
Uremic Myopathy and Mitochondrial Dysfunction in Kidney Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
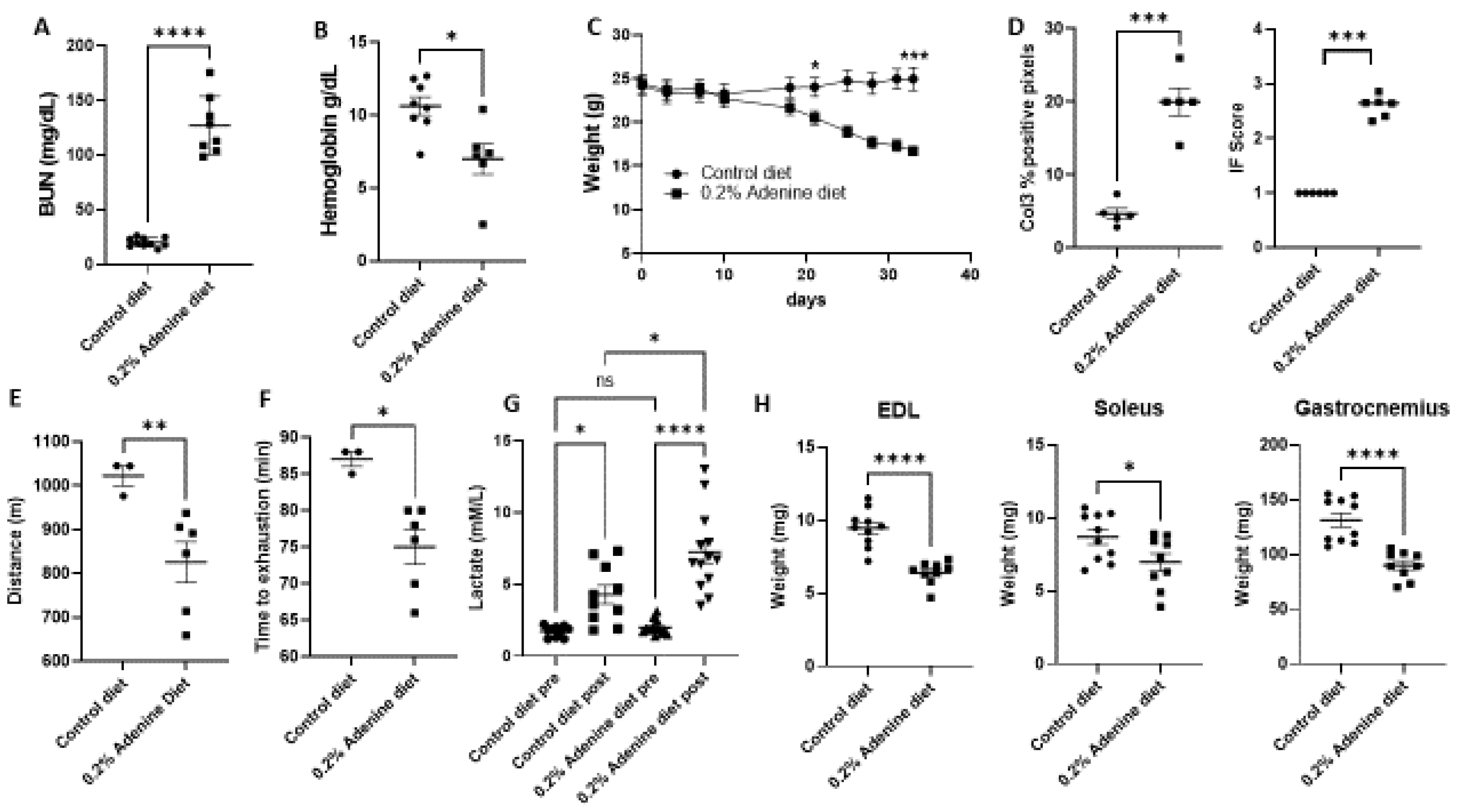
2.1. Characterization of Mouse Kidney Disease Model by Using Adenine Supplementation in Diet
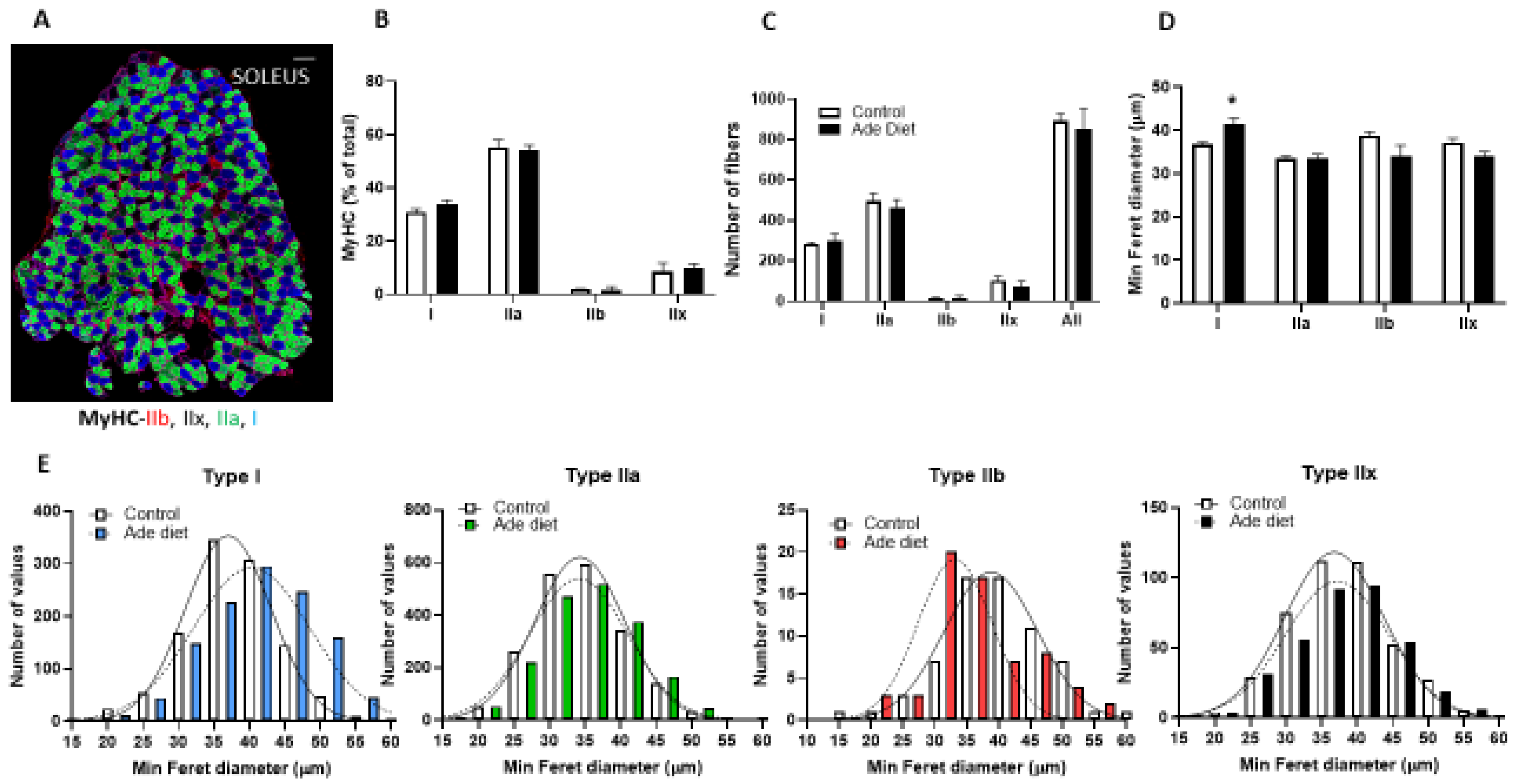
2.2. Muscle Fiber Typing Profile of Mice Exposed to Adenine
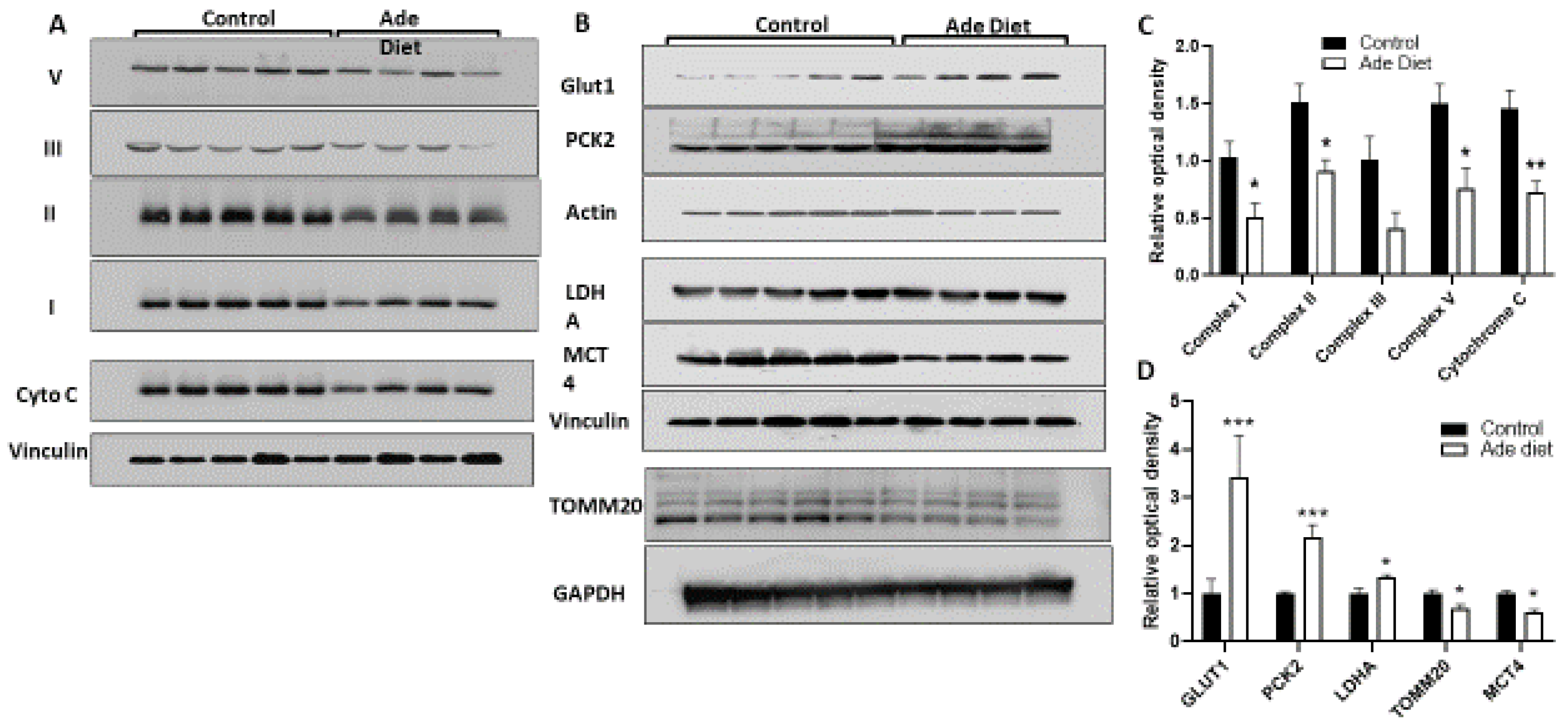
2.3. Muscle of Mice with CKD Have Decreased Expression of OXPHOS Proteins and Increased Expression of Glycolytic Proteins
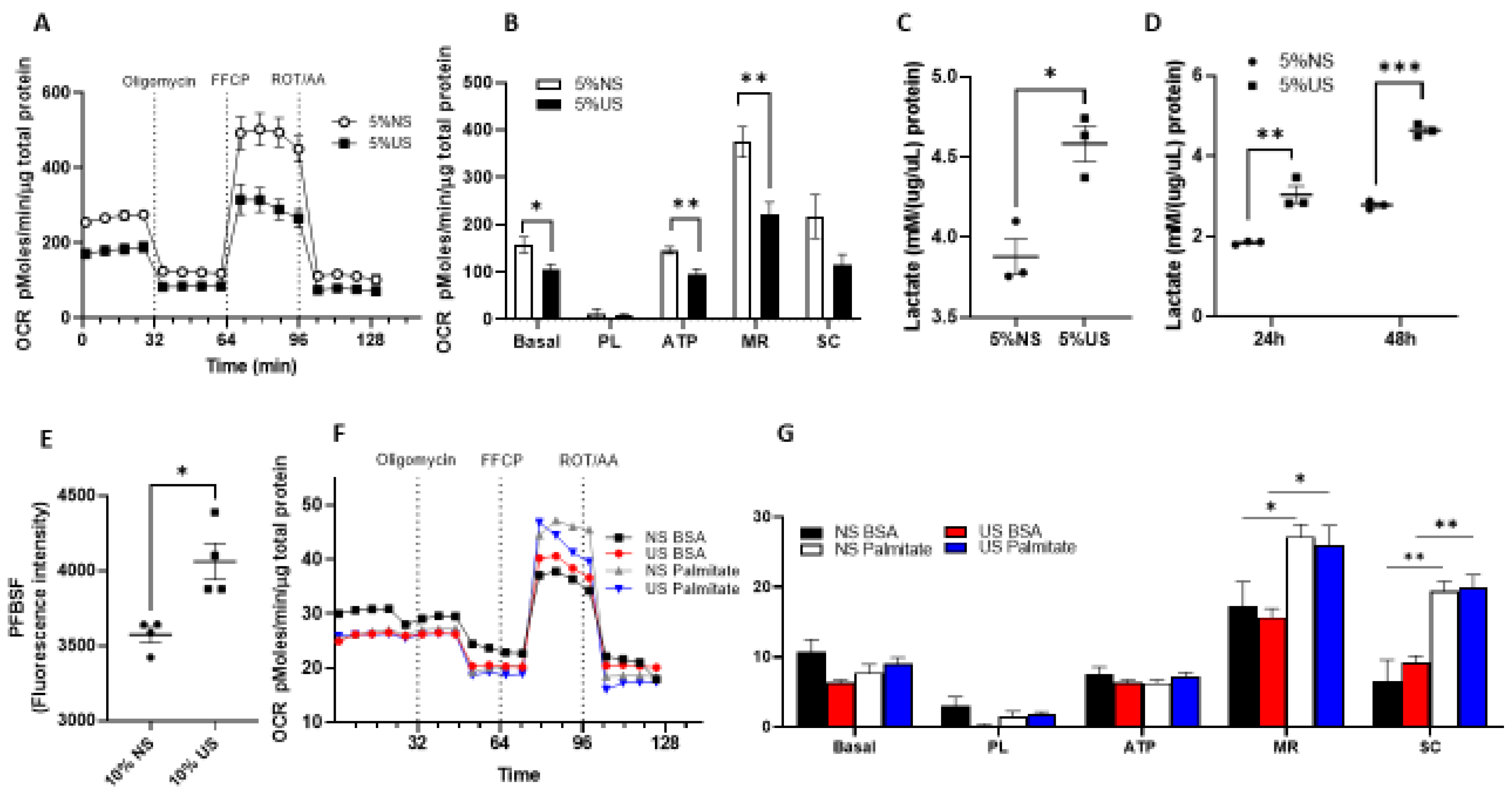
2.4. Cells Exposed to Uremic Serum Have Prominent Glycolytic Metabolism with Decreased Glucose-Derived OXPHOS and Preserved Fatty Acid Oxidation
3. Discussion
4. Materials and Methods
4.1. Exercise Tolerance Test
4.2. Blood Urea Nitrogen (BUN)
4.3. Complete Blood Count (CBC)
4.4. Trichrome Stain and Quantification
4.5. Collagen III Immunohistochemistry and Quantification
4.6. Muscle Fiber Typing and Measurements
4.7. Human Serum Treatment
4.8. Cell Culture
4.9. Oxygen Consumption Rates (OCR) Measurement
4.10. Extracellular Lactate Measurement
4.11. Extracellular H2O2 Production
4.12. Western Blot
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishi, H.; Takemura, K.; Higashihara, T.; Inagi, R. Uremic Sarcopenia: Clinical Evidence and Basic Experimental Approach. Nutrients 2020, 12, 1814. [Google Scholar] [CrossRef]
- Mori, K. Maintenance of Skeletal Muscle to Counteract Sarcopenia in Patients with Advanced Chronic Kidney Disease and Especially Those Undergoing Hemodialysis. Nutrients 2021, 13, 1538. [Google Scholar] [CrossRef]
- Campistol, J.M. Uremic myopathy. Kidney Int. 2002, 62, 1901–1913. [Google Scholar] [CrossRef] [Green Version]
