
Hemizygous Granzyme A Mice Expressing the hSOD1G93A Transgene Show Slightly Extended Lifespan

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
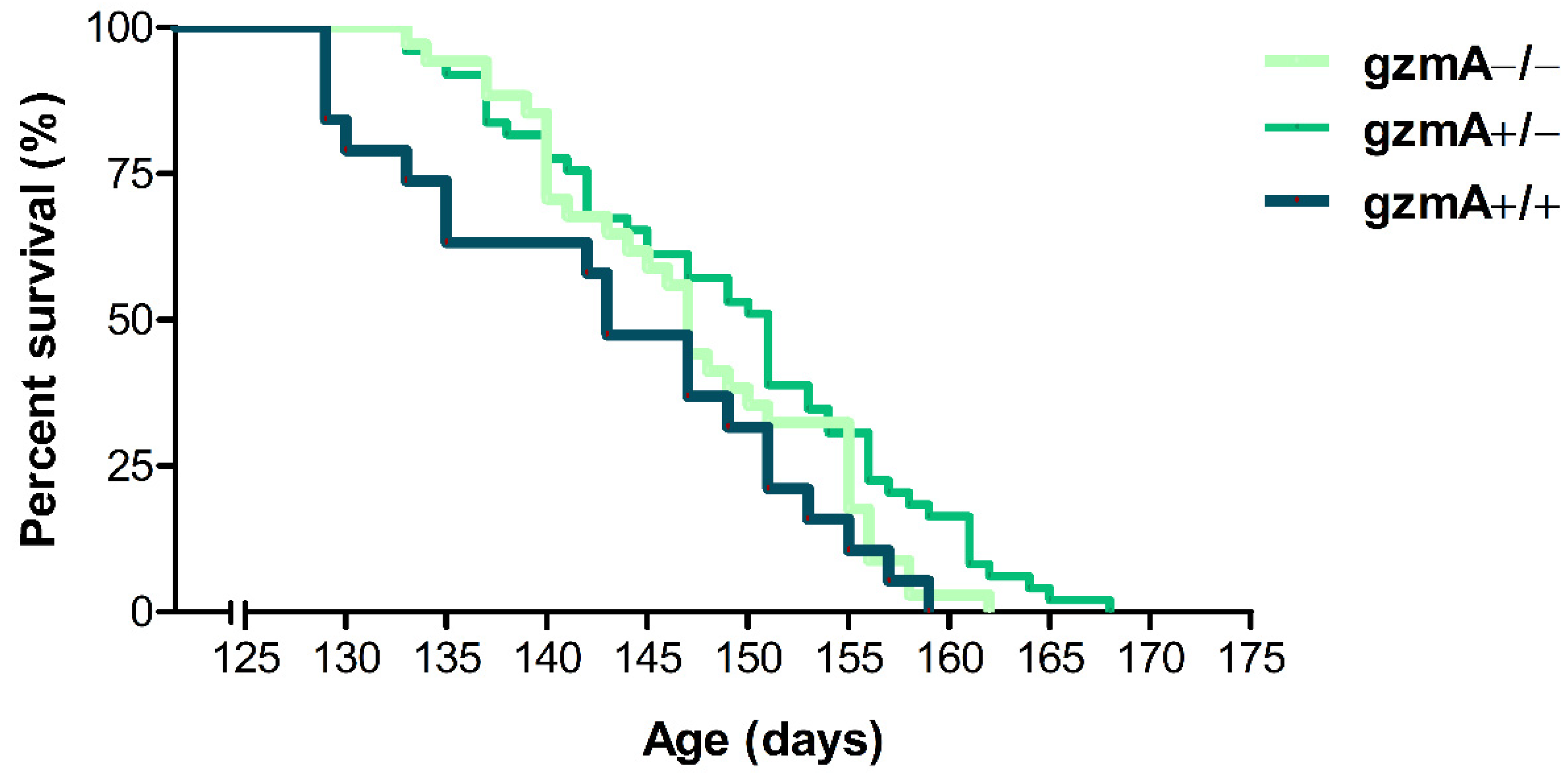
2.1. Lifespan Is Extended in Hemizygous gzmA+/− Mice
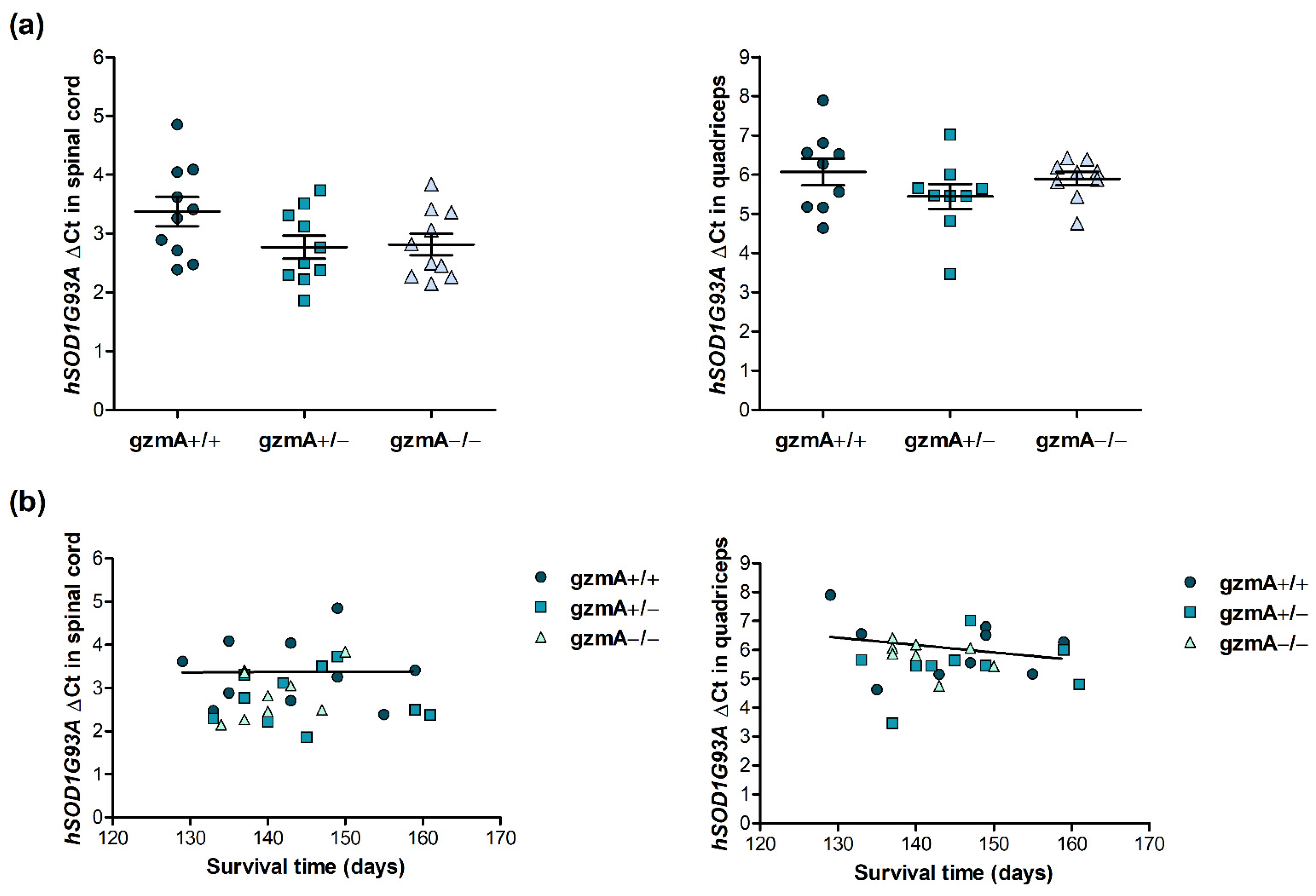
2.2. Human Mutant Superoxide Dismutase 1 (hSOD1G93A) Is Consistently Expressed in All Mice, Regardless of gzmA Genotype
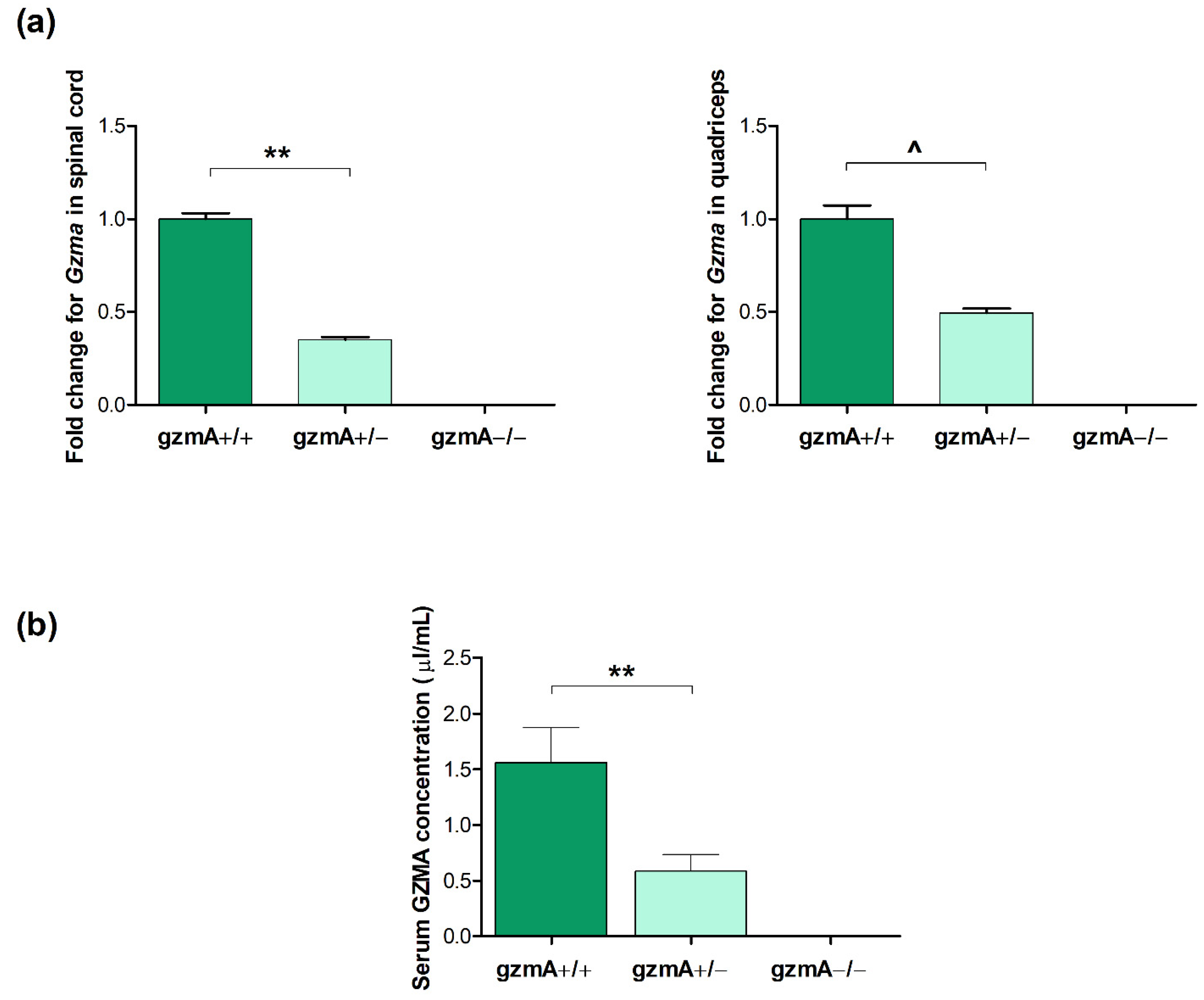
2.3. GzmA Is Expressed According to Each gzmA Genotype
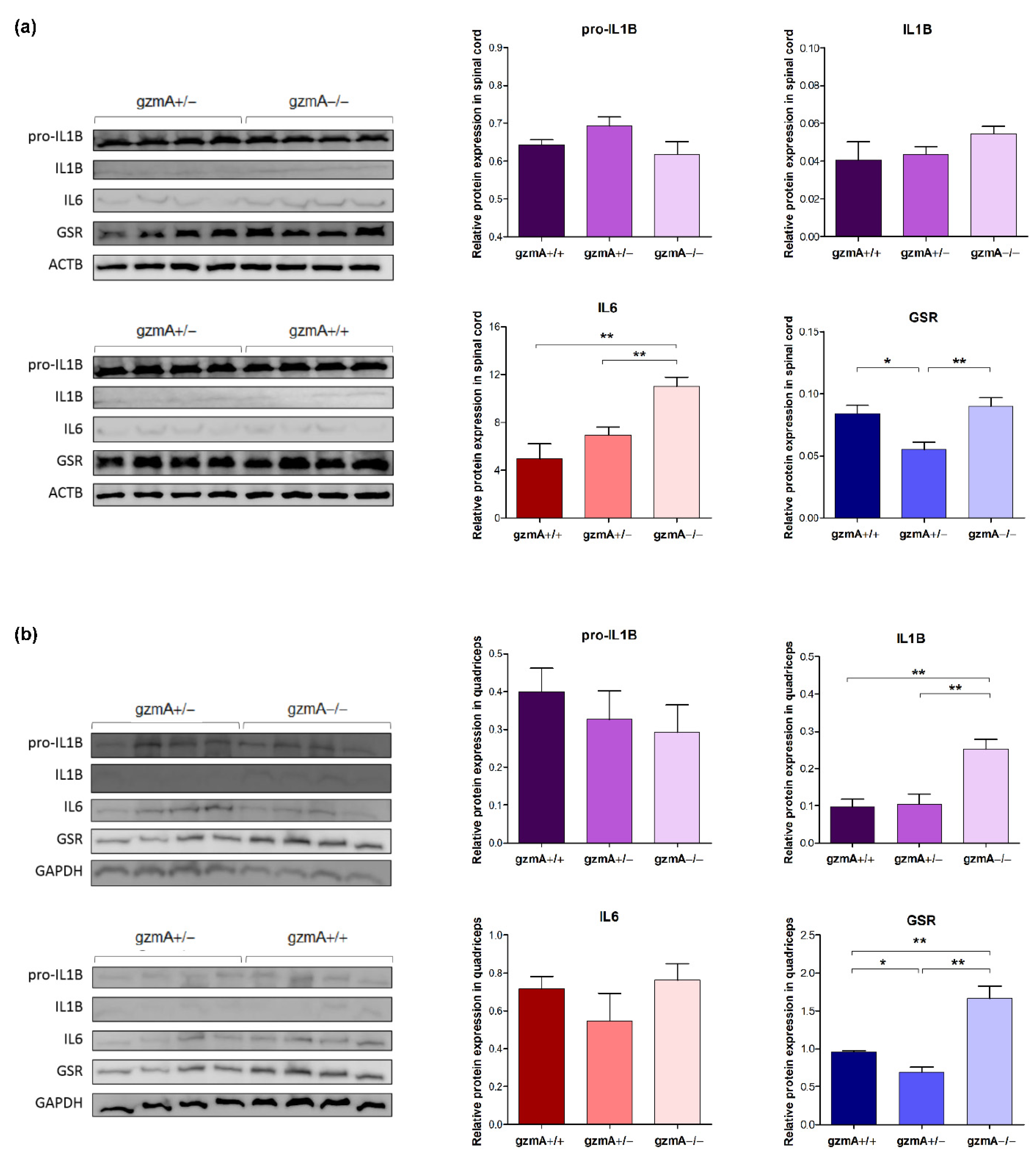
2.4. Gene Expression of Il1b, Il6, Tnfa and Glutathione Reductase (Gsr) Is Reduced in Spinal Cord or Quadricep Muscle of Hemizygous gzmA Mice
2.5. Protein Levels of GSR Are Reduced in Spinal Cord and Quadriceps of Hemizygous gzmA Mice
3. Discussion
4. Materials and Methods
4.1. Animals
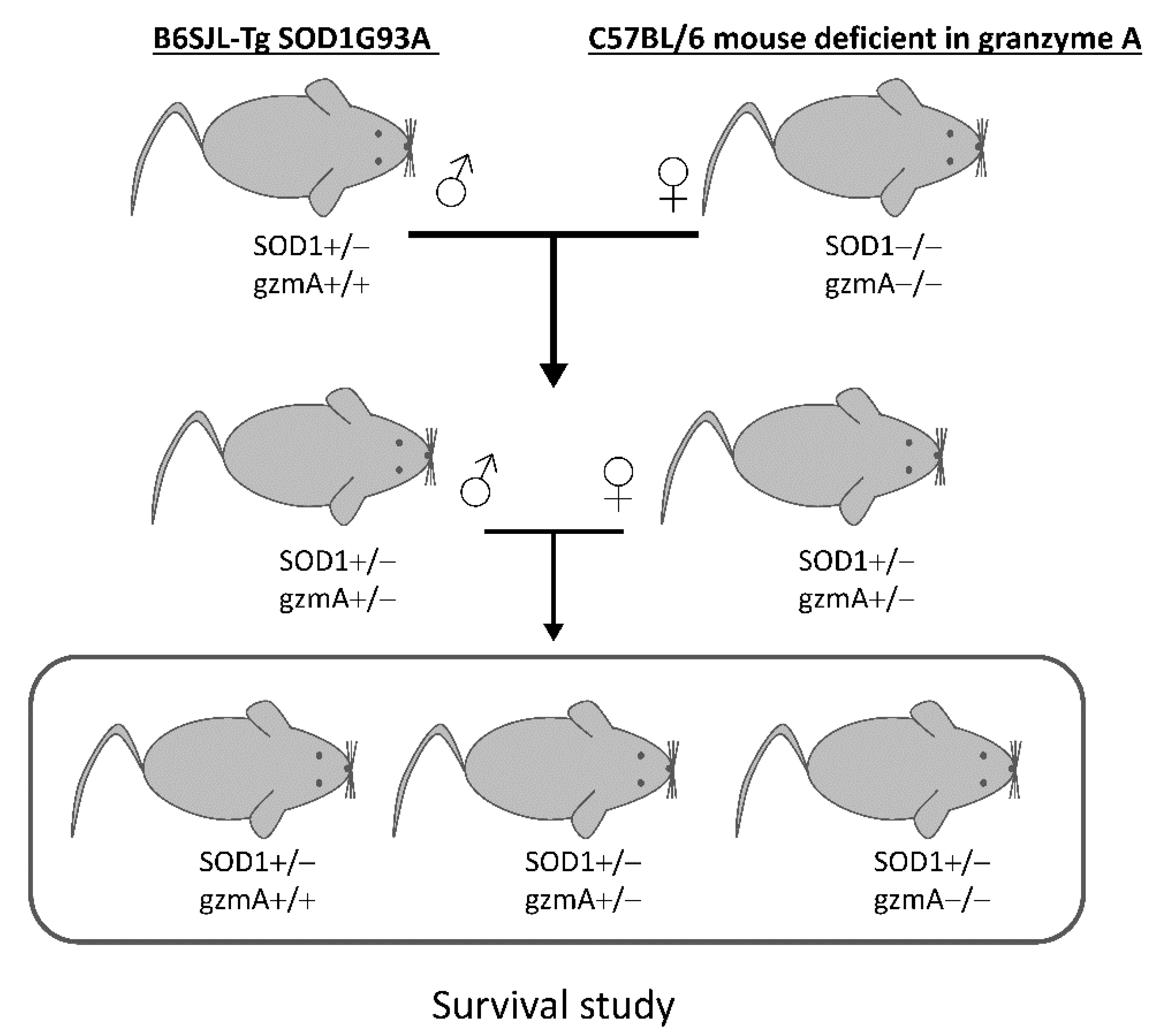
4.2. Crossings and Survival Study
4.3. Enzyme-Linked ImmunoSorbent Assay (ELISA) for gzmA
4.4. RT-qPCR
4.5. Western Blot
4.6. ELISA for TNF-α
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- Phani, S.; Re, D.B.; Przedborski, S. The Role of the Innate Immune System in ALS. Front. Pharmacol. 2012, 3, 150. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues Lima-Junior, J.; Sulzer, D.; Lindestam Arlehamn, C.S.; Sette, A. The Role of Immune-Mediated Alterations and Disorders in ALS Disease. Hum. Immunol. 2021, 82, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Thonhoff, J.R.; Simpson, E.P.; Appel, S.H. Neuroinflammatory Mechanisms in Amyotrophic Lateral Sclerosis Pathogenesis. Curr. Opin. Neurol. 2018, 31, 635–639. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor Neuron Degeneration in Mice That Express a Human Cu, Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Jeyachandran, A.; Mertens, B.; McKissick, E.A.; Mitchell, C.S. Type I vs. Type II Cytokine Levels as a Function of SOD1 G93A Mouse Amyotrophic Lateral Sclerosis Disease Progression. Front. Cell. Neurosci. 