
Characterization of BCMA Expression in Circulating Rare Single Cells of Patients with Plasma Cell Neoplasms
 , , ,
, , ,  , , , and
, , , and 
Abstract
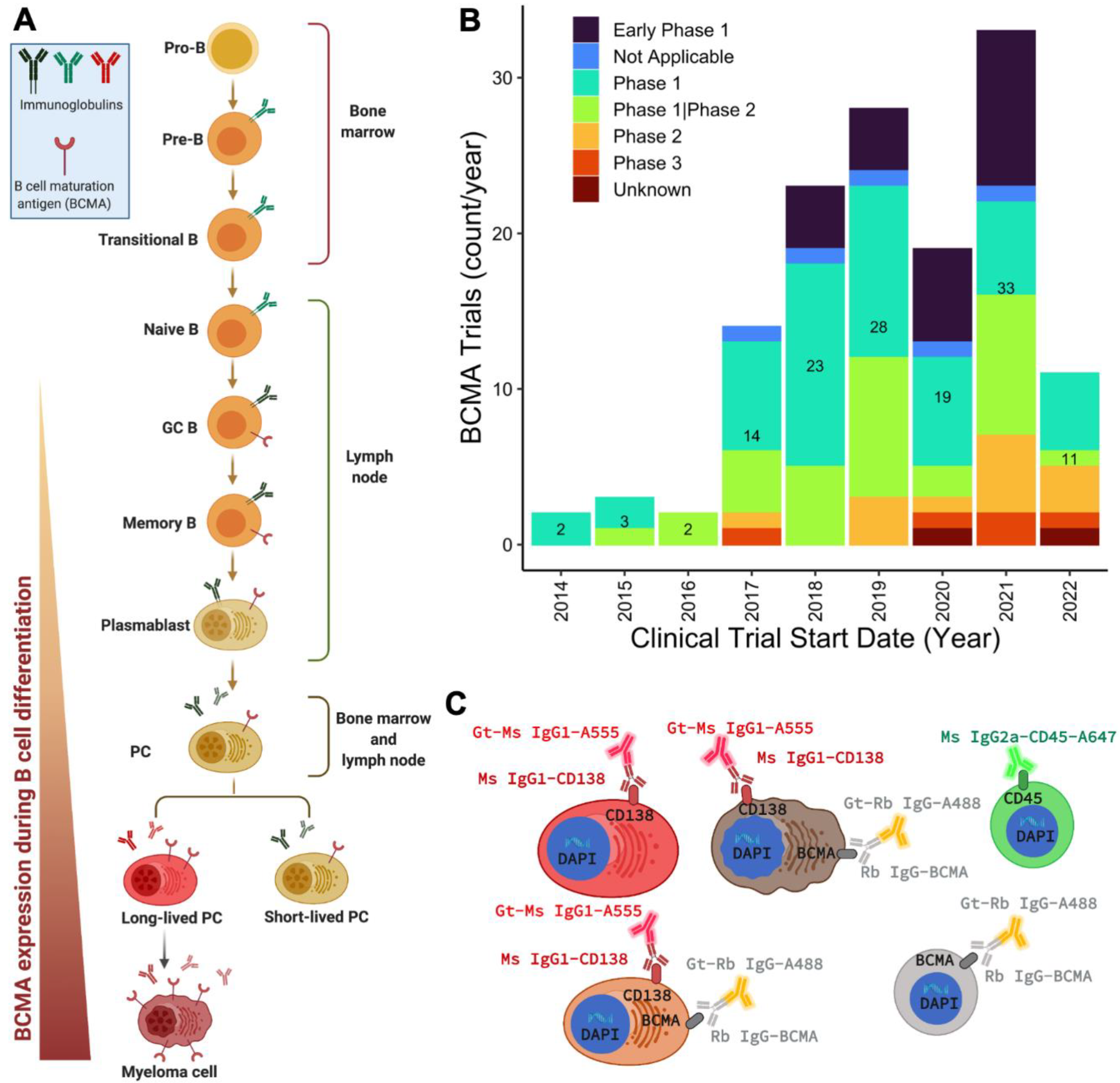
:1. Introduction
2. Results
2.1. Assay Validation and BCMA Expression in Cell Lines and Spiked Normal Donor Samples
2.2. Characteristics of Patients in the Study Cohort
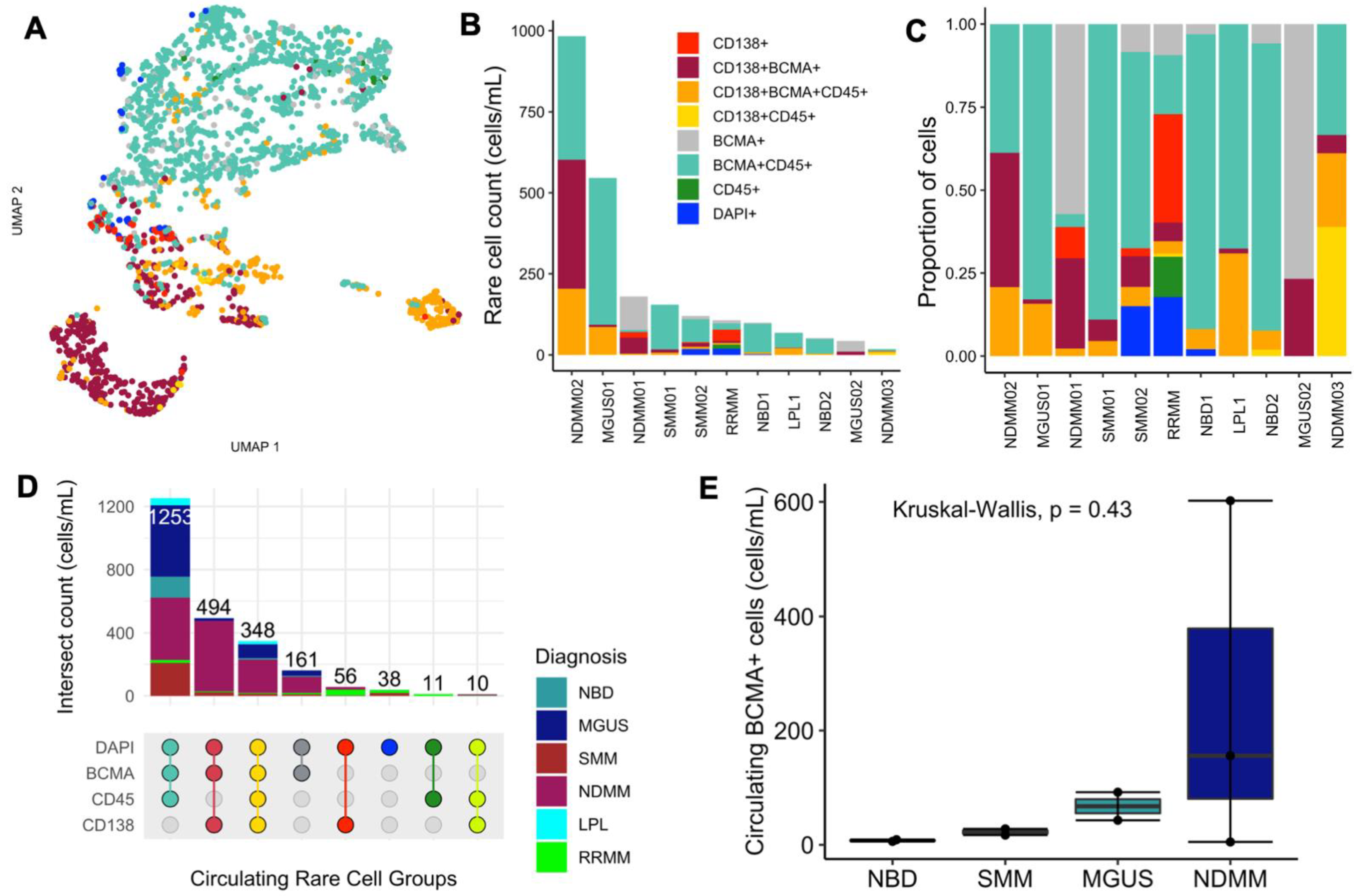
2.3. Morphological Characterization and Enumeration of BCMA+ Cells in Patients’ Peripheral Blood
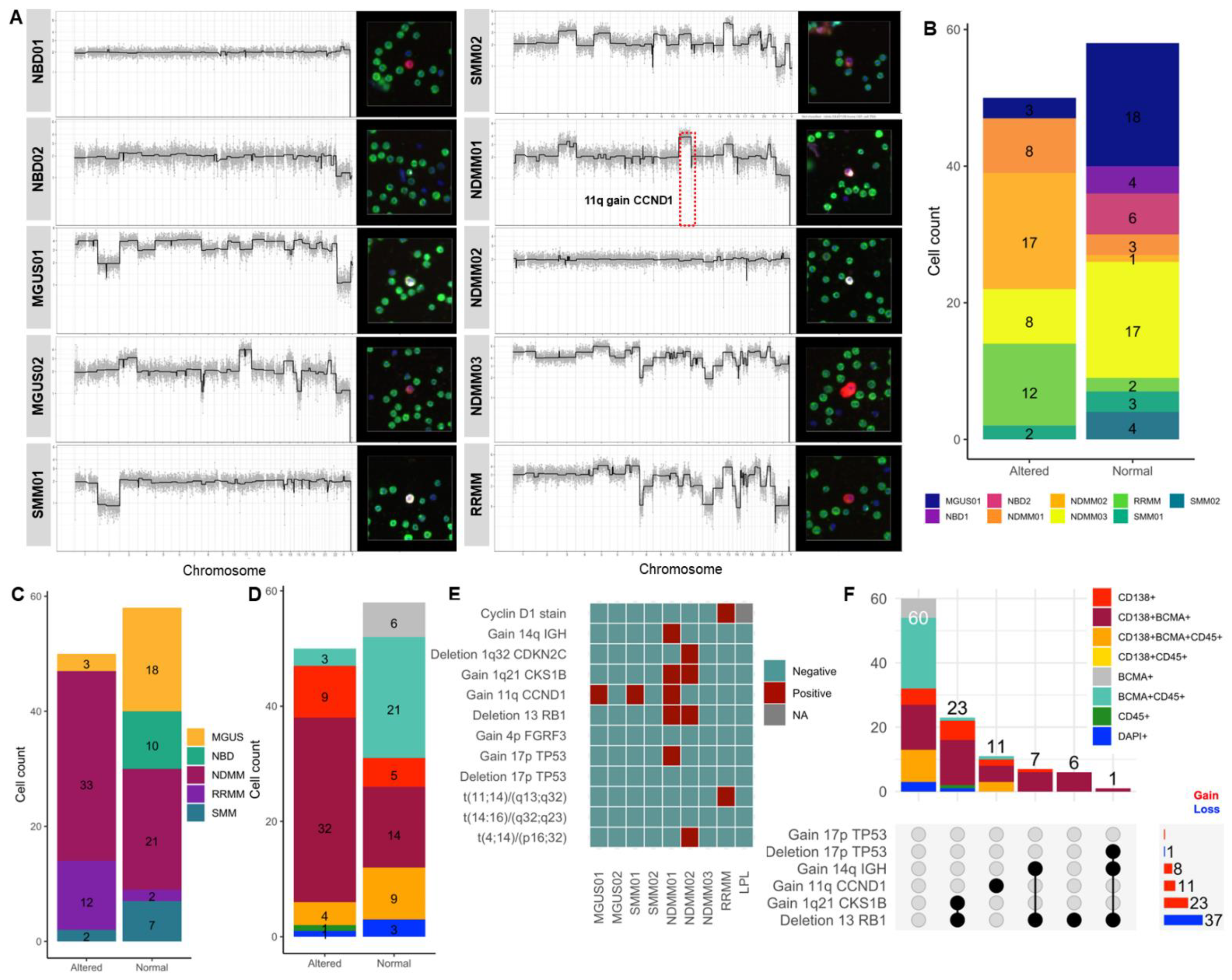
2.4. scCNV for BCMA-Expressing Cells and Correlation with FISH Cytogenetics
3. Discussion
4. Materials and Methods
4.1. Patient Enrollment and Sample Acquisition
4.2. Fluorescent In Situ Hybridization (FISH) and Karyotyping for Clinical Diagnosis
4.3. Selection of Markers and Immunofluorescence Targeting for Assay Development
4.4. BCMA Staining and Validation in Cell Lines and Spiked ND Samples
4.5. BCMA Staining and Validation in Patients’ PB
4.6. Imaging and Technical Analysis for Detection and Classification of Rare Cells
- BCMA+CD138+CD45−
- BCMA+CD138−CD45−
- BCMA+CD138+CD45+
- BCMA−CD138+CD45−
4.7. Single-Cell Sequencing and CNV Analysis for Genomic Validation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.-J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex: Implications for TNF receptor activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef]
