Pattnaik, A.K. The Minor Envelope Glycoproteins Gp2a and Gp4 of Porcine Reproductive and Respiratory Syndrome Virus Interact with the Receptor Cd163. J. Virol. 2010, 84, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkard, C.; Lillico, S.G.; Reid, E.; Jackson, B.; Mileham, A.J.; Ait-Ali, T.; Whitelaw, C.B.; Archibald, A.L. Precision Engineering for Prrsv Resistance in Pigs: Macrophages from Genome Edited Pigs Lacking Cd163 Srcr5 Domain Are Fully Resistant to Both Prrsv Genotypes While Maintaining Biological Function. PLoS Pathog. 2017, 13, e1006206. [Google Scholar] [CrossRef] [Green Version]
- Burkard, C.; Opriessnig, T.; Mileham, A.J.; Stadejek, T.; Ait-Ali, T.; Lillico, S.G.; Whitelaw, C.B.A.; Archibald, A.L. Pigs Lacking the Scavenger Receptor Cysteine-Rich Domain 5 of Cd163 Are Resistant to Porcine Reproductive and Respiratory Syndrome Virus 1 Infection. J. Virol. 2018, 92, 16. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Li, R.; Qiao, S.; Chen, X.X.; Xing, G.; Zhang, G. Glycoprotein 5 Is Cleaved by Cathepsin E During Porcine Reproductive and Respiratory Syndrome Virus Membrane Fusion. J. Virol. 2020, 94, 10. [Google Scholar] [CrossRef]
- Misinzo, G.M.; Delputte, P.L.; Nauwynck, H.J. Involvement of Proteases in Porcine Reproductive and Respiratory Syndrome Virus Uncoating Upon Internalization in Primary Macrophages. Vet. Res. 2008, 39, 55. [Google Scholar] [CrossRef] [Green Version]
- de Vries, A.A.; Chirnside, E.D.; Horzinek, M.C.; Rottier, P.J. Structural Proteins of Equine Arteritis Virus. J. Virol. 1992, 66, 6294–6303. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Dobbe, J.C.; Spaan, W.J. Heterodimerization of the Two Major Envelope Proteins Is Essential for Arterivirus Infectivity. J. Virol. 2003, 77, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Han, X.; Osterrieder, K.; Veit, M. Palmitoylation of the Envelope Membrane Proteins Gp5 and M of Porcine Reproductive and Respiratory Syndrome Virus Is Essential for Virus Growth. PLoS Pathog. 2021, 17, e1009554. [Google Scholar] [CrossRef]
- Snijder, E.J.; Meulenberg, J.J. The Molecular Biology of Arteriviruses. J. Gen. Virol. 1998, 79 Pt 5, 961–979. [Google Scholar] [CrossRef]
- Balasuriya, U.B.; Carossi, M. Reproductive Effects of Arteriviruses: Equine Arteritis Virus and Porcine Reproductive and Respiratory Syndrome Virus Infections. Curr. Opin. Virol. 2017, 27, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Galeota, J.A.; Jar, A.M.; Platt, K.B.; Osorio, F.A.; Lopez, O.J. Identification of Neutralizing and Nonneutralizing Epitopes in the Porcine Reproductive and Respiratory Syndrome Virus Gp5 Ectodomain. J. Virol. 2002, 76, 4241–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plagemann, P.G.; Rowland, R.R.; Faaberg, K.S. The Primary Neutralization Epitope of Porcine Respiratory and Reproductive Syndrome Virus Strain Vr-2332 Is Located in the Middle of the Gp5 Ectodomain. Arch. Virol. 2002, 147, 2327–2347. [Google Scholar] [CrossRef] [PubMed]
- Lopez, O.J.; Osorio, F.A. Role of Neutralizing Antibodies in Prrsv Protective Immunity. Vet. Immunol. Immunopathol. 2004, 102, 155–163. [Google Scholar] [CrossRef]
- Thaa, B.; Sinhadri, B.C.; Tielesch, C.; Krause, E.; Veit, M. Signal Peptide Cleavage from Gp5 of Prrsv: A Minor Fraction of Molecules Retains the Decoy Epitope, a Presumed Molecular Cause for Viral Persistence. PLoS ONE 2013, 8, e65548. [Google Scholar] [CrossRef] [Green Version]
