
The Intratumor Bacterial and Fungal Microbiome Is Characterized by HPV, Smoking, and Alcohol Consumption in Head and Neck Squamous Cell Carcinoma
 , , ,
, , ,
Abstract
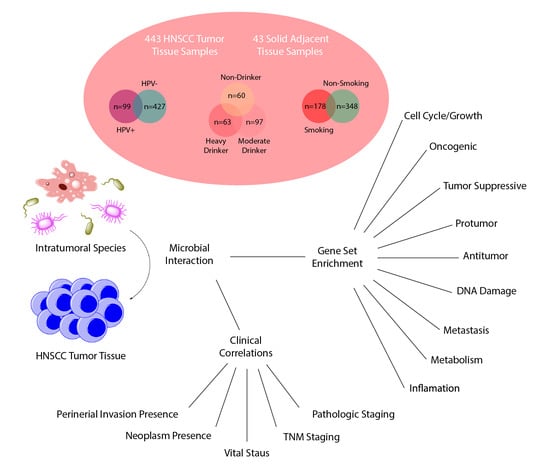
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
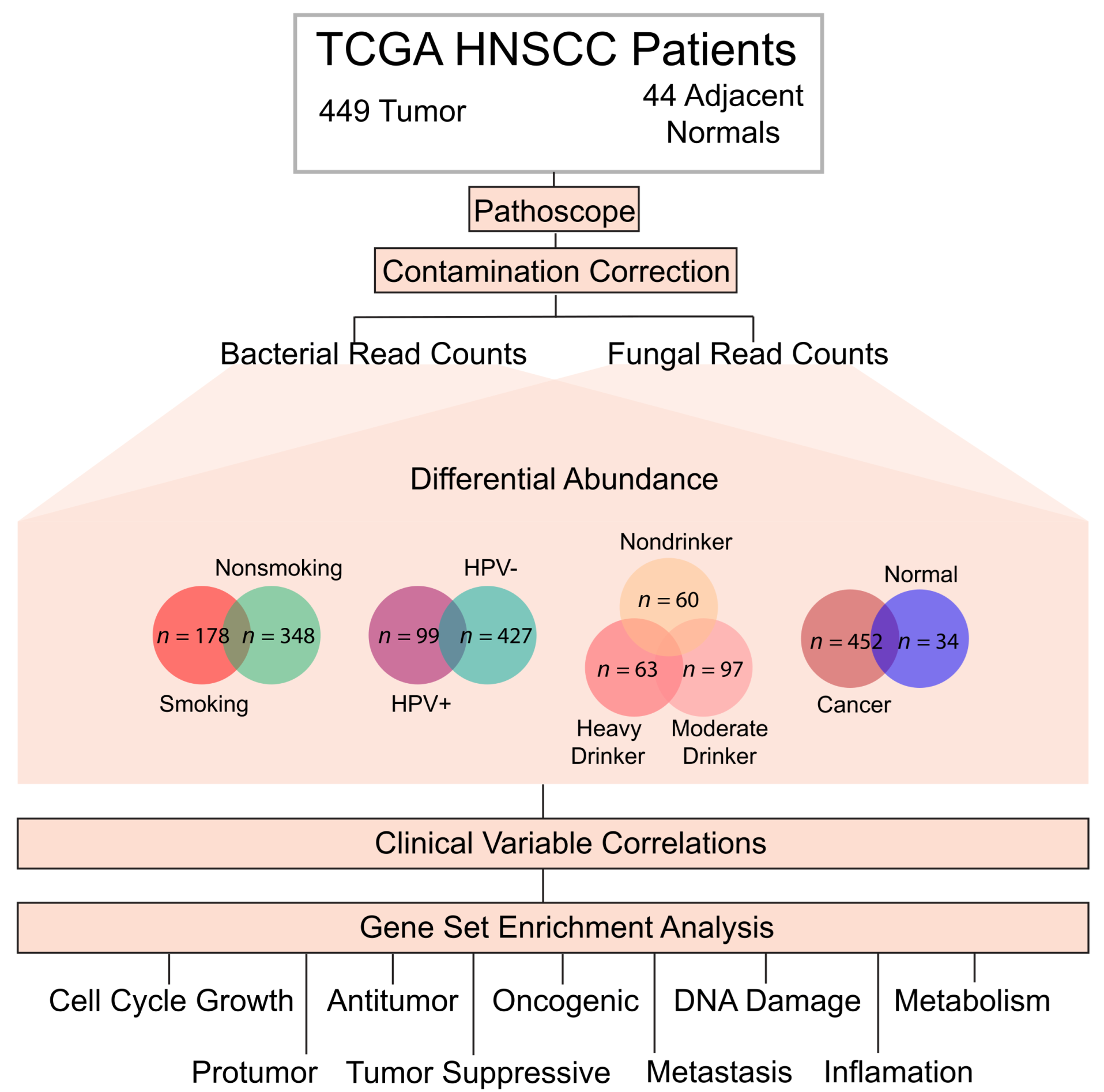
2.1. Data Acquisition and Identification of Intratumor Microbes
2.2. Evaluation of Contamination Correction
2.3. Differential Abundance and Clinical Significance of Intratumor Microbes Associated with HNSCC
2.4. Differential Abundance and Clinical Significance of Intratumor Microbes Associated with HPV Status in HNSCC
2.5. Differential Abundance and Clinical Significance of Intratumor Microbes Associated with HNSCC Smokers
2.6. Smoking and HPV(-)-Associated HNSCC Display Distinct Intratumor Microbial Profiles
2.7. Differential Abundance of Microbes Associated with Alcohol Consumption
2.8. Microbe Abundance Correlation to Oncogenic and Tumor-Suppressive Pathways
2.9. Classification of Microbes as Predictors of Increased and Decreased Carcinogenesis
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Extraction of Bacterial and Fungal Read Counts
4.3. Evaluation of Contamination
4.4. Differential Abundance between Etiologically Defined Cohorts
4.5. Clinical Variable Comparisons to Microbial Abundance in Etiologically Defined Cohorts