- Madry, C.; Laabi, Y.; Callebaut, I.; Roussel, J.; Hatzoglou, A.; Le Coniat, M.; Mornon, J.P.; Berger, R.; Tsapis, A. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int. Immunol. 1998, 10, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coquery, C.M.; Erickson, L.D. Regulatory Roles of the Tumor Necrosis Factor Receptor BCMA. Crit. Rev. Immunol. 2012, 32, 287–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gras, M.-P.; Laâbi, Y.; Linares-Cruz, G.; Blondel, M.-O.; Rigaut, J.-P.; Brouet, J.-C.; Haguenauer-Tsapis, R.; Leca, G.; Tsapis, A. BCMAp: An integral membrane protein in the Golgi apparatus of human mature B lymphocytes. Int. Immunol. 1995, 7, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.-F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF Receptor Family Member BCMA (B Cell Maturation) Associates with TNF Receptor-Associated Factor (TRAF) 1, TRAF2, and TRAF3 and Activates NF-κB, Elk-1, c-Jun N-Terminal Kinase, and p38 Mitogen-Activated Protein Kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.C.; Hering, M.; Peckham, D.; McDonagh, C.F.; Brown, L.; Kim, K.M.; Meyer, D.L.; Zabinski, R.F.; Grewal, I.S.; Carter, P.J. Antibody targeting of B-cell maturation antigen on malignant plasma cells. Mol. Cancer Ther. 2007, 6, 3009–3018. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.-F.; Anderson, K.C.; Tai, Y.-T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef] [Green Version]
- Dogan, A.; Siegel, D.; Tran, N.; Fu, A.; Fowler, J.; Belani, R.; Landgren, O. B-cell maturation antigen expression across hematologic cancers: A systematic literature review. Blood Cancer J. 2020, 10, 73. [Google Scholar] [CrossRef]
