Abstract
Mitochondrion-related organelles (MROs) are loosely defined as degenerated mitochondria in anaerobic and microaerophilic lineages. Opalinids are commonly regarded as commensals in the guts of cold-blooded amphibians. It may represent an intermediate adaptation stage between the conventional aerobic mitochondria and derived anaerobic MROs. In the present study, we sequenced and analyzed the MRO genome of Cepedea longa. It has a linear MRO genome with large inverted repeat gene regions at both ends. Compared to Blastocystis and Proteromonas lacertae, the MRO genome of C. longa has a higher G + C content and repeat sequences near the central region. Although three Opalinata species have different morphological characteristics, phylogenetic analyses based on eight concatenated nad genes indicate that they are close relatives. The phylogenetic analysis showed that C. longa clustered with P. lacertae with strong support. The 18S rRNA gene-based phylogeny resolved the Opalinea clade as a sister clade to Karotomorpha, which then further grouped with Proteromonas. The paraphyly of Proteromonadea needs to be verified due to the lack of MRO genomes for key species, such as Karotomorpha, Opalina and Protoopalina. Besides, our dataset and analyses offered slight support for the paraphyly of Bigyra.
1. Introduction
Mitochondria are generally believed to be evolved from an endosymbiotic α-proteobacterium within an ancestral archaeal-derived host cell [1,2,3,4]. They exhibit diverse forms and are categorized into five classes based on the completeness of the electron transport chain (ETC) and energy metabolism: aerobic mitochondria, anaerobic mitochondria, hydrogen-producing mitochondria, hydrogenosomes and mitosomes [1,5,6]. The mitochondria belonging to the latter three classes are commonly called mitochondrion-related organelles (MROs), and they are often found in anaerobic or low-oxygen niches, such as gastrointestinal tracts, vaginas and anoxic sediments [7,8,9]. However, in recent years, many studies aiming to clarify MROs’ anaerobic metabolism and evolutionary relationships found that MROs might not be a strict classification with clear borders among classes but rather a spectrum of metabolic and functional phenotypes. For example, the flagellate Monocercomonoides sp. completely lost the mitochondrial genome [10,11,12,13,14]. Oxygen-restricted conditions are believed to be the major driving force for the transformation from aerobic mitochondria to MROs [8]. Mitochondria in classes 1–3 contain organelle genomes, but their size and contents are vastly reduced in contrast to the genomes of α-proteobacteria relatives [1].
The stramenopiles are an extraordinarily diverse group of eukaryotes, including photosynthetic lineages that range from diatoms to giant multicellular brown algae and nonphotosynthetic lineages that comprise free-living flagellates, parasites and organisms resembling fungi [15]. It also constitutes one of the most diverse clades of protists, which branches with Rhizaria and Alveolata within the SAR supergroup [15,16,17]. Several stramenopile taxa are parasites/commensals of Metazoa (e.g., Blastocystis, Proteromonas, Cepedea, Opalina, Protoopalina, Aureococcus, etc.) and plants (Phytophthora, etc.) [18]. For example, Blastocystis is a polymorphic and an unusual enteric protozoan parasite of humans and many other animals [19]. However, its pathogenicity is controversial since it is estimated that Blastocystis could be present in more than 1 billion humans, and it is commonly found in healthy individuals [20,21,22,23]. Proteromonas is an obligately anaerobic stramenopile that lives as a commensal in the intestine of urodelans, lizards and rodents [24,25]. Opalinids are commonly regarded as commensals in the guts of cold-blooded vertebrates, especially in the cloacae of amphibians [26,27]. Species belonging to the genus Cepedea are cylindrical and multinucleated, which distinguishes them from the other four genera of opalinids (Protoopalina, Zelleriella, Opalina and Protozelleriella) [26,28]. Cepedea longa was first discovered in the intestines of Fejervarya limnocharis (synonym: Rana limnocharis) and named by Bezzenberger [29]. The three lineages mentioned above exhibit different morphological forms. For example, there are three major forms (vacuolar, granular or ameboid) in Blastocystis without flagella, and the vacuolar form is considered the typical cell form [19,30]; the anterior part of Proteromonas possesses two flagella, one thicker and longer than the other [25,31]; and Cepedea is greatly elongated and cylindrical, as well as thickly flagellated on the cell surface [26]. However, species of the above-mentioned three opalinid lineages are phylogenetically relatively close (on the basis of 18S rRNA sequences) and form a monophyletic group within the Opalinata [27,31,32]. Mitochondria with tubular cristae were observed at the periphery of C. longa cells [26], which resemble the MROs in Blastocystis sp. and P. lacerate. The ancestor of Opalinidae and Blastocystis was most probably free-living since their closest relatives are all free-living [27,33]. In addition, Blastocystis is better adapted to anaerobic conditions than Opalinidae [34]. On this basis, opalinids may represent an intermediate adaptation stage between the conventional aerobic mitochondria and derived anaerobic MROs [34].
Both Blastocystis and Proteromonas can be grown in a man-made medium axenically [25,35,36,37], so it is relatively easy to gain research materials for investigating the genetics and biology of these species [25,38,39]. Although there is no in-vitro culturing method for C. longa, its cells are large and distinguishable in the recta of frogs (F. limnocharis), so it is possible to increase the number of cells via multiple sampling to conduct sequencing and analysis. Herein, we sequenced and analyzed the MRO genome of C. longa and conducted comparative mitogenomic and phylogenetic analyses with other organelle genomes within the Opalinata lineage.
2. Results
2.1. Cepedea longa and Its MRO Architecture
The body of C. longa is greatly elongated, slightly flattened, and thickly flagellated, with the cell surface twisted and coiled when moving (Figure 1B). A more detailed morphological description can be found in Li et al. [26]. Large amounts of MROs and nuclei were identified (Figure 1C,D), but the number of MROs was obviously larger than the number of nuclei (Figure 1E,F). They exhibited a range of shapes (Figure 1F) and double-membranes with highly convoluted cristae (Figure 1G).