- Trible, B.R.; Popescu, L.N.; Monday, N.; Calvert, J.G.; Rowland, R.R. A Single Amino Acid Deletion in the Matrix Protein of Porcine Reproductive and Respiratory Syndrome Virus Confers Resistance to a Polyclonal Swine Antibody with Broadly Neutralizing Activity. J. Virol. 2015, 89, 6515–6520. [Google Scholar] [CrossRef] [Green Version]
- Wissink, E.H.; van Wijk, H.A.; Kroese, M.V.; Weiland, E.; Meulenberg, J.J.; Rottier, P.J.; van Rijn, P.A. The Major Envelope Protein, Gp5, of a European Porcine Reproductive and Respiratory Syndrome Virus Contains a Neutralization Epitope in Its N-Terminal Ectodomain. J. Gen. Virol. 2003, 84, 1535–1543. [Google Scholar] [CrossRef]
- Costers, S.; Lefebvre, D.J.; van Doorsselaere, J.; Vanhee, M.; Delputte, P.L.; Nauwynck, H.J. Gp4 of Porcine Reproductive and Respiratory Syndrome Virus Contains a Neutralizing Epitope That Is Susceptible to Immunoselection in Vitro. Arch. Virol. 2010, 155, 371–378. [Google Scholar] [CrossRef]
- An, T.Q.; Li, J.N.; Su, C.M.; Yoo, D. Molecular and Cellular Mechanisms for Prrsv Pathogenesis and Host Response to Infection. Virus Res. 2020, 286, 197980. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Q.; Feng, W.H. Regulation and Evasion of Antiviral Immune Responses by Porcine Reproductive and Respiratory Syndrome Virus. Virus Res. 2015, 202, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Popescu, L.N.; Trible, B.R.; Chen, N.; Rowland, R.R.R. Gp5 of Porcine Reproductive and Respiratory Syndrome Virus (Prrsv) as a Target for Homologous and Broadly Neutralizing Antibodies. Vet. Microbiol. 2017, 209, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Murtaugh, M.P. Dissociation of Porcine Reproductive and Respiratory Syndrome Virus Neutralization from Antibodies Specific to Major Envelope Protein Surface Epitopes. Virology 2012, 433, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Ansari, I.H.; Kwon, B.; Osorio, F.A.; Pattnaik, A.K. Influence of N-Linked Glycosylation of Porcine Reproductive and Respiratory Syndrome Virus Gp5 on Virus Infectivity, Antigenicity, and Ability to Induce Neutralizing Antibodies. J. Virol. 2006, 80, 3994–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faaberg, K.S.; Hocker, J.D.; Erdman, M.M.; Harris, D.L.; Nelson, E.A.; Torremorell, M.; Plagemann, P.G. Neutralizing Antibody Responses of Pigs Infected with Natural Gp5 N-Glycan Mutants of Porcine Reproductive and Respiratory Syndrome Virus. Viral Immunol. 2006, 19, 294–304. [Google Scholar] [CrossRef]
- Vu, H.L.; Kwon, B.; Yoon, K.J.; Laegreid, W.W.; Pattnaik, A.K.; Osorio, F.A. Immune Evasion of Porcine Reproductive and Respiratory Syndrome Virus through Glycan Shielding Involves Both Glycoprotein 5 as Well as Glycoprotein 3. J. Virol. 2011, 85, 5555–5564. [Google Scholar] [CrossRef] [Green Version]
- Spilman, M.S.; Welbon, C.; Nelson, E.; Dokland, T. Cryo-Electron Tomography of Porcine Reproductive and Respiratory Syndrome Virus: Organization of the Nucleocapsid. J. Gen. Virol. 2009, 90 Pt 3, 527–535. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- AlQuraishi, M. Alphafold at Casp13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Applying and Improving Alphafold at Casp14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with Alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. It Will Change Everything: Deepmind’s Ai Makes Gigantic Leap in Solving Protein Structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. Machine Learning in Protein Structure Prediction. Curr. Opin. Chem. Biol. 2021, 65, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.L.; Berendzen, J.; Stevens, A.O.; He, Y.; Wang, J.; Dokholyan, N.V.; Oprea, T.I. Alphafold Illuminates Half of the Dark Human Proteins. Curr. Opin. Struct. Biol. 2022, 74, 102372. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J. Protein Complex Prediction with Alphafold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cheng, F.; Tuncbag, N. Editorial Overview: Artificial Intelligence (Ai) Methodologies in Structural Biology. Curr. Opin. Struct. Biol. 2022, 74, 102387. [Google Scholar] [CrossRef]