- Ware, C.F. The TNF receptor super family in immune regulation. Immunol. Rev. 2011, 244, 5–8. [Google Scholar] [CrossRef]
- Sanchez, L.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-cell maturation antigen (BCMA) in multiple myeloma: The new frontier of targeted therapies. Ther. Adv. Hematol. 2021, 12, 2040620721989585. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, D.A.; Maric, I.; Yuan, C.M.; Liewehr, D.J.; Venzon, D.J.; Kochenderfer, J.; Stetler-Stevenson, M. Quantification of B-cell maturation antigen, a target for novel chimeric antigen receptor T-cell therapy in Myeloma. Leuk. Res. 2018, 71, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Frigyesi, I.; Adolfsson, J.; Ali, M.; Christophersen, M.K.; Johnsson, E.; Turesson, I.; Gullberg, U.; Hansson, M.; Nilsson, B. Robust isolation of malignant plasma cells in multiple myeloma. Blood 2014, 123, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Welter, L.; Xu, L.; McKinley, D.; Dago, A.E.; Prabakar, R.K.; Restrepo-Vassalli, S.; Xu, K.; Rodriguez-Lee, M.; Kolatkar, A.; Nevarez, R.; et al. Treatment response and tumor evolution: Lessons from an extended series of multianalyte liquid biopsies in a metastatic breast cancer patient. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005819. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.; Li, J.; Luttgen, M.S.; Kolatkar, A.; Kendall, J.T.; Flores, E.; Topp, Z.; E Samlowski, W.; McClay, E.; Bethel, K.; et al. Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys. Biol. 2015, 12, 016008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, S.; Matsumoto, N.; Storgard, R.; Peng, C.