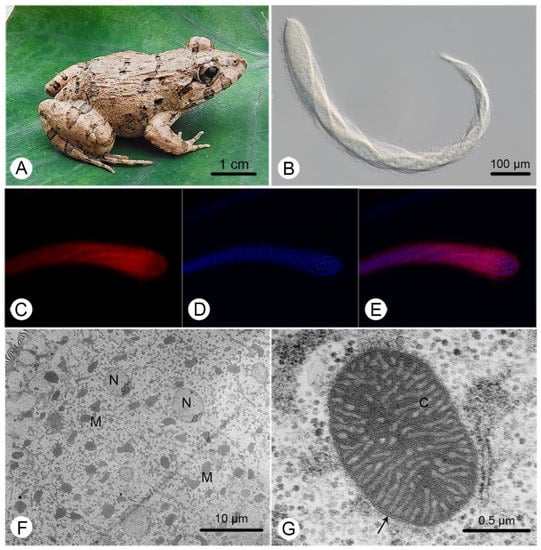
Figure 1.
General characteristics of Cepedea longa, its host and its mitochondrion-related organelles. (A) The photo of the most common host of C. longa, Fejervarya limnocharis; (B) a microscopic image of C. longa; (C) a C. longa specimen stained with Mitotracker Red; (D) a specimen stained with DAPI; (E) a merger of (C,D); (F) a transmission electron microscope (TEM) image of C. longa, showing various MRO (M) shapes and ovoid nuclei (N); (G) a TEM image of the MRO of C. longa, showing the double-membrane (arrow) and cristae (C).
The MRO genome of C. longa is 62,395 bp-long with a linear structure. It contains two large inverted repeat regions with 11 protein-coding genes (PCGs), 20 tRNAs and 2 rRNAs each (Figure 2, Supplementary Table S1). The C. longa MRO genome contains 40 PCGs, 16 of which encode NADH dehydrogenase subunit genes; 13 PCGs encode ribosomal proteins; whereas 11 open reading frames (ORFs) remain unidentified (Figure 2, Supplementary Table S1). Only one ribosomal protein was identified within the two large repeat regions (rps12), whereas all other ribosomal proteins were found within the non-repeat (unique) region (Figure 2). The total length of 40 PCGs is 38,313 bp, with an average length of 958 bp and an average G + C content of 23.3%, varying from 17.24% of orf291 to 31.91% of nad7. All PCGs used the ATG start codon, except for the orf233 and rpl16 genes. As regards stop codons, 27 were TAA, and 13 were TAG (Supplementary Table S1). There are conserved 5′ fragments with ATG start codon in both the pseudo-nad5 gene (443 bp) and pseudo-nad3 gene (333 bp). The pseudo-nad5 has a degraded 3′ end, and the pseudo-nad3 gene possesses an internal stop codon TAG (Supplementary Table S1).
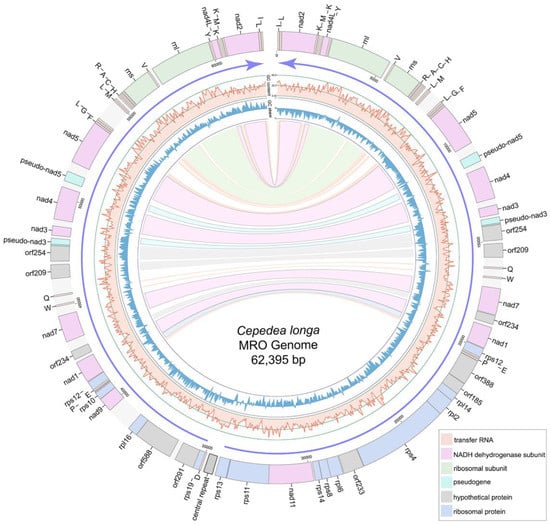
Figure 2.
Gene map of the Cepedea longa mitochondrion-related organellar genome. Light blue blocks are ribosomal proteins; pink blocks are nad genes; gray blocks are unidentified ORFs; light orange blocks are tRNA genes; light green blocks are small/large subunit ribosomal RNA genes of MRO genome; grey block with a black box between the tRNAAsp and rps13 genes is the central repeat region of MRO genome. Purple arrows show the transcriptional direction. GC content and GC skew are shown as light red lines and blue lines, respectively. Repeat genes are indicated by corresponding colored links.
A total of 41 tRNAs were detected in the MRO genome. Among these, 40 were located within the large repeat regions (Figure 2). Genes encoding tRNAThr, tRNASer and tRNAAsn were absent from the MRO genome. Genes rnl and rns are 2764 and 1346 bp in size, with G + C content of 33.36% and 36.48%, respectively (Supplementary Table S1). A set of tRNA genes, tRNAHis-tRNACys-tRNAAla-tRNAArg, was located ahead of rns (Figure 2), which was also detected in P. lacertae. There are 76 intergenic regions (from 1 bp to 1147 bp) interspersed within the MRO genome, with a total of 11,510 bp and an average of 151 bp (Table 1). Repeat regions add up to 1563 bp, accounting for about 2.5% of the MRO genome. The central repeats (several repeat units at the central position of the linear genome) of C. longa are 500 bp long in total, with five repeat units (Supplementary Table S3). Genes are arranged in the opposite transcriptional directions diverging from the central region, and GC skew values switch from negative to positive in the central region (Figure 2).

Table 1.
Comparison of characteristics of MRO genomes of Cepedea longa and other Opalinata species.
2.2. Comparison of MRO Genome Features in Opalinata Species
In accordance with Proteromonas and Blastocystis, no cytochrome b (cob), cytochrome oxidase subunits (cox1-cox3) and F0F1-ATPase subunits (atp) genes were found in the MRO genome of C. longa (Supplementary Table S2). The G + C content (25.7%) of the whole MRO genome is high in comparison to the other Opalinata species (18.9% to 22.7%), while the G + C content (25.4%) of concatenated intergenic regions (IGRs) is more than twice as high as others (8.4% to 12.0%) (Table 1). The Ka/Ks ratios (ω, non-synonymous substitutions/synonymous substitutions) for all PCGs of C. longa vs. P. lacertae ranged from 0.05 to 0.30 (Supplementary Figure S1). The functional constraints (negative selection) on nad3, nad4L, rps4, rps10, rps13 and rps19 genes were relaxed in comparison to other protein-coding genes. The selection pressure analysis indicated that nad5 (0.06), nad7 (0.05) and rps12 (0.05) genes are evolving slowly compared to nad3 (0.30) and rps4 (0.23) genes (Supplementary Figure S1).
