
Molecular Genetics of GLUT1DS Italian Pediatric Cohort: 10 Novel Disease-Related Variants and Structural Analysis
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical and Biochemical Features
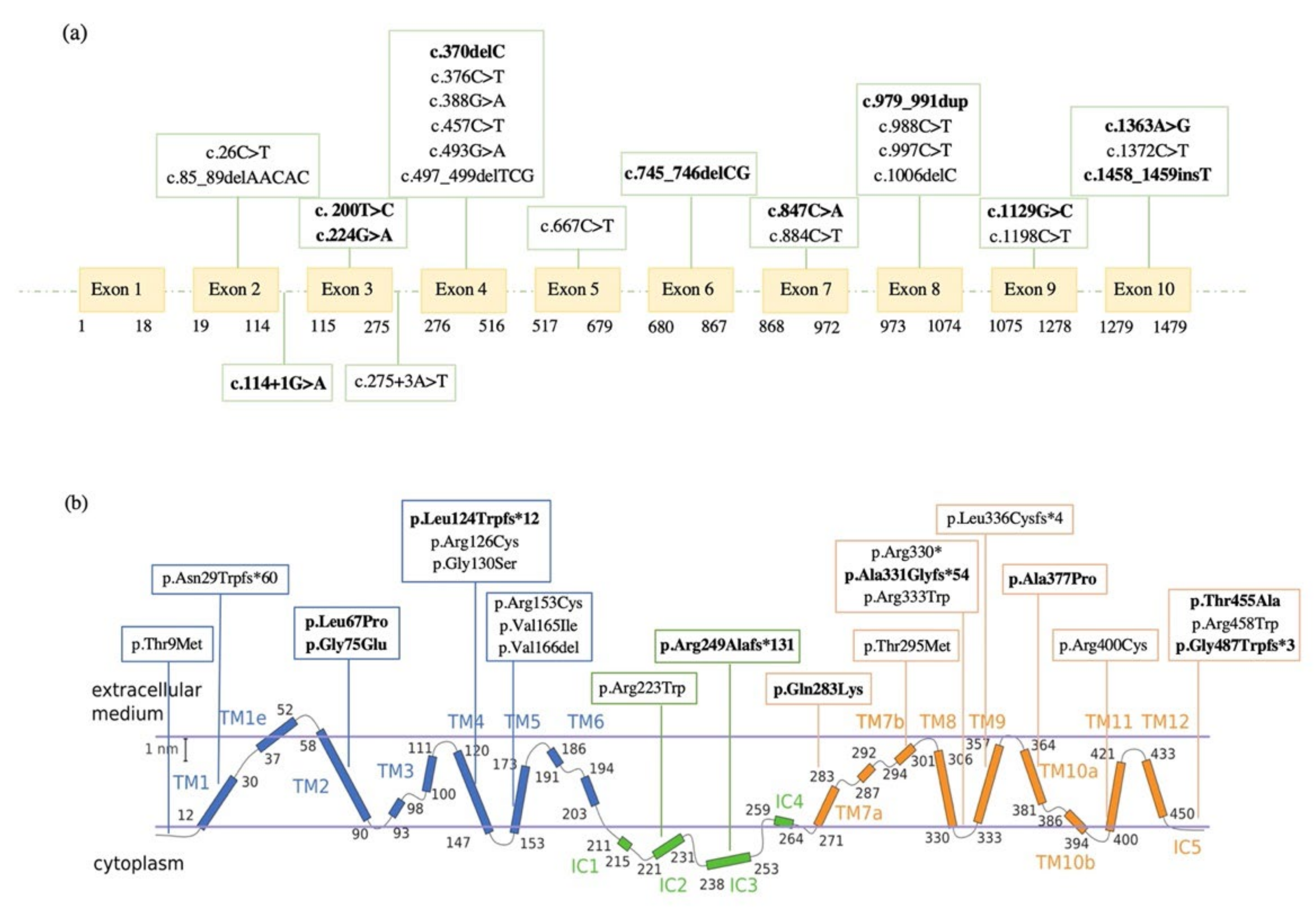
2.2. Molecular Data
2.3. Genetic Variations in Familial Cases
2.4. Genetic Variations in Sporadic Cases
2.5. Novel SLC2A1 Variants and Their Crystallographic Structure
2.6. Genotype, Phenotype and Biochemical Correlations
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. CSF Biochemical Analysis
4.3. Mutation Analysis of the SLC2A1 Gene
4.4. RNA Isolation, RT-qPCR and cDNA Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Vivo, D.C.; Trifiletti, R.R.; Jacobson, R.I.; Ronen, G.M.; Behmand, R.A.; Harik, S.I. Defective Glucose Transport across the Blood-Brain Barrier as a Cause of Persistent Hypoglycorrhachia, Seizures, and Developmental Delay. N. Engl. J. Med. 1991, 325, 703–709. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, D.C.; Leary, L.; Wang, D. Glucose transporter 1 deficiency syndrome and other glycolytic defects. J. Child Neurol. 2002, 17 (Suppl. S3), 3S15-23, discussion 3S24-5. [Google Scholar]
- De Vivo, D.C.; Wang, D.; Pascual, J.M.; Ho, Y.Y. Glucose transporter protein syndromes. Int. Rev. Neurobiol. 2002, 51, 259–288. [Google Scholar] [CrossRef] [PubMed]
- Suls, A.; Dedeken, P.; Goffin, K.; Van Esch, H.; Dupont, P.; Cassiman, D.; Kempfle, J.; Wuttke, T.V.; Weber, Y.; Lerche, H.; et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain 2008, 131, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Klepper, J.; Verbeek, M.M.; Leferink, M.; Hofste, T.; Van Engelen, B.G.; Wevers, R.A.; Arthur, T.; Bahi-Buisson, N.; Ballhausen, D.; et al. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133, 655–670. [Google Scholar] [CrossRef]
- Koch, H.; Weber, Y.G. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2019, 91, 90–93. [Google Scholar] [CrossRef]
- Zorzi, G.; Castellotti, B.; Zibordi, F.; Gellera, C.; Nardocci, N. Paroxysmal movement disorders in GLUT1 deficiency syndrome. Neurology 2008, 71, 146–148. [Google Scholar] [CrossRef]
- Pons, R.; Collins, A.; Rotstein, M.; Engelstad, K.; De Vivo, D.C. The spectrum of movement disorders in Glut-1 deficiency. Mov. Disord. 2010, 25, 275–281. [Google Scholar] [CrossRef]
- Urbizu, A.; Cuenca-León, E.; Raspall-Chaure, M.; Gratacòs, M.; Conill, J.; Redecillas, S.; Roig-Quilis, M.; Macaya, A. Paroxysmal exercise-induced dyskinesia, writer’s cramp, migraine with aura and absence epilepsy in twin brothers with a novel SLC2A1 missense mutation. J. Neurol. Sci. 2010, 295, 110–113. [Google Scholar] [CrossRef]