4.6. Correlation of Microbial Abundance to Oncogenic and Immunological Signatures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Vokes, E.E.; Weichselbaum, R.R.; Lippman, S.M.; Hong, W.K. Head and neck cancer. N. Engl. J. Med. 1993, 328, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Beddok, A.; Vela, A.; Calugaru, V.; Tessonnier, T.; Kubes, J.; Dutheil, P.; Gerard, A.; Vidal, M.; Goudjil, F.; Florescu, C.; et al. Proton therapy for head and neck squamous cell carcinomas: A review of the physical and clinical challenges. Radiother. Oncol. 2020, 147, 30–39. [Google Scholar] [CrossRef]
- Dagan, R.; Uezono, H.; Bryant, C.; Holtzman, A.L.; Morris, C.G.; Mendenhall, W.M. Long-term Outcomes from Proton Therapy for Sinonasal Cancers. Int. J. Part. Ther. 2021, 8, 200–212. [Google Scholar] [CrossRef]
- Kitamura, N.; Sento, S.; Yoshizawa, Y.; Sasabe, E.; Kudo, Y.; Yamamoto, T. Current Trends and Future Prospects of Molecular Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 22, 240. [Google Scholar] [CrossRef]
- Cristina, V.; Herrera-Gomez, R.G.; Szturz, P.; Espeli, V.; Siano, M. Immunotherapies and Future Combination Strategies for Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5399. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute. Cancer Stat Facts: Oral Cavity and Pharynx Cancer; National Cancer Institute: Bethesda, MD, USA, 2019.