Codon usage bias was mostly identical among C. longa, Blastocystis and P. lacertae, except for the most frequent codon for amino acids of Proline and Arginine. The codon TGA for tryptophan and codon CGG/AGG for arginine are unique in C. longa among the three Opalinata species. The effective number of codons (Nc) indicates that the usage of synonymous codons in C. longa is more balanced than in the other two species (Supplementary Table S4). Concatenated alignments of 20 PCGs, 2 rRNAs and 15 tRNAs of C. longa and P. lacertae were used to conduct the sliding window analysis. rnl, rns, nad4, nad5, nad7 and rps12 genes exhibited relatively low sequence variability with Pi values of 0.248, 0.284, 0.364, 0.367, 0.315 and 0.336, respectively. nad3 (0.470), nad11 (0.482) and some ribosomal proteins showed high sequence variability (Figure 3). In general, the nucleotide diversity of ribosomal proteins was higher than that of nad genes.
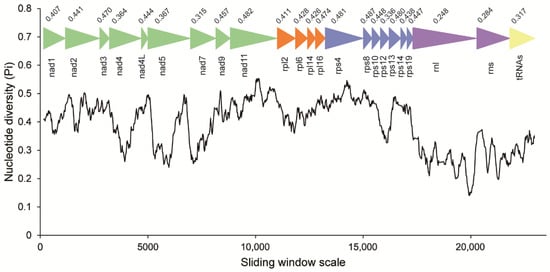
Figure 3.
Sliding window analysis of nucleotide diversity between the MRO genes of Cepedea longa and Proteromonas lacertae. The black curved line represents the value of nucleotide diversity. Gene names, boundaries and average nucleotide diversity values are shown above the line.
2.3. Phylogenetic Analyses
Branch topologies produced by ML and BI phylogenetic analyses of concatenated 8 nad genes were concordant (Figure 4). Opalinata species were divided into two clades: one containing Blastocystis (a representative of Blastocystidae) and the other containing C. longa (Opalinea) and P. lacertae (Proteromonadea). The species of Opalinata were grouped with high bootstrap support values (100 or 98) and Bayesian posterior probabilities (1.0) (Figure 4). However, the Proteromonadea clade was paraphyletic, due to the Karotomorpha species being resolved as close relatives with Opalinea based on 18S rRNA genes of more species (Supplementary Figure S2). We also found that Bigyra might be a paraphyletic (Figure 4).
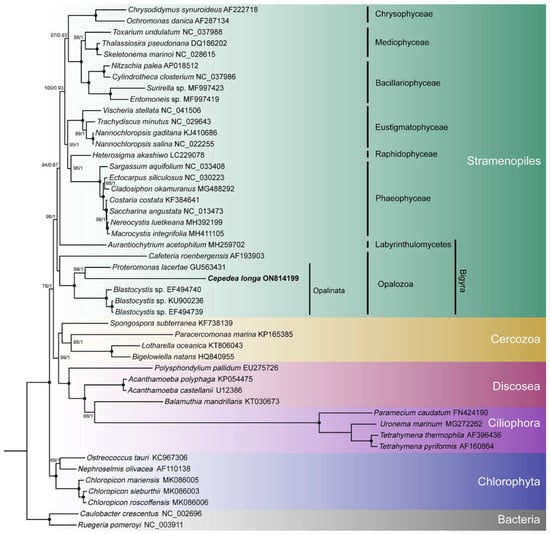
Figure 4.
Phylogenetic tree inferred using eight concatenated nad sequences. Numbers next to nodes are bootstrap values (ML) and posterior probability values (BI). Black dots represent 100% bootstrap support values and 1.00 posterior probability values. The α-proteobacteria species are set as outgroup taxa. GenBank accession numbers of sequences used in the phylogenetic analyses are listed next to the species name.
To explore the topology of the Bigyra, we focused on the phylogenetic relationships on Stramenopiles species. The results showed that Bigyra was paraphyletic (Figure 5). We also removed fast-evolving amino acid sites from the concatenated sequences and performed phylogenetic analysis anew, but the topology of Bigyra in the ML tree did not change (Supplementary Figure S3). Then we tried to remove fast-evolving taxa (Cafeteria) from the dataset, and this resolved monophyletic Bigyra but with a weak support bootstrap value (Supplementary Figure S4).
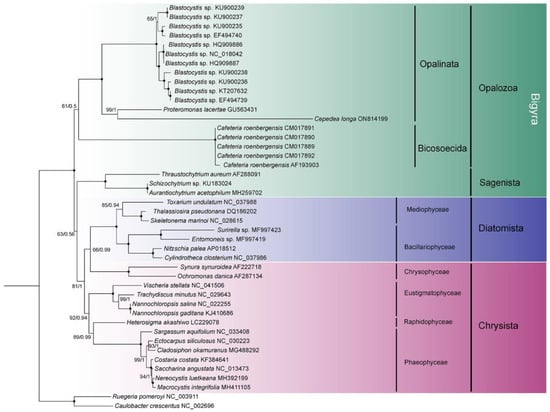
Figure 5.
The phylogenetic tree of Stramenopiles inferred using the dataset comprising concatenated nad sequences of mitochondrial genomes. Black dots represent 100% bootstrap support values and 1.00 posterior probability values. The tree is rooted using two bacterial species.