- De Giorgis, V.; Varesio, C.; Baldassari, C.; Piazza, E.; Olivotto, S.; Macasaet, J.; Balottin, U.; Veggiotti, P. Atypical Manifestations in Glut1 Deficiency Syndrome. J. Child Neurol. 2016, 31, 1174–1180. [Google Scholar] [CrossRef]
- Klepper, J.; Voit, T. Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: Impaired glucose transport into brain—A review. Eur. J. Pediatr. 2002, 161, 295–304. [Google Scholar] [CrossRef]
- Klepper, J.; Akman, C.; Armeno, M.; Auvin, S.; Cervenka, M.; Cross, H.J.; De Giorgis, V.; Della Marina, A.; Engelstad, K.; Heussinger, N.; et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020, 5, 354–365. [Google Scholar] [CrossRef]
- Klepper, J.; Scheffer, H.; Leiendecker, B.; Gertsen, E.; Binder, S.; Leferink, M.; Hertzberg, C.; Näke, A.; Voit, T.; Willemsen, M.A. Seizure Control and Acceptance of the Ketogenic Diet in GLUT1 Deficiency Syndrome: A 2- to 5-Year Follow-Up of 15 Children Enrolled Prospectively. Neuropediatrics 2005, 36, 302–308. [Google Scholar] [CrossRef]
- Friedman, J.R.L.; Thiele, E.A.; Wang, D.; Levine, K.B.; Cloherty, E.K.; Pfeifer, H.H.; De Vivo, D.C.; Carruthers, A.; Natowicz, M.R. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov. Disord. 2006, 21, 241–244. [Google Scholar] [CrossRef]
- Brockmann, K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009, 31, 545–552. [Google Scholar] [CrossRef]
- López-Rivera, J.A.; Pérez-Palma, E.; Symonds, J.; Lindy, A.S.; McKnight, D.A.; Leu, C.; Zuberi, S.; Brunklaus, A.; Møller, R.S.; Lal, D. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain 2020, 143, 1099–1105. [Google Scholar] [CrossRef] [Green Version]
- Seidner, G.; Alvarez, M.G.; Yeh, J.-I.; O’Driscoll, K.R.; Klepper, J.; Stump, T.S.; Wang, D.; Spinner, N.B.; Birnbaum, M.J.; De Vivo, D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998, 18, 188–191. [Google Scholar] [CrossRef]
- Klepper, J.; Scheffer, H.; Elsaid, M.; Kamsteeg, E.-J.; Leferink, M.; Ben-Omran, T. Autosomal Recessive Inheritance of GLUT1 Deficiency Syndrome. Neuropediatrics 2009, 40, 207–210. [Google Scholar] [CrossRef]
- Rotstein, M.; Bs, K.E.; Yang, H.; Wang, D.; Levy, B.; Chung, W.K.; De Vivo, D.C. Glut1 deficiency: Inheritance pattern determined by haploinsufficiency. Ann. Neurol. 2010, 68, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Pascual, J.M.; De Vivo, D. Glucose Transporter Type 1 Deficiency Syndrome. 2002 Jul 30 [Updated 2018 Mar 1]. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1430/ (accessed on 5 September 2022).