2015, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Cao, C.; Qin, X.-Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased Peripheral Blood Inflammatory Cytokine Levels in Amyotrophic Lateral Sclerosis: A Meta-Analysis Study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Martínez, L.; De La Torre, M.; Toivonen, J.M.; Zaragoza, P.; García-Redondo, A.; Calvo, A.C.; Osta, R. Circulating Cytokines Could Not Be Good Prognostic Biomarkers in a Mouse Model of Amyotrophic Lateral Sclerosis. Front. Immunol. 2019, 10, 801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyke, J.M.; Smit-Oistad, I.M.; Macrander, C.; Krakora, D.; Meyer, M.G.; Suzuki, M. Macrophage-Mediated Inflammation and Glial Response in the Skeletal Muscle of a Rat Model of Familial Amyotrophic Lateral Sclerosis (ALS). Exp. Neurol. 2016, 277, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Kim, S.; Cui, X.; Zheng, L.; Oh, S.J.; Anderson, T.; AlSharabati, M.; Kazamel, M.; Volpicelli-Daley, L.; Bamman, M.M.; et al. Transforming Growth Factor Beta (TGF-β) Is a Muscle Biomarker of Disease Progression in ALS and Correlates with Smad Expression. PLoS ONE 2015, 10, e0138425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic Reactions in Amyotrophic Lateral Sclerosis Brain and Spinal Cord Tissue. Am. J. Pathol. 1992, 140, 691. [Google Scholar]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic Infiltrates in the Spinal Cord in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef]
- Melzer, N.; Meuth, S.G.; Wiendl, H. CD8+ T Cells and Neuronal Damage: Direct and Collateral Mechanisms of Cytotoxicity and Impaired Electrical Excitability. FASEB J. 2009, 23, 3659–3673. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.T.; Lim, D.; Granville, D.J. Granzyme B in Skin Inflammation and Disease. Matrix Biol. 2019, 75–76, 126–140. [Google Scholar] [CrossRef]