3. Discussion
Mitochondria are known as “powerhouses” that supply cells with energy [1]. Although their types and functions vary substantially among different eukaryotic lineages [5], they are all derived from endosymbiotic α-proteobacteria within an archaeal host cell closely related to the Asgard archaea. However, mitochondrial genomes are vastly reduced in gene content compared to the genomes of α-proteobacterial relatives [40,41]. Stramenopiles, together with Alveolata and Rhizaria, constitute species-rich clades of the super-group SAR [42,43]. The stramenopiles comprise photosynthetic and nonphotosynthetic lineages. All species in Opalinata live as parasites or commensals in the intestinal tracts of amphibians, lizards, birds and mammals [20,27,34]. Although the ultrastructure of the C. longa MRO is similar to the standard aerobic mitochondria (double-membrane structure and cristae formed by the inner membrane), it lacks genes encoding the complex III-IV of the electron transport chain (ETC) and ATP synthase; this may be because C. longa mainly inhabits the recta with low oxygen concentration [26]. Besides, genes encoding the succinate dehydrogenase (complex II in ETC) were detected in the nuclear genome of C. longa (data not shown). This suggests that C. longa has a highly reduced ETC, akin to Blastocystis [44,45].
Mitochondrial genomes vary extensively in size, structure, organization and gene content among eukaryotes [25]. The IGRs of the MRO genome of C. longa are longer than other Opalinata species. Also, it exhibits an inverted repeats structure that was found in another stramenopile, P. lacertae. This may have been produced by a recombination and gene inversion event. Similar structures have been observed in other lineages, such as nematodes [46], fishes [47] and birds [48]. We speculate that the IGR between tRNAAsp and rps13 genes is the most likely origin of replication, as there are five tandem repeat units with 100 bp each in this region, which we called central tandem repeats, and the direction of gene transcription and GC skew values switch from this region. The central tandem repeats were also found in ciliates with linear mitochondrial genomes (e.g., species in Spirotricha), and they are probably associated with replication and transcription initiation [49,50]. We speculate that the central tandem repeats of C. longa might play an important role in the direction of gene transcription.
Generally, sequences can evolve under negative selection (Ka/Ks < 1), neutral selection (Ka/Ks = 1) or positive selection (Ka/Ks > 1) [51,52]. Ka/Ks values of PCGs in the C. longa MRO genome were all smaller than 1, compared to corresponding genes in Opalinata. This indicates that these PCGs are evolving under negative selection, which is the most prevalent form of selection maintaining the long-term stability of biological structures as it constantly sweeps away deleterious mutations [53]. Biased gene conversion is a recombination-associated evolutionary process that may drive gene evolution, and it tends to increase the G + C content over evolutionary time [54,55]. The G + C content of C. longa is higher than that of P. lacertae and Blastocystis, especially in the G + C content of IGRs (2–3 times as high as in other Opalinata species), which is generally regarded as neutrally evolving positions [52,56].
Pseudogenes are generally defined as nonfunctional sequences originally derived from functional genes [57]. In the C. longa MRO genome, two pseudo-nad3 genes are probably non-functional since the TAG stop codon is located inside the gene, while two pseudo-nad5 genes possess highly degraded 3′ regions. All pseudogenes have conserved 5′ regions exhibiting high identities with the functional nad5 and nad3 genes. We also found the order of genes corresponded to P. lacerate; for example, rps14-rps8-rpl6 and L-H-C-A-R-rns-V. However, the evolutionary routes of gene order rearrangements in Opalinata are still unclear, since the MRO genome data remain scarce or even unavailable for many lineages. To further research this topic, more MRO genomes of Opalinata lineages, such as Karotomorpha, Protoopalina or Opalina, remain to be sequenced, analyzed and compared.
Blastocystis, P. lacerate and C. longa inhabit the intestinal tracts of homeothermic animals, terrestrial ectothermic animals and amphibious ectothermic animals, which represent three different habitats of Opalinata. Although their morphological characteristics and lifestyles are distinctly different, phylogenetic analyses based on eight concatenated nad genes indicate that Cepedea, Proteromonas and Blastocystis are phylogenetically closely related. More specifically, P. lacertae and C. longa were resolved as sister clade with high support based on the MRO nad genes. All available 18S rRNA gene sequences from Opalinea and Proteromonadea in the GenBank database were also downloaded to reconstruct the phylogenetic relationships among Opalinata. The results showed that Proteromonadea is paraphyletic: Proteromonas was monophyletic, while Karotomorpha was closely related to opalinids [32,58]. These relations were also postulated by Patterson [59] via ultrastructural studies on ribbons of microtubules and flagellar transitional regions [24]. Besides, our results also support the monophyly of Opalinea and Blastocystidae.
Phylogenetic analysis of concatenated nad genes extracted from mitochondrial genome indicated the paraphyly of Bigyra, comprising Opalozoa and Sagenista, in our study. The topology was in accordance with that of Noguchi et al. [60], Derelle et al. [15] and Cho et al. [61]. Previously, the monophyly of Bigyra was recovered using the dataset of 339 protein alignments when divergent opalozoan lineages (Blastocystis and Cafeteria) were successively removed, which indicates that long-branch attraction might hamper phylogenetic reconstruction in the Bigyra lineage due to these fast-evolving taxa [15]. Following this evidence, we also attempted to remove Blastocystis or Cafeteria species in phylogenetic analysis and found that the monophyly of Bigyra lineage was recovered when Cafeteria species were removed (Supplementary Figure S4). The topology of the ML tree was congruent with phylogenetic analysis based on a 120-gene dataset [62] and the trees obtained after the removal of divergent taxa [15]. However, it was paraphyletic when Blastocystis was removed (Supplementary Figure S5). Although the fast-evolving amino acid sites in the data matrix were removed (using the threshold of 20% conservation value), the tree topology of Bigyra was still paraphyletic. In conclusion, concatenated mitochondrial genes offer weak support for the paraphyly of Bigyra.