- Mueckler, M.; Makepeace, C. Model of the Exofacial Substrate-Binding Site and Helical Folding of the Human Glut1 Glucose Transporter Based on Scanning Mutagenesis. Biochemistry 2009, 48, 5934–5942. [Google Scholar] [CrossRef] [PubMed]
- Galochkina, T.; Chong, M.N.F.; Challali, L.; Abbar, S.; Etchebest, C. New insights into GluT1 mechanics during glucose transfer. Sci. Rep. 2019, 9, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Pascual, J.M.; Yang, H.; Engelstad, K.; Jhung, S.; Sun, R.P.; De Vivo, D.C. Glut-1 deficiency syndrome: Clinical, genetic, and therapeutic aspects. Ann. Neurol. 2005, 57, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Willemsen, M.; Verrips, A.; Guertsen, E.; Herrmann, R.; Kutzick, C.; Flörcken, A.; Voit, T. Autosomal dominant transmission of GLUT1 deficiency. Hum. Mol. Genet. 2001, 10, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Wang, D.; Korenke, C.G.; Von Moers, A.; Ho, Y.-Y.; Pascual, J.M.; Kuang, K.; Yang, H.; Ma, L.; Kranz-Eble, P.; et al. Autosomal dominant Glut-1 deficiency syndrome and familial epilepsy. Ann. Neurol. 2001, 50, 476–485. [Google Scholar] [CrossRef]
- Gras, D.; Roze, E.; Caillet, S.; Méneret, A.; Doummar, D.; de Villemeur, T.B.; Vidailhet, M.; Mochel, F. GLUT1 deficiency syndrome: An update. Rev. Neurol. 2014, 170, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.M.; Wang, D.; Yang, R.; Shi, L.; Yang, H.; De Vivo, D.C. Structural Signatures and Membrane Helix 4 in GLUT1: Inferences from human blood-brain glucose transport mutants. J. Biol. Chem. 2008, 283, 16732–16742. [Google Scholar] [CrossRef] [Green Version]
- Castellotti, B.; Ragona, F.; Freri, E.; Solazzi, R.; Ciardullo, S.; Tricomi, G.; Venerando, A.; Salis, B.; Canafoglia, L.; Villani, F.; et al. Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: Identification of novel variants and associated phenotypes. J. Neurol. 2019, 266, 1439–1448. [Google Scholar] [CrossRef]
- Graziola, F.; Garone, G.; Stregapede, F.; Bosco, L.; Vigevano, F.; Curatolo, P.; Bertini, E.; Travaglini, L.; Capuano, A. Diagnostic Yield of a Targeted Next-Generation Sequencing Gene Panel for Pediatric-Onset Movement Disorders: A 3-Year Cohort Study. Front. Genet. 2019, 10, 1026. [Google Scholar] [CrossRef]
- Koy, A.; Assmann, B.; Klepper, J.; Mayatepek, E. Glucose transporter type 1 deficiency syndrome with carbohydrate-responsive symptoms but without epilepsy. Dev. Med. Child Neurol. 2011, 53, 1154–1156. [Google Scholar] [CrossRef]