-C.; Aparicio, A.; Ormseth, B.; Rappard, K.; Cunningham, K.; Kolatkar, A.; Nevarez, R.; et al. Platelet-Coated Circulating Tumor Cells Are a Predictive Biomarker in Patients with Metastatic Castrate-Resistant Prostate Cancer. Mol. Cancer Res. 2021, 19, 2036–2045. [Google Scholar] [CrossRef]
- Shishido, S.N.; Sayeed, S.; Courcoubetis, G.; Djaladat, H.; Miranda, G.; Pienta, K.J.; Nieva, J.; Hansel, D.E.; Desai, M.; Gill, I.S.; et al. Characterization of Cellular and Acellular Analytes from Pre-Cystectomy Liquid Biopsies in Patients Newly Diagnosed with Primary Bladder Cancer. Cancers 2022, 14, 758. [Google Scholar] [CrossRef]
- Kolenčík, D.; Narayan, S.; Thiele, J.-A.; McKinley, D.; Gerdtsson, A.S.; Welter, L.; Hošek, P.; Ostašov, P.; Vyčítal, O.; Brůha, J.; et al. Circulating Tumor Cell Kinetics and Morphology from the Liquid Biopsy Predict Disease Progression in Patients with Metastatic Colorectal Cancer Following Resection. Cancers 2022, 14, 642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Beasley, S.; Prigozhina, N.L.; Higgins, R.; Ikeda, S.; Lee, F.Y.; Marrinucci, D.; Jia, S. Detection and Characterization of Circulating Tumour Cells in Multiple Myeloma. J. Circ. Biomarkers 2016, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Ndacayisaba, L.J.; Rappard, K.E.; Shishido, S.N.; Velasco, C.R.; Matsumoto, N.; Navarez, R.; Tang, G.; Lin, P.; Setayesh, S.M.; Naghdloo, A.; et al. Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones. Curr. Oncol. 2022, 29, 2954–2972. [Google Scholar] [CrossRef]