4. Materials and Methods
4.1. Specimen Collection, Identification and Observation
Cepedea longa specimens were collected from the recta of frogs F. limnocharis (Figure 1A) captured in the Meishan, Sichuan Province, China (30°04′–30°16′ N, 103°53′–104°30′ E). Animals were handled in accordance with the recommended guidelines for animal experimentation by the Chinese Association for Laboratory Animals Sciences, and animal procedures were approved by the Animal Care and Ethics Committee of Institute of Hydrobiology, Chinese Academy of Sciences (project identification code: IHB/LL/2019013). Briefly, all frogs were transported alive into the laboratory for further examination, they were anaesthetized and dissected as soon as possible. Opalinids were collected into Petri dishes with sterile 0.65% saline solution after examination of the recta. C. longa cells were transferred to a fresh sterile 0.65% saline solution to remove other opalinids and frog cell contaminants. For MRO fluorescent staining, cells were stained with the MitoTracker Red CMXRos (Meilun Biotechnology, Dalian, China) using the working concentration of 250 nM for 30 min. Then the staining solution was discarded with a micropipette, and cells were again washed with the 0.65% saline solution; finally, cells were stained with DAPI for 10 min. Living specimens and fluorescent-stained cells (579 nm wavelength excitation for MitoTracker Red signals and 359 nm wavelength excitation for DAPI signals) were photographed with a microscope (ZEISS Axio Imager A2, Carl Zeiss, Jena, Germany) equipped with a digital camera (sCMOS PCO, Kelheim, Germany). For transmission electron microscopy, specimens were fixed in 2.5% glutaraldehyde in 0.2 M phosphate-buffered saline (PBS, pH7.4) at 4°C for 24 h; then post-fixed in 2% osmium tetroxide with PBS at 4°C for 3 h; followed by dehydration in a gradient series of acetone; and finally embedded in Araldite. Ultra-thin sections were cut and stained with uranyl acetate and lead citrate before being observed and photographed using an HT-7700 transmission electron microscope (Hitachi, Tokyo, Japan).
4.2. DNA Extraction, MRO Genome Sequencing and Assembly
Cells were preserved in absolute ethanol and stored at −20°C. The total genomic DNA was extracted from preserved C. longa cells (~10,000 cells) for the Illumina sequencing and Sanger sequencing using a standard phenol/chloroform method. The next-generation sequencing library was constructed using the NexteraXT DNA Library Preparation Kit (illumina, CA, USA) and sequenced on the Illumina Novaseq platform (San Diego, CA, USA). For the MRO genome assembly, after trimming the adapters and filtering low-quality reads (reads with 5% unidentified nucleotides and with quality values below 20), the resulting clean reads were assembled de novo using software SPAdes v3.14.1 set as the default parameters. The contigs were aligned to MRO genome sequences of Blastocystis and P. lacerate using blastn v2.9.0 with the e-value < 1 × 10−5. The probable MRO genome sequences of C. longa were filtered, contigs extended using PRICE (Paired-Read Iterative Contig Extension) [63] and mapped with the clean reads using bowtie2 [64]. Finally, the results were assembled de novo again using SPAdes 3.14.0 [65]. The process was repeated until the total size of the draft MRO genome stabilized. To verify the MRO genome sequence of C. longa, “primer walking” and Sanger sequencing were conducted according to the draft MRO genome generated from next-generation sequences assembly.
4.3. MRO Genome Annotation and Analysis
The protein-coding genes (PCGs), transfer RNAs (tRNAs) and large/small rRNA subunit genes (rnl/rns) of the MRO genome of C. longa were initially annotated using the MFannot tool (https://megasun.bch.umontreal.ca/apps/mfannot/ (accessed on 24 May 2020)) using the genetic code 4. The tRNA gene prediction was additionally performed using tRNAscan-SE [66] and RNAweasel (https://megasun.bch.umontreal.ca/cgi-bin/RNAweasel/RNAweaselInterface.pl (accessed on 25 May 2020)). The final tRNA prediction results were confirmed if they were predicted by all three tools. The boundaries of protein open reading frames (ORFs) were inferred with the help of NCBI’s ORF Finder, applying the genetic code 4 (https://www.ncbi.nlm.nih.gov/orffinder/ (accessed on 30 May 2020)). PCGs were identified based on NCBI’s BLAST homology searches with ORFs. We also used the HHpred web server and UniProt BLAST (Pfam and UniProtKB/Swiss-Prot were selected as target databases, respectively) to identify and confirm the PCGs [67,68]. PCGs that could not be identified with confidence were annotated as hypothetical proteins. The rnl and rns genes were verified by aligning them with close relatives Blastocystis and P. lacerate.
The MRO genome map was created using Circos v0.69-8 [69]. Eight completely sequenced mitochondrial genomes of stramenopiles were chosen to compare gene contents. Codon usage was calculated in the MRO genomes of C. longa, Blastocystis sp. and P. lacerate and compared using the CodonW program version 1.4.2. The KaKs_Calculator was used to estimate selective pressure on PCGs [51]. The sliding window analysis was conducted using DnaSP v6 [70], with a sliding window of 300 bp and a step size of 20 bp implemented to estimate the nucleotide divergence between C. longa and P. lacertae.
4.4. Phylogenetic Analyses
Phylogenetic analyses were conducted using the newly sequenced C. longa MRO genome and 44 mitochondrial genome sequences from other stramenopiles, as well as some other eukaryotic and prokaryotic organisms, which were retrieved from the Genbank. Two species of prokaryotes, Caulobacter crescentus and Ruegeria pomeroyi, were set as outgroups. Eight nad subunit protein sequences were aligned using the MAFFT v7.313 [71] plugin in PhyloSuite v1.2.1 [72] and then concatenated in the order nad1, nad2, nad3, nad4, nad4L, nad5, nad7, nad9 using the “Concatenate Sequence” function in PhyloSuite. The best partitioning scheme and the most appropriate evolutionary models were selected by PartitionFinder2 [73] with a greedy algorithm and AICc criterion. Phylogenetic analyses were conducted using two programs implemented in PhyloSuite: IQ-Tree v1.6.8 [74] was used for the Maximum likelihood (ML) analysis, and Bayesian inference (BI) analysis was performed using MrBayes v3.2.6 [75]. The ML analysis was conducted with the LG + I + G + F model of amino acid evolution and 5000 ultrafast bootstrap replicates, whereas the BI analysis was performed with 2,000,000 MCMC generations where trees were sampled every 1000 generations and the burn-in set at 25% (500,000 samples). Finally, trees were visualized by Figtree v1.4.0 and further edited in iTOL [76].