- Wang, D.; Kranz-Eble, P.; De Vivo, D.C. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum. Mutat. 2000, 16, 224–231. [Google Scholar] [CrossRef]
- Lindy, A.S.; Stosser, M.B.; Butler, E.; Downtain-Pickersgill, C.; Shanmugham, A.; Retterer, K.; Brandt, T.; Richard, G.; McKnight, D.A. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018, 59, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Bao, X.; Wang, D.; Fu, N.; Zhang, X.; Cao, G.; Song, F.; Wang, S.; Zhang, Y.; Qin, J.; et al. Allelic variations of glut-1 deficiency syndrome: The chinese experience. Pediatr. Neurol. 2012, 47, 30–34. [Google Scholar] [CrossRef]
- Arsov, T.; Mullen, S.A.; Rogers, S.; Phillips, A.M.; Lawrence, K.M.; Damiano, J.A.; Goldberg-Stern, H.; Afawi, Z.; Kivity, S.; Trager, C.; et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann. Neurol. 2012, 72, 807–815. [Google Scholar] [CrossRef]
- De Giorgis, V.; Teutonico, F.; Cereda, C.; Balottin, U.; Bianchi, M.; Giordano, L.; Olivotto, S.; Ragona, F.; Tagliabue, A.; Zorzi, G.; et al. Sporadic and familial glut1ds Italian patients: A wide clinical variability. Seizure 2015, 24, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, H.; Shi, L.; Ma, L.; Fujii, T.; Engelstad, K.; Pascual, J.M.; De Vivo, D.C. Functional Studies of the T295M Mutation Causing Glut1 Deficiency: Glucose Efflux Preferentially Affected by T295M. Pediatr. Res. 2008, 64, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Custódio, T.F.; Paulsen, P.A.; Frain, K.M.; Pedersen, B.P. Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family. Life Sci. Alliance 2021, 4, e202000858. [Google Scholar] [CrossRef]
- Deng, D.; Sun, P.; Yan, C.; Ke, M.; Jiang, X.; Xiong, L.; Ren, W.; Hirata, K.; Yamamoto, M.; Fan, S.; et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. [Google Scholar] [CrossRef]
- Sun, L.; Zeng, X.; Yan, C.; Sun, X.; Gong, X.; Rao, Y.; Yan, N. Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature 2012, 490, 361–366. [Google Scholar] [CrossRef]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Müller, T.; Siebeneicher, H.; et al. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, D.; Ms, K.E.; Bagay, L.; Wei, Y.; Rotstein, M.; Aggarwal, V.; Levy, B.; Ma, L.; Chung, W.K.; et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann. Neurol. 