- Pérez-Persona, E.; Vidriales, M.-B.; Mateo, G.; García-Sanz, R.; Mateos, M.-V.; de Coca, A.G.; Galende, J.; Martín-Nuñez, G.; Alonso, J.M.; de Las Heras, N.; et al. New Criteria to Identify Risk of Progression in Monoclonal Gammopathy of Uncertain Significance and Smoldering Multiple Myeloma Based on Multiparameter Flow Cytometry Analysis of Bone Marrow Plasma Cells. Blood 2007, 110, 2586–2592. [Google Scholar] [CrossRef]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.-H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef] [Green Version]
- Bujarski, S.; Soof, C.; Chen, H.; Li, M.; Sanchez, E.; Wang, C.S.; Emamy-Sadr, M.; Swift, R.A.; Rahbari, K.J.; Patil, S.; et al. Serum b-cell maturation antigen levels to predict progression free survival and responses among relapsed or refractory multiple myeloma patients treated on the phase I IRUX trial. J. Clin. Oncol. 2018, 36, e24313. [Google Scholar] [CrossRef]
- Visram, A.; Soof, C.; Rajkumar, S.V.; Kumar, S.K.; Bujarski, S.; Spektor, T.M.; Kyle, R.A.; Berenson, J.R.; Dispenzieri, A. Serum BCMA levels predict outcomes in MGUS and smoldering myeloma patients. Blood Cancer J. 2021, 11, 120. [Google Scholar] [CrossRef]
- Secretase Inhibition Increases Efficacy of BCMA-Specific Chimeric Antigen Receptor T Cells in Multiple Myeloma|Blood|American Society of Hematology. Available online: https://ashpublications.org/blood/article/134/19/1585/374996/Secretase-inhibition-increases-efficacy-of-BCMA (accessed on 9 June 2022).