- Arias, M.; Martínez-Lostao, L.; Santiago, L.; Ferrandez, A.; Granville, D.J.; Pardo, J. The Untold Story of Granzymes in Oncoimmunology: Novel Opportunities with Old Acquaintances. Trends Cancer 2017, 3, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.A.; Franks, Z.; Bhatnagar, A.; Rowley, J.W.; Manne, B.K.; Supiano, M.A.; Schwertz, H.; Weyrich, A.S.; Rondina, M.T. Granzyme A in Human Platelets Regulates the Synthesis of Proinflammatory Cytokines by Monocytes in Aging. J. Immunol. 2018, 200, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metkar, S.S.; Menaa, C.; Pardo, J.; Wang, B.; Wallich, R.; Freudenberg, M.; Kim, S.; Raja, S.M.; Shi, L.; Simon, M.M.; et al. Human and Mouse Granzyme A Induce a Proinflammatory Cytokine Response. Immunity 2008, 29, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Daalen, K.R.; Reijneveld, J.F.; Bovenschen, N. Modulation of Inflammation by Extracellular Granzyme A. Front. Immunol. 2020, 11, 931. [Google Scholar] [CrossRef]
- Garzón-Tituaña, M.; Sierra-Monzón, J.L.; Comas, L.; Santiago, L.; Khaliulina-Ushakova, T.; Uranga-Murillo, I.; Ramirez-Labrada, A.; Tapia, E.; Morte-Romea, E.; Algarate, S.; et al. Granzyme A Inhibition Reduces Inflammation and Increases Survival during Abdominal Sepsis. Theranostics 2021, 11, 3781–3795. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8 + T Lymphocytes Expressing ALS-Causing SOD1 Mutant Selectively Trigger Death of Spinal Motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef] [Green Version]
- Iłżecka, J. Granzymes A and B Levels in Serum of Patients with Amyotrophic Lateral Sclerosis. Clin. Biochem. 2011, 44, 650–653. [Google Scholar] [CrossRef]
- Van Eck, J.A.; Shan, L.; Meeldijk, J.; Hack, C.E.; Bovenschen, N. A Novel Proinflammatory Role for Granzyme A. Cell Death Dis. 2017, 8, e2630. [Google Scholar] [CrossRef] [Green Version]
- Duvall, M.G.; Barnig, C.; Cernadas, M.; Ricklefs, I.; Krishnamoorthy, N.; Grossman, N.L.; Bhakta, N.R.; Fahy, J.V.; Bleecker, E.R.; Castro, M.; et al. Natural Killer Cell–Mediated Inflammation Resolution Is Disabled in Severe Asthma. Sci. Immunol. 2017, 2, eaam5446. [Google Scholar] [CrossRef] [Green Version]