- Wlodawer, A. Stereochemistry and Validation of Macromolecular Structures. Methods Mol. Biol. 2017, 1607, 595–610. [Google Scholar]
- Li, J.; Tao, S.; Orlando, R.; Murtaugh, M.P. N-Glycosylation Profiling of Porcine Reproductive and Respiratory Syndrome Virus Envelope Glycoprotein 5. Virology 2015, 478, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Liu, Z.; Tong, X.; Wang, Z.; Xu, S.; Chen, Q.; Zhou, J.; Fang, L.; Wang, D.; Xiao, S. Evolutionary Dynamics of Type 2 Porcine Reproductive and Respiratory Syndrome Virus by Whole-Genome Analysis. Viruses 2021, 13, 2469. [Google Scholar] [CrossRef] [PubMed]
- Liaci, A.M.; Steigenberger, B.; de Souza, P.C.T.; Tamara, S.; Grollers-Mulderij, M.; Ogrissek, P.; Marrink, S.J.; Scheltema, R.A.; Forster, F. Structure of the Human Signal Peptidase Complex Reveals the Determinants for Signal Peptide Cleavage. Mol. Cell 2021, 81, 3934–3948.e11. [Google Scholar] [CrossRef] [PubMed]
- Owji, H.; Nezafat, N.; Negahdaripour, M.; Hajiebrahimi, A.; Ghasemi, Y. A Comprehensive Review of Signal Peptides: Structure, Roles, and Applications. Eur. J. Cell Biol. 2018, 97, 422–441. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Andres, M.; Copic, A.; Antonny, B. The Many Faces of Amphipathic Helices. Biomolecules 2018, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Rossman, J.S.; Lamb, R.A. Viral Membrane Scission. Annu. Rev. Cell Dev. Biol. 2013, 29, 551–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plagemann, P.G. Gp5 Ectodomain Epitope of Porcine Reproductive and Respiratory Syndrome Virus, Strain Lelystad Virus. Virus Res. 2004, 102, 225–230. [Google Scholar] [CrossRef]
- Thaa, B.; Kaufer, S.; Neumann, S.A.; Peibst, B.; Nauwynck, H.; Krause, E.; Veit, M. The Complex Co-Translational Processing of Glycoprotein Gp5 of Type 1 Porcine Reproductive and Respiratory Syndrome Virus. Virus Res. 2017, 240, 112–120. [Google Scholar] [CrossRef]
- Lizak, C.; Gerber, S.; Numao, S.; Aebi, M.; Locher, K.P. X-ray Structure of a Bacterial Oligosaccharyltransferase. Nature 2011, 474, 350–355. [Google Scholar] [CrossRef]
- Nilsson, I.M.; von Heijne, G. Determination of the Distance between the Oligosaccharyltransferase Active Site and the Endoplasmic Reticulum Membrane. J. Biol. Chem. 1993, 268, 5798–5801. [Google Scholar] [CrossRef]
- Tan, Y.; Schneider, T.; Shukla, P.K.; Chandrasekharan, M.B.; Aravind, L.; Zhang, D. Unification and Extensive Diversification of M/Orf3-Related Ion Channel Proteins in Coronaviruses and Other Nidoviruses. Virus Evol. 2021, 7, veab014. [Google Scholar] [CrossRef]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-Em Structure of Sars-Cov-2 Orf3a in Lipid Nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of Sars-Cov-2 Membrane Protein Essential for Virus Assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Understanding the Role of Sars-Cov-2 Orf3a in Viral Pathogenesis and COVID-19. Front Microbiol. 2022, 13, 854567. [Google Scholar] [CrossRef]