- Fahal, I.H. Uraemic sarcopenia: Aetiology and implications. Nephrol. Dial. Transplant. 2014, 29, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Carrero, J.J.; von Walden, F.; Ikizler, T.A.; Nader, G.A. Muscle wasting in end-stage renal disease promulgates premature death: Established, emerging and potential novel treatment strategies. Nephrol. Dial. Transplant. 2016, 31, 1070–1077. [Google Scholar] [CrossRef] [Green Version]
- Sinkeler, S.J.; Kwakernaak, A.J.; Bakker, S.J.; Shahinfar, S.; Esmatjes, E.; De Zeeuw, D.; Navis, G.; Lambers Heerspink, H.J. Creatinine excretion rate and mortality in type 2 diabetes and nephropathy. Diabetes Care 2013, 36, 1489–1494. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.A.; Cordeiro, A.C.; Avesani, C.M. Sarcopenia in chronic kidney disease on conservative therapy: Prevalence and association with mortality. Nephrol. Dial. Transplant. 2015, 30, 1718–1725. [Google Scholar] [CrossRef] [Green Version]
- Foley, R.N.; Wang, C.; Ishani, A.; Collins, A.J.; Murray, A.M. Kidney function and sarcopenia in the United States general population: NHANES III. Am. J. Nephrol. 2007, 27, 279–286. [Google Scholar] [CrossRef]
- Chu, N.M.; Chen, X.; Norman, S.P.; Fitzpatrick, J.; Sozio, S.M.; Jaar, B.G.; Frey, A.; Estrella, M.M.; Xue, Q.L.; Parekh, R.S.; et al. Frailty Prevalence in Younger End-Stage Kidney Disease Patients Undergoing Dialysis and Transplantation. Am. J. Nephrol. 2020, 51, 501–510. [Google Scholar] [CrossRef]
- Carrero, J.J.; Johansen, K.L.; Lindholm, B.; Stenvinkel, P.; Cuppari, L.; Avesani, C.M. Screening for muscle wasting and dysfunction in patients with chronic kidney disease. Kidney Int. 2016, 90, 53–66. [Google Scholar]
- Wilhelm-Leen, E.R.; Hall, Y.N.; Tamura, M.K.; Chertow, G.M. Frailty and chronic kidney disease: The Third National Health and Nutrition Evaluation Survey. Am. J. Med. 2009, 122, 664–671.e2. [Google Scholar] [CrossRef]