We also conducted phylogenetic analyses on Opalinata using 18S rRNA sequences: 32 sequences of Opalinata and 3 sequences of Cafeteria were obtained from GenBank (among these, the 18S rRNA sequence of C. longa was submitted by us in our previous study). The phylogenetic trees were reconstructed using ML and BI methods.
To verify the topology of the Bigyra lineage, a total of 44 stramenopiles affiliated to the Bigyra (Opalozoa + Sagenista) and Gyrista (Diatomista + Chrysista), with a bacterial outgroup, were selected to reconstruct the phylogenetic tree using the ML method in IQ-Tree. Bayesian inference was performed using the site-heterogeneous mixture model in PhyloBayes-mpi version 1.8 [77]. Two independent runs were performed until the two chains converged (a threshold of maxdiff < 0.1). Fast-evolving sites were removed using the trimAl v1.2 [78] by setting the threshold to the conservation value of 80%.
5. Conclusions
In this study, the opalinid C. longa inhabiting the recta of F. limnocharis were collected to sequence their MRO genome, which is the first MRO genome reported within the opalinid lineage. Its MRO genome was the longest in Opalinata (to date), with two inverted repeat structures. Gene rearrangements seem to exist within the Opalinata, but this needs to be further clarified by sequencing further MRO genomes in this lineage. Although three Opalinata species have different morphological characteristics, phylogenetic analyses based on eight concatenated nad genes indicate that they are close relatives. Phylogenetic analyses based on the 18S rRNA gene also support the monophyly of Opalinea and Blastocystidae and the paraphyly of Proteromonadea. Besides, our dataset and analyses offered weak support for the paraphyly of Bigyra.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232113472/s1.
Author Contributions
Formal analysis, investigation and writing—original draft, W.Z.; methodology and validation, X.B.; investigation and visualization, H.Z.; validation and writing—review and editing, W.L. and S.W.; data curation, project administration and funding acquisition, M.L.; supervision, funding acquisition, G.W. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (No. 32170437), the China Postdoctoral Science Foundation (No. 2021M703435), the Second Tibetan Plateau Scientific Expedition and Research Program (STEP) (No. 2019QZKK0304), the earmarked fund for CARS (No. CARS-45), the Protist 10,000 Genomics Project (P10K) Consortium and the National Aquatic Biological Resource Center (NABRC).
Institutional Review Board Statement
The animal study protocol was approved by the Animal Care and Ethics Committee of Institute of Hydrobiology, Chinese Academy of Sciences (project identification code: IHB/LL/2019013; date of approval: 18 June 2020).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw paired-end reads in this study were deposited in the NCBI SRA with BioProject ID: PRJNA850552. MRO genome sequence of Cepedea longa was deposited in Genbank under the accession number ON814199.
Acknowledgments
We are grateful to Ivan Jakovlić for polishing the language and revising the manuscript and to Dong Zhang for his constructive suggestions on phylogeny.
Conflicts of Interest
All the authors declare that they have no conflicts of interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The origin and diversification of mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Müller, M. The hydrogen hypothesis for the first eukaryote. Nature 1998, 392, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Oyaizu, Y.; Oyaizu, H.; Olsen, G.J.; Woese, C.R. Mitochondrial origins. Proc. Natl. Acad. Sci. USA 1985, 82, 4443–4447. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.G.; Kurland, C.G. Origins of mitochondria and hydrogenosomes. Curr. Opin. Microbiol. 1999, 2, 535–541. [Google Scholar] [CrossRef]
- Müller, M.; Mentel, M.; van Hellemond, J.J.; Henze, K.; Woehle, C.; Gould, S.B.; Yu, R.Y.; van der Giezen, M.; Tielens, A.G.; Martin, W.F. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 2012, 76, 444–495. [Google Scholar] [CrossRef]
- Hjort, K.; Goldberg, A.V.; Tsaousis, A.D.; Hirt, R.P.; Embley, T.M. Diversity and reductive evolution of mitochondria among microbial eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.A.; Hayes, A.J.; Suller, M.T. Hydrogenosomes of Metopus contortus physiologically resemble mitochondria. Microbiology 1997, 143, 1623–1629. [Google Scholar] [CrossRef]
- Santos, H.J.; Makiuchi, T.; Nozaki, T. Reinventing an organelle: The reduced mitochondrion in parasitic protists. Trends Parasitol. 2018, 34, 1038–1055. [Google Scholar] [CrossRef]
- Leger, M.M.; Kolisko, M.; Kamikawa, R.; Stairs, C.W.; Kume, K.; Cepicka, I.; Silberman, J.D.; Andersson, J.O.; Xu, F.; Yabuki, A.; et al. Organelles that illuminate the origins of Trichomonas hydrogenosomes and Giardia mitosomes. Nat. Ecol. Evol. 2017, 1, 0092. [Google Scholar] [CrossRef]
- Karnkowska, A.; Vacek, V.; Zubacova, Z.; Treitli, S.C.; Petrzelkova, R.; Eme, L.; Novak, L.; Zarsky, V.; Barlow, L.D.; Herman, E.K.; et al. A eukaryote without a mitochondrial organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef]
- Stairs, C.W.; Leger, M.M.; Roger, A.J. Diversity and origins of anaerobic metabolism in mitochondria and related organelles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140326. [Google Scholar] [CrossRef] [PubMed]