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Tsai, J.C.; Casteneda, G.; Lee, A.; Dereschuk, K.; Li, W.T.; Chakladar, J.; Lombardi, A.F.; Ongkeko, W.M.; Chang, E.Y. Identification and characterization of the intra-articular microbiome in the osteoarthritic knee. Int. J. Mol. Sci. 2020, 21, 8618. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [Green Version]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craciun, C.I.; Neag, M.A.; Catinean, A.; Mitre, A.O.; Rusu, A.; Bala, C.; Roman, G.; Buzoianu, A.D.; Muntean, D.M.; Craciun, A.E. The Relationships between Gut Microbiota and Diabetes Mellitus, and Treatments for Diabetes Mellitus. Biomedicines 2022, 10, 308. [Google Scholar] [CrossRef] [PubMed]
- Frost, F.; Kacprowski, T.; Rühlemann, M.; Pietzner, M.; Bang, C.; Franke, A.; Nauck, M.; Völker, U.; Völzke, H.; Dörr, M.; et al. Long-term instability of the intestinal microbiome is associated with metabolic liver disease, low microbiota diversity, diabetes mellitus and impaired exocrine pancreatic function. Gut 2021, 70, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Rossi, T.; Vergara, D.; Fanini, F.; Maffia, M.; Bravaccini, S.; Pirini, F. Microbiota-Derived Metabolites in Tumor Progression and Metastasis. Int. J. Mol. Sci. 2020, 21, 5786. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, B.; Wei, Y.; Kuang, D.-M. Influence of gut and intratumoral microbiota on the immune microenvironment and anti-cancer therapy. Pharmacol. Res. 2021, 174, 105966. [Google Scholar] [CrossRef]
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef] [Green Version]
- Gnanasekar, A.; Castaneda, G.; Iyangar, A.; Magesh, S.; Perez, D.; Chakladar, J.; Li, W.T.; Bouvet, M.; Chang, E.Y.; Ongkeko, W.M. The intratumor microbiome predicts prognosis across gender and subtypes in papillary thyroid carcinoma. Comput. Struct. Biotechnol. J. 2021, 19, 1986–1997. [Google Scholar] [CrossRef]
- Li, W.T.; Iyangar, A.S.; Reddy, R.; Chakladar, J.; Bhargava, V.; Sakamoto, K.K.; Ongkeko, W.M.; Rajasekaran, M. The Bladder Microbiome Is Associated with Epithelial-Mesenchymal Transition in Muscle Invasive Urothelial Bladder Carcinoma. Cancers 2021, 13, 3649. [Google Scholar] [CrossRef]
- Chakladar, J.; Kuo, S.Z.; Castaneda, G.; Li, W.T.; Gnanasekar, A.; Yu, M.A.; Chang, E.Y.; Wang, X.Q.; Ongkeko, W.M. The Pancreatic Microbiome is Associated with Carcinogenesis and Worse Prognosis in Males and Smokers. Cancers 2020, 12, 2672. [Google Scholar] [CrossRef]
- Wong, L.M.; Shende, N.; Li, W.T.; Castaneda, G.; Apostol, L.; Chang, E.Y.; Ongkeko, W.M. Comparative Analysis of Age- and Gender-Associated Microbiome in Lung Adenocarcinoma and Lung Squamous Cell Carcinoma. Cancers 2020, 12, 1447. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gnanasekar, A.; Lee, A.; Li, W.T.; Haas, M.; Wang-Rodriguez, J.; Chang, E.Y.; Rajasekaran, M.; Ongkeko, W.M. Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer. Cancers 2020, 12, 2524. [Google Scholar] [CrossRef] [PubMed]
- Binder Gallimidi, A.; Fischman, S.; Revach, B.; Bulvik, R.; Maliutina, A.; Rubinstein, A.M.; Nussbaum, G.; Elkin, M. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget 2015, 6, 22613–22623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Wang, J. A comprehensive analysis of intratumor microbiome in head and neck squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 2022, 279, 4127–4136. [Google Scholar] [CrossRef]