- Johansen, K.L.; Delgado, C.; Bao, Y.; Kurella Tamura, M. Frailty and dialysis initiation. Semin. Dial. 2013, 26, 690–696. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.C.; Huang, S.H.; Kao, C.L.; Hsu, P.C. Muscle Wasting in Chronic Kidney Disease: Mechanism and Clinical Implications-A Narrative Review. Int. J. Mol. Sci. 2022, 23, 6047. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Kaltsatou, A.; Sakkas, G.K.; Poulianiti, K.P.; Koutedakis, Y.; Tepetes, K.; Christodoulidis, G.; Stefanidis, I.; Karatzaferi, C. Uremic myopathy: Is oxidative stress implicated in muscle dysfunction in uremia? Front. Physiol. 2015, 6, 102. [Google Scholar] [CrossRef] [Green Version]
- Delano, M.J.; Moldawer, L.L. The origins of cachexia in acute and chronic inflammatory diseases. Nutr. Clin. Pract. 2006, 21, 68–81. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Alvestrand, A. Inflammation in end-stage renal disease: Sources, consequences, and therapy. Semin. Dial. 2002, 15, 329–337. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Billings, I.V.F.T.; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J.; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Roshanravan, B.; Towse, T.; Keller, C.A.; Falck, A.M.; Yu, C.; Frontera, W.R.; Brown, N.J.; Ikizler, T.A. Skeletal Muscle Mitochondrial Dysfunction Is Present in Patients with CKD before Initiation of Maintenance Hemodialysis. Clin. J. Am. Soc. Nephrol. 2020, 15, 926–936. [Google Scholar] [CrossRef]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef]
- Marrades, R.M.; Alonso, J.; Roca, J.; de Suso, J.G.; Campistol, J.M.; Barberà, J.A.; Diaz, O.; Torregrosa, J.V.; Masclans, J.R.; Rodríguez-Roisin, R.; et al. Cellular bioenergetics after erythropoietin therapy in chronic renal failure. J. Clin. Investig. 1996, 97, 2101–2110. [Google Scholar] [CrossRef] [Green Version]