- Johnsen, H.E.; Bøgsted, M.; Schmitz, A.; Bødker, J.S.; El-Galaly, T.C.; Johansen, P.; Valent, P.; Zojer, N.; Van Valckenborgh, E.; Vanderkerken, K.; et al. The myeloma stem cell concept, revisited: From phenomenology to operational terms. Haematologica 2016, 101, 1451–1459. [Google Scholar] [CrossRef]
- Gao, M.; Bai, H.; Jethava, Y.; Wu, Y.; Zhu, Y.; Yang, Y.; Xia, J.; Cao, H.; Franqui-Machin, R.; Nadiminti, K.; et al. Identification and Characterization of Tumor-Initiating Cells in Multiple Myeloma. JNCI J. Natl. Cancer Inst. 2019, 112, 507–515. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Fernández de Larrea, C.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.-G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 146–154. [Google Scholar] [CrossRef]
- Elkins, K.; Zheng, B.; Go, M.; Slaga, D.; Du, C.; Scales, S.J.; Yu, S.-F.; McBride, J.; de Tute, R.; Rawstron, A.; et al. FcRL5 as a Target of Antibody–Drug Conjugates for the Treatment of Multiple Myeloma. Mol. Cancer Ther. 2012, 11, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.K.; Krishnan, A.Y.; Singhal, S.; Boccia, R.V.; Patel, M.R.; Niesvizky, R.; Chanan-Khan, A.A.; Ailawadhi, S.; Brumm, J.; Mundt, K.E.; et al. Phase I study of the anti-FcRH5 antibody-drug conjugate DFRF4539A in relapsed or refractory multiple myeloma. Blood Cancer J. 2019, 9, 17. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Du, T.; Buon, L.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Identification and validation of ecto-5′ nucleotidase as an immunotherapeutic target in multiple myeloma. Blood Cancer J. 2022, 12, 50. [Google Scholar] [CrossRef]
- Leow, C.; Low, M. Targeted Therapies for Multiple Myeloma. J. Pers. Med. 2021, 11, 334. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.N.; Welter, L.; Rodriguez-Lee, M.; Kolatkar, A.; Xu, L.; Ruiz, C.; Gerdtsson, A.S.; Restrepo-Vassalli, S.; Carlsson, A.; Larsen, J.; et al. Preanalytical Variables for the Genomic Assessment of the Cellular and Acellular Fractions of the Liquid Biopsy in a Cohort of Breast Cancer Patients. J. Mol. Diagn. 2020, 22, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Baslan, T.; Kendall, J.; Ward, B.; Cox, H.; Leotta, A.; Rodgers, L.; Riggs, M.; D’Italia, S.; Sun, G.; Yong, M.; et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015, 25, 714–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dago, A.E.; Stepansky, A.; Carlsson, A.; Luttgen, M.; Kendall, J.; Baslan, T.; Kolatkar, A.; Wigler, M.; Bethel, K.; Gross, M.E.; et al. Rapid Phenotypic and Genomic Change in Response to Therapeutic Pressure in Prostate Cancer Inferred by High Content Analysis of Single Circulating Tumor Cells. PLoS ONE 2014, 9, e101777. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MGUS1 | MGUS2 | SMM1 | SMM2 | NDMM1 | NDMM2 | NDMM3 | RRMM | LPL | |
|---|---|---|---|---|---|---|---|---|---|
| Age | 78 | 38 | 72 | 55 | 63 | 67 | 58 | 50 | 84 |
| Sex | Male | Female | Female | Female | Female | Female | Female | Male | Male |
| Diagnosis | MGUS | MGUS | SMM | SMM | NDMM | NDMM | NDMM | RRMM | LPL |
| Ig isotype | IgGk | IgMλ | IgGk | IgGk | IgGk | IgAk | IgGk | IgDλ | IgGλ |
| PCs in BM aspirate (%) | 1 | 4 | 10 | 10 | 15 | 76 | 7 | 27 | 2 |
| Aberrant PCs from the total PC BM compartment (%) | 92 | 0 | 78.9 | 98 | 95.2 | 100 | 99.6 | 99.9 | 94 |