- Elhalawani, H.; Mohamed, A.S.R.; Elgohari, B.; Lin, T.A.; Sikora, A.G.; Lai, S.Y.; Abusaif, A.; Phan, J.; Morrison, W.H.; Gunn, G.B.; et al. Tobacco exposure as a major modifier of oncologic outcomes in human papillomavirus (HPV) associated oropharyngeal squamous cell carcinoma. BMC Cancer 2020, 20, 912. [Google Scholar] [CrossRef]
- Liu, X.; Liu, P.; Chernock, R.D.; Kuhs, K.A.L.; Lewis, J.S., Jr.; Luo, J.; Gay, H.A.; Thorstad, W.L.; Wang, X. A prognostic gene expression signature for oropharyngeal squamous cell carcinoma. EBioMedicine 2020, 61, 102805. [Google Scholar] [CrossRef]
- Rotsides, J.M.; Oliver, J.R.; Moses, L.E.; Tam, M.; Li, Z.; Schreiber, D.; Jacobson, A.S.; Hu, K.S.; Givi, B. Socioeconomic and Racial Disparities and Survival of Human Papillomavirus-Associated Oropharyngeal Squamous Cell Carcinoma. Otolaryngol. Head Neck Surg. 2021, 164, 131–138. [Google Scholar] [CrossRef]
- White, R.; Abel, S.; Hasan, S.; Verma, V.; Greenberg, L.; Colonias, A.; Wegner, R.E. Practice patterns and outcomes following radiation dose de-escalation for oropharyngeal cancer. Laryngoscope 2020, 130, E171–E176. [Google Scholar] [CrossRef]
- Powell, S.F.; Vu, L.; Spanos, W.C.; Pyeon, D. The Key Differences between Human Papillomavirus-Positive and -Negative Head and Neck Cancers: Biological and Clinical Implications. Cancers 2021, 13, 5206. [Google Scholar] [CrossRef]
- Sawabe, M.; Ito, H.; Oze, I.; Hosono, S.; Kawakita, D.; Tanaka, H.; Hasegawa, Y.; Murakami, S.; Matsuo, K. Heterogeneous impact of alcohol consumption according to treatment method on survival in head and neck cancer: A prospective study. Cancer Sci. 2017, 108, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osazuwa-Peters, N.; Adjei Boakye, E.; Chen, B.Y.; Tobo, B.B.; Varvares, M.A. Association between Head and Neck Squamous Cell Carcinoma Survival, Smoking at Diagnosis, and Marital Status. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Logan, H.L.; Mendenhall, W.M. Human papillomavirus, smoking, and head and neck cancer. Am. J. Otolaryngol. 2012, 33, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Ferraguti, G.; Terracina, S.; Petrella, C.; Greco, A.; Minni, A.; Lucarelli, M.; Agostinelli, E.; Ralli, M.; de Vincentiis, M.; Raponi, G.; et al. Alcohol and Head and Neck Cancer: Updates on the Role of Oxidative Stress, Genetic, Epigenetics, Oral Microbiota, Antioxidants, and Alkylating Agents. Antioxidants 2022, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Näsman, A.; Holzhauser, S.; Kostopoulou, O.N.; Zupancic, M.; Ährlund-Richter, A.; Du, J.; Dalianis, T. Prognostic Markers and Driver Genes and Options for Targeted Therapy in Human-Papillomavirus-Positive Tonsillar and Base-of-Tongue Squamous Cell Carcinoma. Viruses 2021, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, J.V.; Slebos, R.J.C.; Martin-Gomez, L.; Wang, X.; Teer, J.K.; Tan, A.C.; Gerke, T.A.; Aden-Buie, G.; van Veen, T.; Masannat, J.; et al. Effects of Tobacco Smoking on the Tumor Immune Microenvironment in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2020, 26, 1474–1485. [Google Scholar] [CrossRef] [Green Version]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Rampelotto, P.H.; Sereia, A.F.R.; de Oliveira, L.F.V.; Margis, R. Exploring the Hospital Microbiome by High-Resolution 16S rRNA Profiling. Int. J. Mol. Sci. 2019, 20, 3099. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.A.; Bellile, E.L.; Wolf, G.T.; Virani, S.; Shuman, A.G.; Taylor, J.M.; Rozek, L.S. Cigarette use, comorbidities, and prognosis in a prospective head and neck squamous cell carcinoma population. Head Neck 2016, 38, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Hisamatsu, K.; Suzui, N.; Hara, A.; Tomita, H.; Miyazaki, T. A Review of HPV-Related Head and Neck Cancer. J. Clin. Med. 2018, 7, 241. [Google Scholar] [CrossRef]