- García-Laorden, M.I.; Stroo, I.; Terpstra, S.; Florquin, S.; Medema, J.P.; Vańt Veer, C.; De Vos, A.F.; Van Der Poll, T. Expression and Function of Granzymes A and B in Escherichia Coli Peritonitis and Sepsis. Mediat. Inflamm. 2017, 2017, 4137563. [Google Scholar] [CrossRef] [Green Version]
- Arias, M.A.; JiménezdeBagües, M.P.; Aguiló, N.; Menao, S.; Hervás-Stubbs, S.; deMartino, A.; Alcaraz, A.; Simon, M.M.; Froelich, C.J.; Pardo, J. Elucidating Sources and Roles of Granzymes A and B during Bacterial Infection and Sepsis. Cell Rep. 2014, 8, 420–429. [Google Scholar] [CrossRef]
- Santiago, L.; Menaa, C.; Arias, M.; Martin, P.; Jaime-Sánchez, P.; Metkar, S.; Comas, L.; Erill, N.; Gonzalez-Rumayor, V.; Esser, E.; et al. Granzyme A Contributes to Inflammatory Arthritis in Mice Through Stimulation of Osteoclastogenesis. Arthritis Rheumatol. 2017, 69, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, G.M.; Erwin, K.L.; Byers, N.; Deitch, J.S.; Augelli, B.J.; Blankenhorn, E.P.; Heiman-Patterson, T.D. Effect of Transgene Copy Number on Survival in the G93A SOD1 Transgenic Mouse Model of ALS. Mol. Brain Res. 2004, 130, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Arozena, A.; Kalmar, B.; Essa, S.; Ricketts, T.; Joyce, P.; Kent, R.; Rowe, C.; Parker, A.; Gray, A.; Hafezparast, M.; et al. A Comprehensive Assessment of the SOD1G93A Low-Copy Transgenic Mouse, Which Models Human Amyotrophic Lateral Sclerosis. Dis. Model. Mech. 2011, 4, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, P.; Braeken, D.; Callewaert, G.; Robberecht, W.; Van Den Bosch, L. GluR2 Deficiency Accelerates Motor Neuron Degeneration in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Lev, N.; Barhum, Y.; Lotan, I.; Steiner, I.; Offen, D. DJ-1 Knockout Augments Disease Severity and Shortens Survival in a Mouse Model of ALS. PLoS ONE 2015, 10, e0117190. [Google Scholar] [CrossRef]