| FC CD138 | Positive | NA | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| FC CD38 | Positive | NA | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| FC CD56 | Positive | NA | Positive | Negative | Positive | Positive | Positive | Positive | Negative |
| FC CD45 | Negative | NA | Negative | Negative | Negative | Positive | Negative | Negative | Positive |
| FC CD19 | Negative | NA | Negative | Negative | Negative | Negative | Negative | Negative | Positive |
| FC CD27 | Positive | NA | Positive | Positive | Negative | Negative | Negative | Negative | Positive |
| M-Spike (g/dL) | 0.7 | 0.4 | 3.9 | 1.5 | 0.4 | 0.2 | 2.1 | 0.3 | 0.2 |
| sFLC ratio | 8.14 | 1.06 | 37.58 | 8.45 | 186.84 | 1362.11 | 8.7 | 691.27 | 44.24 |
| Karyotype | Normal | Normal | Normal | Normal | Normal | Abnormal * | Normal | Normal | Normal |
| FISH | Three copies of CCND1 | Normal | Trisomy 11 | Negative | Three copies of CCND1 | t(4;14) and monosomy 13 | Normal | t(11:14) | Normal |
| Clinical presentation | Low-risk MGUS for progression to MM by PETHEMA [21] criteria | Low-risk MGUS by PETHEMA criteria | High-risk SMM by PETHEMA criteria | High-risk SMM by PETHEMA criteria | Standard-risk NDMM | High-risk NDMM | Standard-risk NDMM | Standard-risk RRMM | Newly diagnosed Waldenström’s macroglobulinemia |
| Fixation: 2% PFA for 20 min |
| Wash: 1 × TBS 2 × 3 min |
| Blocking: 10% filtered GS in TBS for 30 min |
| Primary Mix: |
| CD138 mouse IgG1 (2 µg/mL) |
| BCMA rabbit IgG (2.5 µg/mL) |
| CD45-Alexa647 mouse IgG2a (1.6 µg/mL) incubated 1 h RT |
| Wash: 1 × TBS 2 × 3 min |
| 10% filtered GS in TBS for 30 min |
| Secondary Mix: Alexa555 (1:500) + AlexaPLUS488 (1:500) + DAPI incubated 40 min RT |
| Wash and finish: 1 × TBS 2 × 3 min, dip ddH2O, coverslip with live cell media, seal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndacayisaba, L.J.; Rappard, K.E.; Shishido, S.N.; Setayesh, S.M.; Tang, G.; Lin, P.; Matsumoto, N.; Hsu, C.-J.; Nevarez, R.; Velasco, C.R.; et al. Characterization of BCMA Expression in Circulating Rare Single Cells of Patients with Plasma Cell Neoplasms. Int. J. Mol. Sci. 2022, 23, 13427. https://doi.org/10.3390/ijms232113427
Ndacayisaba LJ, Rappard KE, Shishido SN, Setayesh SM, Tang G, Lin P, Matsumoto N, Hsu C-J, Nevarez R, Velasco CR, et al. Characterization of BCMA Expression in Circulating Rare Single Cells of Patients with Plasma Cell Neoplasms. International Journal of Molecular Sciences. 2022; 23(21):13427. https://doi.org/10.3390/ijms232113427
Chicago/Turabian StyleNdacayisaba, Libere J., Kate E. Rappard, Stephanie N. Shishido, Sonia M. Setayesh, Guilin Tang, Pei Lin, Nicholas Matsumoto, Ching-Ju Hsu, Rafael Nevarez, Carmen Ruiz Velasco, and et al. 2022. "Characterization of BCMA Expression in Circulating Rare Single Cells of Patients with Plasma Cell Neoplasms" International Journal of Molecular Sciences 23, no. 21: 13427. https://doi.org/10.3390/ijms232113427
APA StyleNdacayisaba, L. J., Rappard, K. E., Shishido, S. N., Setayesh, S. M., Tang, G., Lin, P., Matsumoto, N., Hsu, C.-J., Nevarez, R., Velasco, C. R., Naghdloo, A., Yang, E., Kelly, K., Hicks, J., Mason, J., Orlowski, R. Z., Manasanch, E. E., & Kuhn, P. (2022). Characterization of BCMA Expression in Circulating Rare Single Cells of Patients with Plasma Cell Neoplasms. International Journal of Molecular Sciences, 23(21), 13427. https://doi.org/10.3390/ijms232113427