- Kemp, G.J.; Crowe, A.V.; Anijeet, H.K.; Gong, Q.Y.; Bimson, W.E.; Frostick, S.P.; Bone, J.M.; Bell, G.M.; Roberts, J.N. Abnormal mitochondrial function and muscle wasting, but normal contractile efficiency, in haemodialysed patients studied non-invasively in vivo. Nephrol. Dial. Transplant. 2004, 19, 1520–1527. [Google Scholar] [CrossRef] [Green Version]
- Kestenbaum, B.; Gamboa, J.; Liu, S.; Ali, A.S.; Shankland, E.; Jue, T.; Giulivi, C.; Smith, L.R.; Himmelfarb, J.; de Boer, I.H.; et al. Impaired skeletal muscle mitochondrial bioenergetics and physical performance in chronic kidney disease. JCI Insight 2020, 5, e133289. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, M.; Miyashita, K.; Wakino, S.; Mitsuishi, M.; Hayashi, K.; Itoh, H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight 2020, 6, e139826. [Google Scholar] [CrossRef]
- Desai, K.H.; Bernstein, D. Exercise and Oxygen Consumption in the Mouse. In Cardiovascular Physiology in the Genetically Engineered Mouse; Hoit, B.D., Walsh, R.A., Eds.; Springer: Boston, MA, USA, 2002; pp. 277–302. [Google Scholar]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62.e8. [Google Scholar] [CrossRef]
- Martinez Cantarin, M.P.; Whitaker-Menezes, D.; Lin, Z.; Falkner, B. Uremia induces adipose tissue inflammation and muscle mitochondrial dysfunction. Nephrol. Dial. Transplant. 2017, 32, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Johansen, K.L.; Shubert, T.; Doyle, J.; Soher, B.; Sakkas, G.K.; Kent-Braun, J.A. Muscle atrophy in patients receiving hemodialysis: Effects on muscle strength, muscle quality, and physical function. Kidney Int. 2003, 63, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Coen, P.M.; Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Ferrell, R.E.; Toledo, F.G.; Goodpaster, B.H. Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 2010, 59, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Higashihara, T.; Nishi, H.; Takemura, K.; Watanabe, H.; Maruyama, T.; Inagi, R.; Tanaka, T.; Nangaku, M. Beta2-adrenergic receptor agonist counteracts skeletal muscle atrophy and oxidative stress in uremic mice. Sci. Rep. 2021, 11, 9130. [Google Scholar] [CrossRef]
- Souweine, J.S.; Gouzi, F.; Badia, É.; Pomies, P.; Garrigue, V.; Morena, M.; Hayot, M.; Mercier, J.; Ayoub, B.; Le Quintrec, M.; et al. Skeletal Muscle Phenotype in Patients Undergoing Long-Term Hemodialysis Awaiting Kidney Transplantation. Clin. J. Am. Soc. Nephrol. 2021, 16, 1676–1685. [Google Scholar] [CrossRef]
- Lewis, M.I.; Fournier, M.; Wang, H.; Storer, T.W.; Casaburi, R.; Cohen, A.H.; Kopple, J.D. Metabolic and morphometric profile of muscle fibers in chronic hemodialysis patients. J. Appl. Physiol. 2012, 112, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Choksi, K.B.; Nuss, J.E.; Boylston, W.H.; Rabek, J.P.; Papaconstantinou, J. Age-related increases in oxidatively damaged proteins of mouse kidney mitochondrial electron transport chain complexes. Free Radic. Biol. Med. 2007, 43, 1423–1438. [Google Scholar] [CrossRef] [Green Version]
- Quoilin, C.; Mouithys-Mickalad, A.; Lecart, S.; Fontaine-Aupart, M.P.; Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Biophys. Acta 2014, 1837, 1790–1800. [Google Scholar] [CrossRef] [Green Version]