- Staats, K.A.; Humblet-Baron, S.; Bento-Abreu, A.; Scheveneels, W.; Nikolaou, A.; Deckers, K.; Lemmens, R.; Goris, A.; Van Ginderachter, J.A.; Van Damme, P.; et al. Genetic Ablation of IP3 Receptor 2 Increases Cytokines and Decreases Survival of SOD1G93A Mice. Hum. Mol. Genet. 2016, 25, 3491–3499. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.-H.; Allen, K.; Oei, F.; Leoni, E.; Kuhle, J.; Tree, T.; Fratta, P.; Sharma, N.; Sidle, K.; Howard, R.; et al. Systemic Inflammatory Response and Neuromuscular Involvement in Amyotrophic Lateral Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e244. [Google Scholar] [CrossRef] [Green Version]
- Andrés-Benito, P.; Moreno, J.; Domínguez, R.; Aso, E.; Povedano, M.; Ferrer, I. Inflammatory Gene Expression in Whole Peripheral Blood at Early Stages of Sporadic Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 546. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Yang, X.; Gao, L.; Zang, D. Evaluating the Levels of CSF and Serum Factors in ALS. Brain Behav. 2017, 7, e00637. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant Superoxide Dismutase 1-Induced IL-1β Accelerates ALS Pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; McDonald, T.S.; Fung, J.N.T.; Woodruff, T.M. Absence of Receptor for Advanced Glycation End Product (RAGE) Reduces Inflammation and Extends Survival in the HSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2020, 57, 4143–4155. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-Hydroxynonenal in Cerebrospinal Fluid of Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased Lipid Peroxidation in Sera of ALS Patients. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Santella, R.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Gonzalo, I.; Ruiz-Soto, M.; Berciano, J. ¿Por Qué Degeneran Las Motoneuronas? Actualización En La Patogenia de La Esclerosis Lateral Amiotrófica. Neurología 2019, 34, 27–37. [Google Scholar] [CrossRef]