- Koo, H.Y.; Han, K.; Shin, D.W.; Yoo, J.E.; Cho, M.H.; Jeon, K.H.; Kim, D.; Hong, S.; Jun, J.K. Alcohol Drinking Pattern and Risk of Head and Neck Cancer: A Nationwide Cohort Study. Int. J. Environ. Res. Public Health 2021, 18, 11204. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Freedman, N.D.; Alekseyenko, A.V.; Wu, J.; Yang, L.; Pei, Z.; et al. Drinking alcohol is associated with variation in the human oral microbiome in a large study of American adults. Microbiome 2018, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranzani, M.; Iyer, V.; Ibarra-Soria, X.; Del Castillo Velasco-Herrera, M.; Garnett, M.; Logan, D.; Adams, J.D. Revisiting olfactory receptors as putative drivers of cancer. Wellcome Open Res. 2017, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Benbernou, N.; Esnault, S.; Galibert, F. Activation of SRE and AP1 by olfactory receptors via the MAPK and Rho dependent pathways. Cell Signal 2013, 25, 1486–1497. [Google Scholar] [CrossRef]
- Kim, S.Y.; Mammen, A.; Yoo, S.J.; Cho, B.; Kim, E.K.; Park, J.I.; Moon, C.; Ronnett, G.V. Phosphoinositide and Erk signaling pathways mediate activity-driven rodent olfactory sensory neuronal survival and stress mitigation. J. Neurochem. 2015, 134, 486–498. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Yu, D. Molecular mechanisms of erbB2-mediated breast cancer chemoresistance. In Breast Cancer Chemosensitivity; Springer: Berlin, Germany, 2007; pp. 119–129. [Google Scholar]
- Farrell, F.; Lee, A. The erythropoietin receptor and its expression in tumor cells and other tissues. Oncologist 2004, 9 (Suppl. 5), 18–30. [Google Scholar] [CrossRef] [Green Version]
- Weidner, K.M.; Sachs, M.; Birchmeier, W. The Met receptor tyrosine kinase transduces motility, proliferation, and morphogenic signals of scatter factor/hepatocyte growth factor in epithelial cells. J. Cell Biol. 1993, 121, 145–154. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, T.; Li, S.; Henry, L.E.; Liu, S.; Sartor, M.A. Molecular Tumor Subtypes of HPV-Positive Head and Neck Cancers: Biological Characteristics and Implications for Clinical Outcomes. Cancers 2021, 13, 2721. [Google Scholar] [CrossRef]
- Farah, C.S. Molecular landscape of head and neck cancer and implications for therapy. Ann. Transl. Med. 2021, 9, 915. [Google Scholar] [CrossRef]
- Irimie, A.I.; Braicu, C.; Cojocneanu, R.; Magdo, L.; Onaciu, A.; Ciocan, C.; Mehterov, N.; Dudea, D.; Buduru, S.; Berindan-Neagoe, I. Differential Effect of Smoking on Gene Expression in Head and Neck Cancer Patients. Int. J. Environ. Res. Public Health 2018, 15, 1558. [Google Scholar] [CrossRef] [Green Version]
- Avissar, M.; McClean, M.D.; Kelsey, K.T.; Marsit, C.J. MicroRNA expression in head and neck cancer associates with alcohol consumption and survival. Carcinogenesis 2009, 30, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.A.; Kuo, S.Z.; Rahimy, E.; Zou, A.E.; Korrapati, A.; Rahimy, M.; Kim, E.; Zheng, H.; Yu, M.A.; Wang-Rodriguez, J.; et al. Alcohol-dysregulated miR-30a and miR-934 in head and neck squamous cell carcinoma. Mol. Cancer 2015, 14, 181. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; D’Souza, G.; Fakhry, C.; Bigelow, E.O.; Usyk, M.; Burk, R.D.; Zhao, N. Oral Human Papillomavirus Associated with Differences in Oral Microbiota Beta Diversity and Microbiota Abundance. J. Infect. Dis. 2022, 226, 1098–1108. [Google Scholar] [CrossRef]