- Stairs, C.W.; Eme, L.; Brown, M.W.; Mutsaers, C.; Susko, E.; Dellaire, G.; Soanes, D.M.; van der Giezen, M.; Roger, A.J. A SUF Fe-S cluster biogenesis system in the mitochondrion-related organelles of the anaerobic protist Pygsuia. Curr. Biol. 2014, 24, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Maguire, F.; Richards, T.A. Organelle evolution: A mosaic of ‘mitochondrial’ functions. Curr. Biol. 2014, 24, R518–R520. [Google Scholar] [CrossRef] [PubMed]
- Rotterova, J.; Salomaki, E.; Panek, T.; Bourland, W.; Zihala, D.; Taborsky, P.; Edgcomb, V.P.; Beinart, R.A.; Kolisko, M.; Cepicka, I. Genomics of new ciliate lineages provides insight into the evolution of obligate anaerobiosis. Curr. Biol. 2020, 30, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Derelle, R.; Lopez-Garcia, P.; Timpano, H.; Moreira, D. A phylogenomic framework to study the diversity and evolution of stramenopiles (=heterokonts). Mol. Biol. Evol. 2016, 33, 2890–2898. [Google Scholar] [CrossRef]
- Adl, S.M.; Simpson, A.G.; Lane, C.E.; Lukes, J.; Bass, D.; Bowser, S.S.; Brown, M.W.; Burki, F.; Dunthorn, M.; Hampl, V.; et al. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 2012, 59, 429–493. [Google Scholar] [CrossRef]
- Burki, F. The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb. Perspect. Biol. 2014, 6, a016147. [Google Scholar] [CrossRef]
- Walker, G.; Dorrell, R.G.; Schlacht, A.; Dacks, J.B. Eukaryotic systematics: A user’s guide for cell biologists and parasitologists. Parasitology 2011, 138, 1638–1663. [Google Scholar] [CrossRef]
- Stenzel, D.J.; Boreham, P.F. Blastocystis hominis revisited. Clin. Microbiol. Rev. 1996, 9, 563–584. [Google Scholar] [CrossRef]
- Stensvold, C.R.; Tan, K.S.W.; Clark, C.G. Blastocystis. Trends Parasitol. 2020, 36, 315–316. [Google Scholar] [CrossRef]
- Eme, L.; Gentekaki, E.; Curtis, B.; Archibald, J.M.; Roger, A.J. Lateral gene transfer in the adaptation of the anaerobic parasite Blastocystis to the gut. Curr. Biol. 2017, 27, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Audebert, C.; Even, G.; Cian, A.; Blastocystis Investigation, G.; Loywick, A.; Merlin, S.; Viscogliosi, E.; Chabe, M. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci. Rep. 2016, 6, 25255. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.L.; Gilchrist, C.A.; Lynn, T.C.; Petri, W.A. Parasitic protozoa and interactions with the host intestinal microbiota. Infect. Immun. 2017, 85, e00101-17. [Google Scholar] [CrossRef] [PubMed]
- Kostka, M.; Cepicka, I.; Hampl, V.; Flegr, J. Phylogenetic position of Karotomorpha and paraphyly of Proteromonadidae. Mol. Phylogenet. Evol. 2007, 43, 1167–1170. [Google Scholar] [CrossRef]
- Perez-Brocal, V.; Shahar-Golan, R.; Clark, C.G. A linear molecule with two large inverted repeats: The mitochondrial genome of the stramenopile Proteromonas lacertae. Genome Biol. Evol. 2010, 2, 257–266. [Google Scholar] [CrossRef]
- Li, C.; Jin, X.; Li, M.; Wang, G.; Zou, H.; Li, W.; Wu, S. Light and transmission electron microscopy of Cepedea longa (Opalinidae) from Fejervarya limnocharis. Parasite 2017, 24, 6. [Google Scholar] [CrossRef]
- Li, M.; Ponce-Gordo, F.; Grim, J.N.; Li, C.; Zou, H.; Li, W.; Wu, S.; Wang, G. Morphological redescription of Opalina undulata nie 1932 from Fejervarya limnocharis with molecular phylogenetic study of opalinids (Heterokonta, Opalinea). J. Eukaryot. Microbiol. 2018, 65, 783–791. [Google Scholar] [CrossRef]
- Metcalf, M.M. Opalina and the origin of the Ciliata. J. Wash. Acad. Sci. 1918, 8, 427–431. [Google Scholar]
- Bezzenberger, E. Über Infusorien aus asiatischen Anuren. Archiv Für Protistenkd. 1904, 3, 138–174. [Google Scholar]
- Boreham, P.F.L.; Stenzel, D.J. Blastocystis in humans and animals: Morphology, biology, and epizootiology. Adv. Parasitol. 1993, 32, 1–70. [Google Scholar] [CrossRef]
- Kostka, M. Opalinata. In Handbook of the Protists; Springer: Cham, Switzerland, 2017; pp. 543–565. [Google Scholar]
- Wang, R.Q.; Zhao, W.S.; Hu, G.R.; Ponce-Gordo, F.; Zou, H.; Li, W.X.; Wu, S.G.; Wang, G.T.; Li, M. Redescription of Opalina triangulata (Heterokonta, Opalinea) from Fejervarya limnocharis based on morphological and molecular data. Eur. J. Protistol. 2019, 71, 125639. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, T.; Thakur, R.; Ishida, K.-I. Pseudophyllomitus vesiculosus (Larsen and Patterson 1990) Lee, 2002, a poorly studied phagotrophic biflagellate is the first characterized member of stramenopile environmental clade MAST-6. Protist 2017, 168, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Yubuki, N.; Galindo, L.J.; Reboul, G.; Lopez-Garcia, P.; Brown, M.W.; Pollet, N.; Moreira, D. Ancient adaptive lateral gene transfers in the symbiotic Opalina-Blastocystis stramenopile lineage. Mol. Biol. Evol. 2020, 37, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Lanuza, M.D.; Carbajal, J.A.; Villar, J.; Borrás, R. Description of an improved method for Blastocystis hominis culture and axenization. Parasitol. Res. 1997, 83, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Zierdt, C.H.; Williams, R.L. Blastocystis hominis: Axenic cultivation. Exp. Parasitol. 1974, 36, 233–243. [Google Scholar] [CrossRef]
- Ho, L.C.; Singh, M.; Suresh, G.; Ng, G.C.; Yap, E.H. Axenic culture of Blastocystis hominis in Iscove’s modified Dulbecco’s medium. Parasitol. Res. 1993, 79, 614. [Google Scholar] [CrossRef] [PubMed]
- Denoeud, F.; Roussel, M.; Noel, B. Genome sequence of the stramenopile Blastocystis, a human anaerobic parasite. Genome Biol. 2011, 12, R29. [Google Scholar] [CrossRef]