- Antoun, G.; McMurray, F.; Thrush, A.; Patten, D.A.; Peixoto, A.C.; Slack, R.S.; McPherson, R.; Dent, R.; Harper, M.E. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia 2015, 58, 2861–2866. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Anderson, E.M.; Thome, T.; Lu, G.; Salyers, Z.R.; Cort, T.A.; O’Malley, K.A.; Scali, S.T.; Ryan, T.E. Skeletal myopathy in CKD: A comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am. J. Physiol. Renal Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Wright, K.J.; Romestaing, C.; Thomas, M.M.; Rowan, S.L.; Taivassalo, T.; Hepple, R.T. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell 2010, 9, 1032–1046. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Faivre, A.; Verissimo, T.; Auwerx, H.; Legouis, D.; de Seigneux, S. Tubular Cell Glucose Metabolism Shift During Acute and Chronic Injuries. Front. Med. 2021, 8, 742072. [Google Scholar] [CrossRef]
- Dickman, K.G.; Mandel, L.J. Differential effects of respiratory inhibitors on glycolysis in proximal tubules. Am. J. Physiol. 1990, 258, F1608–F1615. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology Glycolytic Shift during Proximal Tubule Atrophy after Ischemic, A.K.I. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Johnson, A.C.; Becker, K. Renal cortical pyruvate depletion during AKI. J. Am. Soc. Nephrol. 2014, 25, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef]
- Lamb, G.D.; Westerblad, H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J. Physiol. 2011, 589, 2119–2127. [Google Scholar] [CrossRef]
- Samouilidou, E.; Grapsa, E. Effect of dialysis on plasma total antioxidant capacity and lipid peroxidation products in patients with end-stage renal failure. Blood Purif. 2003, 21, 209–212. [Google Scholar] [CrossRef]
- Filiopoulos, V.; Hadjiyannakos, D.; Takouli, L.; Metaxaki, P.; Sideris, V.; Vlassopoulos, D. Inflammation and oxidative stress in end-stage renal disease patients treated with hemodialysis or peritoneal dialysis. Int. J. Artif. Organs. 2009, 32, 872–882. [Google Scholar] [CrossRef]