- Wu, J.; Peters, B.A.; Dominianni, C.; Zhang, Y.; Pei, Z.; Yang, L.; Ma, Y.; Purdue, M.P.; Jacobs, E.J.; Gapstur, S.M.; et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016, 10, 2435–2446. [Google Scholar] [CrossRef]
- Sambrani, R.; Abdolalizadeh, J.; Kohan, L.; Jafari, B. Saccharomyces cerevisiae inhibits growth and metastasis and stimulates apoptosis in HT-29 colorectal cancer cell line. Comp. Clin. Pathol. 2019, 28, 985–995. [Google Scholar] [CrossRef]
- Li, J.Q.; Li, J.L.; Xie, Y.H.; Wang, Y.; Shen, X.N.; Qian, Y.; Han, J.X.; Chen, Y.X.; Fang, J.Y. Saccharomyces cerevisiae may serve as a probiotic in colorectal cancer by promoting cancer cell apoptosis. J. Dig. Dis. 2020, 21, 571–582. [Google Scholar] [CrossRef]
- Lin, D.; Kouzy, R.; Abi Jaoude, J.; Noticewala, S.S.; Delgado Medrano, A.Y.; Klopp, A.H.; Taniguchi, C.M.; Colbert, L.E. Microbiome factors in HPV-driven carcinogenesis and cancers. PLoS Pathog. 2020, 16, e1008524. [Google Scholar] [CrossRef]
- Liao, Y.; Tong, X.T.; Jia, Y.J.; Liu, Q.Y.; Wu, Y.X.; Xue, W.Q.; He, Y.Q.; Wang, T.M.; Zheng, X.H.; Zheng, M.Q.; et al. The Effects of Alcohol Drinking on Oral Microbiota in the Chinese Population. Int. J. Environ. Res. Public Health 2022, 19, 5729. [Google Scholar] [CrossRef]
- Musher, D.M. Haemophilus Species; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 2011. [Google Scholar]
- Zhang, M.; Zhang, Y.; Sun, Y.; Wang, S.; Liang, H.; Han, Y. Intratumoral Microbiota Impacts the First-Line Treatment Efficacy and Survival in Non-Small Cell Lung Cancer Patients Free of Lung Infection. J. Healthc. Eng. 2022, 2022, 5466853. [Google Scholar] [CrossRef]
- King, P. Haemophilus influenzae and the lung (Haemophilus and the lung). Clin. Transl. Med. 2012, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Urashima, M.; Hama, T.; Suda, T.; Suzuki, Y.; Ikegami, M.; Sakanashi, C.; Akutsu, T.; Amagaya, S.; Horiuchi, K.; Imai, Y.; et al. Distinct effects of alcohol consumption and smoking on genetic alterations in head and neck carcinoma. PLoS ONE 2013, 8, e80828. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakladar, J.; John, D.; Magesh, S.; Uzelac, M.; Li, W.T.; Dereschuk, K.; Apostol, L.; Brumund, K.T.; Rodriguez, J.-W.; Ongkeko, W.M. The Intratumor Bacterial and Fungal Microbiome Is Characterized by HPV, Smoking, and Alcohol Consumption in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 13250. https://doi.org/10.3390/ijms232113250
Chakladar J, John D, Magesh S, Uzelac M, Li WT, Dereschuk K, Apostol L, Brumund KT, Rodriguez J-W, Ongkeko WM. The Intratumor Bacterial and Fungal Microbiome Is Characterized by HPV, Smoking, and Alcohol Consumption in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2022; 23(21):13250. https://doi.org/10.3390/ijms232113250
Chicago/Turabian StyleChakladar, Jaideep, Daniel John, Shruti Magesh, Matthew Uzelac, Wei Tse Li, Kypros Dereschuk, Lauren Apostol, Kevin T. Brumund, Jessica-Wang Rodriguez, and Weg M. Ongkeko. 2022. "The Intratumor Bacterial and Fungal Microbiome Is Characterized by HPV, Smoking, and Alcohol Consumption in Head and Neck Squamous Cell Carcinoma" International Journal of Molecular Sciences 23, no. 21: 13250. https://doi.org/10.3390/ijms232113250