2011, 70, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, S.; Duse, A.; Bova, S.M.; Leonardi, V.; Biganzoli, E.; Milanese, A.; Cereda, C.; Bertoli, S.; Previtali, R.; Veggiotti, P. Glut1 deficiency syndrome throughout life: Clinical phenotypes, intelligence, life achievements and quality of life in familial cases. Orphanet J. Rare Dis. 2002, 17, 365. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sporadic/ Familial | Sex | Age | Ratio | MCP | ID | Spasticity | Seizure: Onset/Type | MD: Onset/Type | EEG | KD | Other | Clinical Phenotypes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (F1) | Familial | F | NA | 0.52 | − | nor | − | 6y/ABS | - | NA | + | Late-onset classical phenotype | |
| 2 | Sporadic | M | 5m | 0.25 | − | sev | − | 3m/FS, MS | 3m/NA | NA | + | Early-onset classical phenotype | |
| 3 | Sporadic | F | 24m | 0.37 | + | sev | + | 3m/FS, ES | - | BGS, FD, EE | + | Early-onset classical phenotype | |
| 4 (F2) | Familial | M | 10y | 0.50 | − | nor | − | 24m/ABS | 7y/PED | N | + | Early-onset classical phenotype | |
| 5 | Sporadic | F | 8y | 0.39 | − | sev | − | 6m/GTC | 24m/PD | BGS, FD | + | Early-onset classical phenotype | |
| 6 | Sporadic | M | 11y | 0.30 | − | sev | − | - | 11y/PED | NA | − | Non-classical phenotype | |
| 7 | Sporadic | F | 10y | 0.36 | − | sev | − | 5m/MS | 24m/C | BGS, G | − | Early-onset classical phenotype | |
| 8 | Sporadic | F | 7y | 0.34 | − | sev | − | 24m/MAS, DS, ABS | 6y/PED | BGS, G, FD | − | stroke-like episode | Early-onset classical phenotype |
| 9 (F3) | Familial | F | 7y | 0.39 | − | mild | + | 3y/GTC | NA/no PED | BGS, G, FD | + | Late-onset classical phenotype | |
| 10 | Sporadic | F | NA | 0.39 | − | nor | − | 12m/DS, GTC, ABS | 16m/C | BGS, G, FD | − | stroke-like episode | Early-onset classical phenotype |
| 11 (F4) | Familial | F | 7y | 0.38 | − | sev | − | - | 7y/PED | N | − | Non-classical phenotype | |
| 12 | Sporadic | F | NA | 0.50 | − | mild | − | 3y/ABS | 7m/PED | BGS, G | − | Late-onset classical phenotype | |
| 13 | Sporadic | M | 24m | 0.39 | − | mild | − | 15m/MAS | 5m/NA | G | + | Early-onset classical phenotype | |
| 14 | Sporadic | F | 11y | 0.37 | − | sev | + | 9m/MAS | - | BGS, G | − | Early-onset classical phenotype | |
| 15 | Sporadic | F | 7y | 0.43 | − | sev | − | 4m/MS, AS | 18m/C | G | + | Early-onset classical phenotype | |
| 16 (F5) | Familial | F | 7y | NA | − | bord | − | 6m/ABS | 24m/PED, PND | NA | + | Early-onset classical phenotype | |
| 17 | Sporadic | F | NA | 0,42 | − | mild | − | 24m/ABS | 5y/PED | NA | + | Early-onset classical phenotype | |