- Zhang, J.; Wong, M.G.; Wong, M. A cationic-independent mannose 6-phosphate receptor inhibitor (PXS64) ameliorates kidney fibrosis by inhibiting activation of transforming growth factor-beta1. PLoS ONE 2015, 10, e0116888. [Google Scholar]
- Zhao, J.; Meng, M.; Zhang, J. Astaxanthin ameliorates renal interstitial fibrosis and peritubular capillary rarefaction in unilateral ureteral obstruction. Mol. Med. Rep. 2019, 19, 3168–3178. [Google Scholar] [CrossRef] [Green Version]
- Stangenberg, S.; Saad, S.; Schilter, H.C. Lysyl oxidase-like 2 inhibition ameliorates glomerulosclerosis and albuminuria in diabetic nephropathy. Sci. Rep. 2018, 8, 9423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhu, Y.; Zhen, T. Transcriptomic analysis of the mechanisms of alleviating renal interstitial fibrosis using the traditional Chinese medicine Kangxianling in a rat model. Sci. Rep. 2020, 10, 10682. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano, E.; Whitaker-Menezes, D.; Lin, Z.; Roche, M.; Martinez Cantarin, M.P. Uremic Myopathy and Mitochondrial Dysfunction in Kidney Disease. Int. J. Mol. Sci. 2022, 23, 13515. https://doi.org/10.3390/ijms232113515
Serrano E, Whitaker-Menezes D, Lin Z, Roche M, Martinez Cantarin MP. Uremic Myopathy and Mitochondrial Dysfunction in Kidney Disease. International Journal of Molecular Sciences. 2022; 23(21):13515. https://doi.org/10.3390/ijms232113515
Chicago/Turabian StyleSerrano, Eurico, Diana Whitaker-Menezes, Zhao Lin, Megan Roche, and Maria Paula Martinez Cantarin. 2022. "Uremic Myopathy and Mitochondrial Dysfunction in Kidney Disease" International Journal of Molecular Sciences 23, no. 21: 13515. https://doi.org/10.3390/ijms232113515