- Wawrzyniak, I.; Roussel, M.; Diogon, M.; Couloux, A.; Texier, C.; Tan, K.S.; Vivares, C.P.; Delbac, F.; Wincker, P.; El Alaoui, H. Complete circular DNA in the mitochondria-like organelles of Blastocystis hominis. Int. J. Parasitol. 2008, 38, 1377–1382. [Google Scholar] [CrossRef]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Bäckström, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 2015, 5, 7949. [Google Scholar] [CrossRef]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G.B. The new tree of eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Adl, S.M.; Bass, D.; Lane, C.E.; Lukes, J.; Schoch, C.L.; Smirnov, A.; Agatha, S.; Berney, C.; Brown, M.W.; Burki, F.; et al. Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 2019, 66, 4–119. [Google Scholar] [CrossRef] [PubMed]
- Gentekaki, E.; Curtis, B.A.; Stairs, C.W.; Klimes, V.; Elias, M.; Salas-Leiva, D.E.; Herman, E.K.; Eme, L.; Arias, M.C.; Henrissat, B.; et al. Extreme genome diversity in the hyper-prevalent parasitic eukaryote Blastocystis. PLoS Biol. 2017, 15, e2003769. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.S.; Andersen, L.O.; Bitar, P.P.; Richards, V.P.; Shah, S.; Stanhope, M.J.; Stensvold, C.R.; Clark, C.G. Blastocystis mitochondrial genomes appear to show multiple independent gains and losses of start and stop codons. Genome Biol. Evol. 2016, 8, 3340–3350. [Google Scholar] [CrossRef]
- Zou, H.; Jakovlic, I.; Chen, R.; Zhang, D.; Zhang, J.; Li, W.X.; Wang, G.T. The complete mitochondrial genome of parasitic nematode Camallanus cotti: Extreme discontinuity in the rate of mitogenomic architecture evolution within the Chromadorea class. BMC Genom. 2017, 18, 1–17. [Google Scholar] [CrossRef]
- Ciborowski, K.L.; Consuegra, S.; Garcia de Leaniz, C.; Beaumont, M.A.; Wang, J.; Jordan, W.C. Rare and fleeting: An example of interspecific recombination in animal mitochondrial DNA. Biol. Lett. 2007, 3, 554–557. [Google Scholar] [CrossRef][Green Version]
- Sammler, S.; Bleidorn, C.; Tiedemann, R. Full mitochondrial genome sequences of two endemic Philippine hornbill species (Aves: Bucerotidae) provide evidence for pervasive mitochondrial DNA recombination. BMC Genom. 2011, 12, 35. [Google Scholar] [CrossRef]
- Zhang, T.; Li, C.; Zhang, X.; Wang, C.; Roger, A.J.; Gao, F. Characterization and comparative analyses of mitochondrial genomes in single-celled eukaryotes to shed light on the diversity and evolution of linear molecular architecture. Int. J. Mol. Sci. 2021, 22, 2546. [Google Scholar] [CrossRef]
- Hauth, A.M.; Maier, U.G.; Lang, B.F.; Burger, G. The Rhodomonas salina mitochondrial genome: Bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res. 2005, 33, 4433–4442. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Barth, D.; Berendonk, T.U. The mitochondrial genome sequence of the ciliate Paramecium caudatum reveals a shift in nucleotide composition and codon usage within the genus Paramecium. BMC Genomics 2011, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, C.D.; Montooth, K.L.; Rand, D.M. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kostka, D.; Hubisz, M.J.; Siepel, A.; Pollard, K.S. The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol. 2012, 29, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Pessia, E.; Popa, A.; Mousset, S.; Rezvoy, C.; Duret, L.; Marais, G.A. Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol. Evol. 2012, 4, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Lee, R.W. Mitochondrial genome of the colorless green alga Polytomella capuana: A linear molecule with an unprecedented GC content. Mol. Biol. Evol. 2008, 25, 487–496. [Google Scholar] [CrossRef]
- Balakirev, E.S.; Ayala, F.J. Pseudogenes: Are they “junk” or functional DNA? Annu. Rev. Genet. 2003, 37, 123–151. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, G.; Ponce-Gordo, F.; Zou, H.; Li, W.; Wu, S.; Li, M.; Wang, G. Morphological description of Opalina obtrigonoidea Metcalf, 1923 (Heterokonta, Opalinea) from Duttaphrynus melanostictus and evaluation of the ITS region as a suitable genetic marker for inter-species identification in Opalina. Parasitol. Int. 2020, 76, 102103. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D. The fine strusture of Opalina ranarum (Family Opalinidae): Opalinid phylogeny and classification. Protistologica 1985, 21, 413–428. [Google Scholar]
- Noguchi, F.; Tanifuji, G.; Brown, M.W.; Fujikura, K.; Takishita, K. Complex evolution of two types of cardiolipin synthase in the eukaryotic lineage stramenopiles. Mol. Phylogenet. Evol. 2016, 101, 133–141. [Google Scholar] [CrossRef]
- Cho, A.; Tikhonenkov, D.V.; Hehenberger, E.; Karnkowska, A.; Mylnikov, A.P.; Keeling, P.J. Monophyly of diverse Bigyromonadea and their impact on phylogenomic relationships within stramenopiles. Mol. Phylogenet. Evol. 2022, 171, 107468. [Google Scholar]
- Thakur, R.; Shiratori, T.; Ishida, K.I. Taxon-rich multigene phylogenetic analyses resolve the phylogenetic relationship among deep-branching stramenopiles. Protist 2019, 170, 125682. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Bellare, P.; DeRisi, J.L. PRICE: Software for the targeted assembly of components of (Meta) genomic sequence data. G3 Genes Genomes Genet. 2013, 3, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Humana: New York, NY, USA, 2019; pp. 1–14. [Google Scholar]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar]
- Krzywinski, M.I.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).