- Ramaraj, T.; Angel, T.; Dratz, E.A.; Jesaitis, A.J.; Mumey, B. Antigen-Antibody Interface Properties: Composition, Residue Interactions, and Features of 53 Non-Redundant Structures. Biochim. Biophys. Acta 2012, 1824, 520–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.R.; Abrahante, J.E.; Johnson, C.R.; Murtaugh, M.P. Purifying Selection in Porcine Reproductive and Respiratory Syndrome Virus Orf5a Protein Influences Variation in Envelope Glycoprotein 5 Glycosylation. Infect. Genet. Evol. 2013, 11, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogozheva, I.D.; Tristram-Nagle, S.; Mosberg, H.I.; Lomize, A.L. Structural Adaptations of Proteins to Different Biological Membranes. Biochim. Biophys. Acta 2013, 1828, 2592–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Trible, B.R.; Kerrigan, M.A.; Tian, K.; Rowland, R.R.R. Orf5 of Porcine Reproductive and Respiratory Syndrome Virus (Prrsv) Is a Target of Diversifying Selection as Infection Progresses from Acute Infection to Virus Rebound. Infect. Genet. Evol. 2016, 40, 167–175. [Google Scholar] [CrossRef]
- Matczuk, A.K.; Kunec, D.; Veit, M. Co-Translational Processing of Glycoprotein 3 from Equine Arteritis Virus: N-Glycosylation Adjacent to the Signal Peptide Prevents Cleavage. J. Biol. Chem. 2013, 288, 35396–35405. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Pelkmans, L. A New Paradigm for Membrane-Organizing and -Shaping Scaffolds. FEBS Lett 2006, 580, 5559–5564. [Google Scholar] [CrossRef] [Green Version]
- McMahon, H.T.; Boucrot, E. Membrane Curvature at a Glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Zimmerberg, J.; Kozlov, M.M. How Proteins Produce Cellular Membrane Curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza Virus M2 Protein Mediates Escrt-Independent Membrane Scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaa, B.; Tielesch, C.; Moller, L.; Schmitt, A.O.; Wolff, T.; Bannert, N.; Herrmann, A.; Veit, M. Growth of Influenza a Virus Is Not Impeded by Simultaneous Removal of the Cholesterol-Binding and Acylation Sites in the M2 Protein. J. Gen. Virol. 2012, 93 Pt 2, 282–292. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Sergeeva, O.; Turelli, P.; Qing, E.; Kunz, B.; Raclot, C.; Montoya, J.P.; Abriata, L.A.; Gallagher, T.; et al. S-Acylation Controls Sars-Cov-2 Membrane Lipid Organization and Enhances Infectivity. Dev. Cell 2021, 56, 2790–2807.e8. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid Rafts as a Membrane-Organizing Principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, X.; Bai, J.; Gao, Y.; Song, Z.; Nauwynck, H.; Wang, X.; Yang, Y.; Jiang, P. Glyceraldehyde-3-Phosphate Dehydrogenase Restricted in Cytoplasmic Location by Viral Gp5 Facilitates Porcine Reproductive and Respiratory Syndrome Virus Replication Via Its Glycolytic Activity. J. Virol. 2021, 95, e0021021. [Google Scholar] [CrossRef]
- Hanada, K.; Suzuki, Y.; Nakane, T.; Hirose, O.; Gojobori, T. The Origin and Evolution of Porcine Reproductive and Respiratory Syndrome Viruses. Mol. Biol. Evol. 2005, 22, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Garoff, H. The Budding Mechanisms of Enveloped Animal Viruses. J. Gen. Virol. 1980, 50, 1–21. [Google Scholar]
- Doan, D.N.; Dokland, T. Structure of the Nucleocapsid Protein of Porcine Reproductive and Respiratory Syndrome Virus. Structure 2003, 11, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Krabben, L.; Wang, F.; Veit, M. Glycoprotein 3 of Porcine Reproductive and Respiratory Syndrome Virus Exhibits an Unusual Hairpin-Like Membrane Topology. J. Virol. 2018, 92, 15. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Yan, X.; Li, Y.; Cui, J.; Misra, S.; Firth, A.E.; Snijder, E.J.; Fang, Y. A Swine Arterivirus Deubiquitinase Stabilizes Two Major Envelope Proteins and Promotes Production of Viral Progeny. PLoS Pathog. 2021, 17, e1009403. [Google Scholar] [CrossRef] [PubMed]