| 18 | Sporadic | F | 24m | 0.31 | − | mod | − | 20m/ABS | 20m/C | G | + | Early-onset classical phenotype | |
| 19 | Sporadic | M | 4y | 0.41 | + | mild | − | - | - | N | + | Non-classical phenotype | |
| 20 | Sporadic | M | 4y | 0.32 | + | sev | − | - | 24m/PND | N | + | Non-classical phenotype | |
| 21 | Sporadic | F | 12m | 0.40 | − | mild | − | 2m/FS | 3m/PND | N | + | Early-onset classical phenotype | |
| 22 | Sporadic | F | 3y | 0.38 | − | sev | − | 3y/GTC | NA/PND | NA | + | Late-onset classical phenotype | |
| 23 | Sporadic | M | 19y | NA | − | NA | NA | 8m/NA | - | N | + | Early-onset classical phenotype | |
| 24 | Sporadic | F | 7y | 0.38 | − | sev | − | 3y/ABS, MAS | 3m/PED | BGS, G | + | Late-onset classical phenotype | |
| 25 (F6) | Familial | M | 9m | 0.37 | − | mod | − | 4m/FS | - | FD | + | Early-onset classical phenotype | |
| 26 | Sporadic | M | NA | 0.51 | − | sev | − | 4y/CFS | 14y/PED | FD | − | stroke-like episode | Late-onset classical phenotype |
| 27 | Sporadic | M | 5y | 0.37 | − | mild | − | 10m/FS, GTC | 8y/PED | G | + | stroke-like episode | Early-onset classical phenotype |
| Patient | HGVS (Coding) | HGVS (Protein) | Exon/ Intron | Location | RefSNP | ACMG Classification | Literature Data |
|---|---|---|---|---|---|---|---|
| 1 (F1) | c.26C > T | p.Thr9Met | 2 | TMH1 | rs1570601100 | VUS (PM2 PP2 PP3) | Castellotti et al., 2019 [29] |
| 2 | c.85_89delAACAC | p.Asn29Trpfs*60 | 2 | TMH1 | - | Pathogenic (PVS1 PM2 PM6) | Castellotti et al., 2019 [29] |
| 3 | c.114+1G > A | - | Intron 2 | - | - | Pathogenic (PVS1 PM2 PM6) | Novel |
| 4 (F2) | c. 200T > C | p.Leu67Pro | 3 | TMH2 | - | VUS (PM2 PP2 PP3) | Novel |
| 5 | c.224G > A | p.Gly75Glu | 3 | TMH2 | - | Likely Pathogenic(PM2 PP2 PP3 PM6) | Novel |
| 6 | c.275+3A > T | - | Intron 3 | - | - | VUS (PM2 PP3) | Graziola et al., 2019 [30] |
| 7 | c.370delC | p.Leu124Trpfs*12 | 4 | TMH4 | - | Pathogenic (PVS1 PM2 PM6) | Novel |
| 8 | c.376C > T | p.Arg126Cys | 4 | TMH4 | rs80359818 | Pathogenic (PP5 PM1 PM2 PM5 PM6 PP2 PP3) | Pascual et al., 2002 [3] |
| 9 (F3) | c.388G > A | p.Gly130Ser | 4 | TMH4 | rs80359819 | Pathogenic (PP5 PM1 PM2 PM5 PM6 PP2 PP3) | Wang et al., 2005 [24] |
| 10 | c.457C > T | p.Arg153Cys | 4 | TMH5 | - | Pathogenic (PP5 PM1 PM2 PM5 PP2 PP3) | Pascual et al., 2002 [3] |
| 11 (F4) | c.493G > A | p.Val165Ile | 4 | TMH5 | rs1057520545 | Pathogenic (PP5 PM1 PM2 PM6 PP2) | Urbizu et al., 2010 [9] |
| 12 | c.497_499delTCG | p.Val166del | 4 | TMH5 | - | Likely Pathogenic (PM1 PM2 PM4 PM6) | Koy et al., 2011 [31] |