- Pardo, J.; Bosque, A.; Brehm, R.; Wallich, R.; Naval, J.; Müllbacher, A.; Anel, A.; Simon, M.M. Apoptotic Pathways Are Selectively Activated by Granzyme A and/or Granzyme B in CTL-Mediated Target Cell Lysis. J. Cell Biol. 2004, 167, 457. [Google Scholar] [CrossRef]
- Lieberman, J. Granzyme A Activates Another Way to Die. Immunol. Rev. 2010, 235, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Chico, L.; Ienco, E.C.; Bisordi, C.; Lo Gerfo, A.; Petrozzi, L.; Petrucci, A.; Mancuso, M.; Siciliano, G. Amyotrophic Lateral Sclerosis and Oxidative Stress: A Double-Blind Therapeutic Trial After Curcumin Supplementation. CNS Neurol. Disord.-Drug Targets 2018, 17, 767–779. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094. [Google Scholar] [CrossRef] [Green Version]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 2616. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Nomura, E.; Shang, J.; Feng, T.; Huang, Y.; Liu, X.; Shi, X.; Nakano, Y.; Hishikawa, N.; Sato, K.; et al. Enhanced Oxidative Stress and the Treatment by Edaravone in Mice Model of Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2019, 97, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, H. Edaravone for the Treatment of Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2019, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Wosiski-Kuhn, M.; Lyon, M.S.; Caress, J.; Milligan, C. Inflammation, Immunity, and Amyotrophic Lateral Sclerosis: II. Immune-Modulating Therapies. Muscle Nerve 2019, 59, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Mifflin, L.; Hu, Z.; Dufort, C.; Hession, C.C.; Walker, A.J.; Niu, K.; Zhu, H.; Liu, N.; Liu, J.S.; Levin, J.Z.; et al. A RIPK1-Regulated Inflammatory Microglial State in Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2025102118. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-Paralysis Tyrosine Kinase Inhibition with Masitinib Abrogates Neuroinflammation and Slows Disease Progression in Inherited Amyotrophic Lateral Sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef] [Green Version]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an Add-on Therapy to Riluzole in Patients with Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Zeldich, E.; Chen, C.D.; Boden, E.; Howat, B.; Nasse, J.S.; Zeldich, D.; Lambert, A.G.; Yuste, A.; Cherry, J.D.; Mathias, R.M.; et al. Klotho Is Neuroprotective in the Superoxide Dismutase (SOD1G93A) Mouse Model of ALS. J. Mol. Neurosci. 2019, 69, 264. [Google Scholar] [CrossRef]
- Ravnik-Glavač, M.; Goričar, K.; Vogrinc, D.; Koritnik, B.; Lavrenčič, J.G.; Glavač, D.; Dolžan, V. Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis. Genes 2022, 13, 757. [Google Scholar] [CrossRef]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in Amyotrophic Lateral Sclerosis: Blurred Lines between Excessive Inflammation and Inefficient Immune Responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef]
- Simon, M.M.; Hausmann, M.; Tran, T.; Ebnet, K.; Tschopp, J.; ThaHla, R.; Müllbacher, A. In Vitro- and Ex Vivo-Derived Cytolytic Leukocytes from Granzyme A X B Double Knockout Mice Are Defective in Granule-Mediated Apoptosis but Not Lysis of Target Cells. J. Exp. Med. 1997, 186, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transgenic Mice Genotype | Mean (Days) | Median (Days) | SEM 1 (Days) | SD 2 (Days) | Minimum (Days) | Maximum (Days) |
|---|---|---|---|---|---|---|
| SOD1+/−, gzmA+/+ | 143.00 | 143.00 | 2.34 | 10.21 | 129.00 | 159.00 |
| SOD1+/−, gzmA+/− | 149.12 | 151.00 | 1.34 | 9.41 | 133.00 | 168.00 |
| SOD1+/−, gzmA−/− | 147.24 | 147.00 | 1.33 | 7.78 | 133.00 | 162.00 |
| Genes | Type | Sequences/TaqMan® Probe ID |
|---|---|---|
| Gzma | TaqMan® | Mm01304452_m1 |
| Il1b | TaqMan® | Mm00434228_m1 |
| Il6 | TaqMan® | Mm00446190_m1 |
| Gsr | TaqMan® | Mm00833903_m1 |
| Tnfa | SYBR Green | Forward: 5′ TAT GGC CCA GAC CCT CAC A 3′ |
| Reverse: 5′ GGA GTA GAC AAG GTA CAA CCC ATC 3′ | ||
| hSOD1G93A | TaqMan® | Hs00533490_m1 |
| Gapdh | TaqMan® | 4352932E |