- Bedi, S.; Ono, A. OFriend or Foe: The Role of the Cytoskeleton in Influenza a Virus Assembly. Viruses 2019, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.E. Membrane Bending by Actin Polymerization. Curr. Opin. Cell Biol. 2018, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.; Dong, H.; Wang, L.; Peng, J.; An, T.; Yang, X.; Tian, Z.; Cai, X. Identification of Host Cellular Proteins That Interact with the M Protein of a Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus Vaccine Strain. Virol. J. 2017, 14, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zakhartchouk, A. Characterization of the Interactome of the Porcine Reproductive and Respiratory Syndrome Virus Glycoprotein-5. Arch. Virol. 2018, 163, 1595–1605. [Google Scholar] [CrossRef]
- Ouzounis, C.A. A Recent Origin of Orf3a from M Protein across the Coronavirus Lineage Arising by Sharp Divergence. Comput. Struct. Biotechnol. J. 2020, 18, 4093–4102. [Google Scholar] [CrossRef]
- Abdulrahman, D.A.; Meng, X.; Veit, M. S-Acylation of Proteins of Coronavirus and Influenza Virus: Conservation of Acylation Sites in Animal Viruses and Dhhc Acyltransferases in Their Animal Reservoirs. Pathogens 2021, 10, 669. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe Acute Respiratory Syndrome-Associated Coronavirus 3a Protein Forms an Ion Channel and Modulates Virus Release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [Green Version]
- Kojer, K.; Riemer, J. Balancing Oxidative Protein Folding: The Influences of Reducing Pathways on Disulfide Bond Formation. Biochim. Biophys. Acta 2014, 1844, 1383–1390. [Google Scholar] [CrossRef]
- Brinton, M.A.; Di, H.; Vatter, H.A. Simian Hemorrhagic Fever Virus: Recent Advances. Virus Res. 2015, 202, 112–119. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. Colabfold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Steinegger, M.; Soding, J. Mmseqs2 Desktop and Local Web Server App for Fast, Interactive Sequence Searches. Bioinformatics 2019, 35, 2856–2858. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. Pdbsum: Structural Summaries of Pdb Entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the Apbs Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. Opm Database and Ppm Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The Helical Hydrophobic Moment: A Measure of the Amphiphilicity of a Helix. Nature 1982, 299, 371–374. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. Heliquest: A Web Server to Screen Sequences with Specific Alpha-Helical Properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The Hdock Server for Integrated Protein-Protein Docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veit, M.; Gadalla, M.R.; Zhang, M. Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus. Int. J. Mol. Sci. 2022, 23, 13209. https://doi.org/10.3390/ijms232113209
Veit M, Gadalla MR, Zhang M. Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus. International Journal of Molecular Sciences. 2022; 23(21):13209. https://doi.org/10.3390/ijms232113209
Chicago/Turabian StyleVeit, Michael, Mohamed Rasheed Gadalla, and Minze Zhang. 2022. "Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus" International Journal of Molecular Sciences 23, no. 21: 13209. https://doi.org/10.3390/ijms232113209
APA StyleVeit, M., Gadalla, M. R., & Zhang, M. (2022). Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus. International Journal of Molecular Sciences, 23(21), 13209. https://doi.org/10.3390/ijms232113209