| 13 | c.667C > T | p.Arg223Trp | 5 | IC2 | rs796053248 | Pathogenic (PP5 PM2 PM5 PM6 PP2 PP3) | Leen et al., 2010 [5] |
| 14 | c.745_746delCG | p.Arg249Alafs*131 | 6 | IC2 | - | Pathogenic (PVS1 PM2 PM6) | Novel |
| 15 | c.847C > A | p.Gln283Lys | 6 | TMH7 | - | Likely Pathogenic (PM1 PM2 PM6 PP2 PP3) | Novel |
| 16 (F5), 17 | c.884C > T | p.Thr295Met | 7 | TMH7 | rs80359823 | Pathogenic (PP5 PM2 PM5 PP2 PP3) | Wang et al., 2005 [24] |
| 18 | c.979_991dup | p.Ala331Glyfs*54 | 8 | TMH8 | - | Pathogenic (PVS1 PM2 PM6) | Novel |
| 19 | c.988C > T | p.Arg330* | 8 | TMH8 | rs80359826 | Pathogenic (PVS1 PM2 PM6 PP5) | Wang et al., 2000 [32] |
| 20 | c.997C > T | p.Arg333Trp | 8 | TMH8 | rs80359825 | Pathogenic (PP5 PM1 PM2 PM6 PP2 PP3) | Wang et al., 2000 [32] |
| 21 | c.1006delC | p.Leu336Cysfs*4 | 8 | TMH9 | rs796053271 | Pathogenic (PVS1 PM2 PM6 PP5) | Lindy et al., 2018 [33] |
| 22 | c.1129G > C | p.Ala377Pro | 9 | TMH10 | - | Likely Pathogenic (PM1 PM2 PM6 PP2 PP3) | Novel |
| 23, 24 | c.1198C > T | p.Arg400Cys | 9 | TMH10 | rs796053263 | Pathogenic (PP5 PM1 PM2 PM5 PM6 PP2 PP3) | Liu et al., 2012 [34] |
| 25 (F6) | c.1363A > G | p.Thr455Ala | 10 | IC5 | - | VUS (PM2 PP2 PP3) | Novel |
| 26 | c.1372C > T | p.Arg458Trp | 10 | IC5 | rs13306758 | Pathogenic (PP5 PM2 PM5 PM6 PP2 PP3) | Arsov et al., 2012 [35] |
| 27 | c.1458_1459insT | p.Gly487Trpfs*3 | 10 | IC5 | - | Likely Pathogenic (PVS1 PM2 PM6) | Novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauri, A.; Duse, A.; Palm, G.; Previtali, R.; Bova, S.M.; Olivotto, S.; Benedetti, S.; Coscia, F.; Veggiotti, P.; Cereda, C. Molecular Genetics of GLUT1DS Italian Pediatric Cohort: 10 Novel Disease-Related Variants and Structural Analysis. Int. J. Mol. Sci. 2022, 23, 13560. https://doi.org/10.3390/ijms232113560
Mauri A, Duse A, Palm G, Previtali R, Bova SM, Olivotto S, Benedetti S, Coscia F, Veggiotti P, Cereda C. Molecular Genetics of GLUT1DS Italian Pediatric Cohort: 10 Novel Disease-Related Variants and Structural Analysis. International Journal of Molecular Sciences. 2022; 23(21):13560. https://doi.org/10.3390/ijms232113560
Chicago/Turabian StyleMauri, Alessia, Alessandra Duse, Giacomo Palm, Roberto Previtali, Stefania Maria Bova, Sara Olivotto, Sara Benedetti, Francesca Coscia, Pierangelo Veggiotti, and Cristina Cereda. 2022. "Molecular Genetics of GLUT1DS Italian Pediatric Cohort: 10 Novel Disease-Related Variants and Structural Analysis" International Journal of Molecular Sciences 23, no. 21: 13560. https://doi.org/10.3390/ijms232113560