| Primary Antibody | Reference | Dilution | Secondary Antibody | Reference | Dilution | Tissue |
|---|---|---|---|---|---|---|
| ACTB | PA1-183, Thermo Fisher Scientific | 1:1000 | goat anti-Rabbit IgG | 31466, Thermo Fisher Scientific | 1:3000 | Spinal cord |
| GAPDH | PK-AB718-3781, Promocell | 1:1000 | goat anti-Rabbit IgG | 31466, Thermo Fisher Scientific | 1:3000 | Quadriceps |
| IL1B | sc-7884, Santa Cruz Biotechnology | 1:250 | m-IgG Fc BP-HRP | sc-525409, Santa Cruz Biotechnology | 1:3000 | Spinal cord, Quadriceps |
| IL6 | PK-AB815-61632M, Promocell | 1:1000 | goat anti-Rabbit IgG | 31466, Thermo Fisher Scientific | 1:3000 | Spinal cord, Quadriceps |
| GSR | sc-32886 | 1:500 | goat anti-Rabbit IgG | 31466, Thermo Fisher Scientific | 1:3000 | Spinal cord, Quadriceps |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Martinez, L.; Santiago, L.; de la Torre, M.; Calvo, A.C.; Pardo, J.; Osta, R. Hemizygous Granzyme A Mice Expressing the hSOD1G93A Transgene Show Slightly Extended Lifespan. Int. J. Mol. Sci. 2022, 23, 13554. https://doi.org/10.3390/ijms232113554
Moreno-Martinez L, Santiago L, de la Torre M, Calvo AC, Pardo J, Osta R. Hemizygous Granzyme A Mice Expressing the hSOD1G93A Transgene Show Slightly Extended Lifespan. International Journal of Molecular Sciences. 2022; 23(21):13554. https://doi.org/10.3390/ijms232113554
Chicago/Turabian StyleMoreno-Martinez, Laura, Llipsy Santiago, Miriam de la Torre, Ana Cristina Calvo, Julián Pardo, and Rosario Osta. 2022. "Hemizygous Granzyme A Mice Expressing the hSOD1G93A Transgene Show Slightly Extended Lifespan" International Journal of Molecular Sciences 23, no. 21: 13554. https://doi.org/10.3390/ijms232113554
APA StyleMoreno-Martinez, L., Santiago, L., de la Torre, M., Calvo, A. C., Pardo, J., & Osta, R. (2022). Hemizygous Granzyme A Mice Expressing the hSOD1G93A Transgene Show Slightly Extended Lifespan. International Journal of Molecular Sciences, 23(21), 13554. https://doi.org/10.3390/ijms232113554