
Systemic Bile Acids Affect the Severity of Acute Pancreatitis in Mice Depending on Their Hydrophobicity and the Disease Pathogenesis

 , , ,
, , ,  ,
,  , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
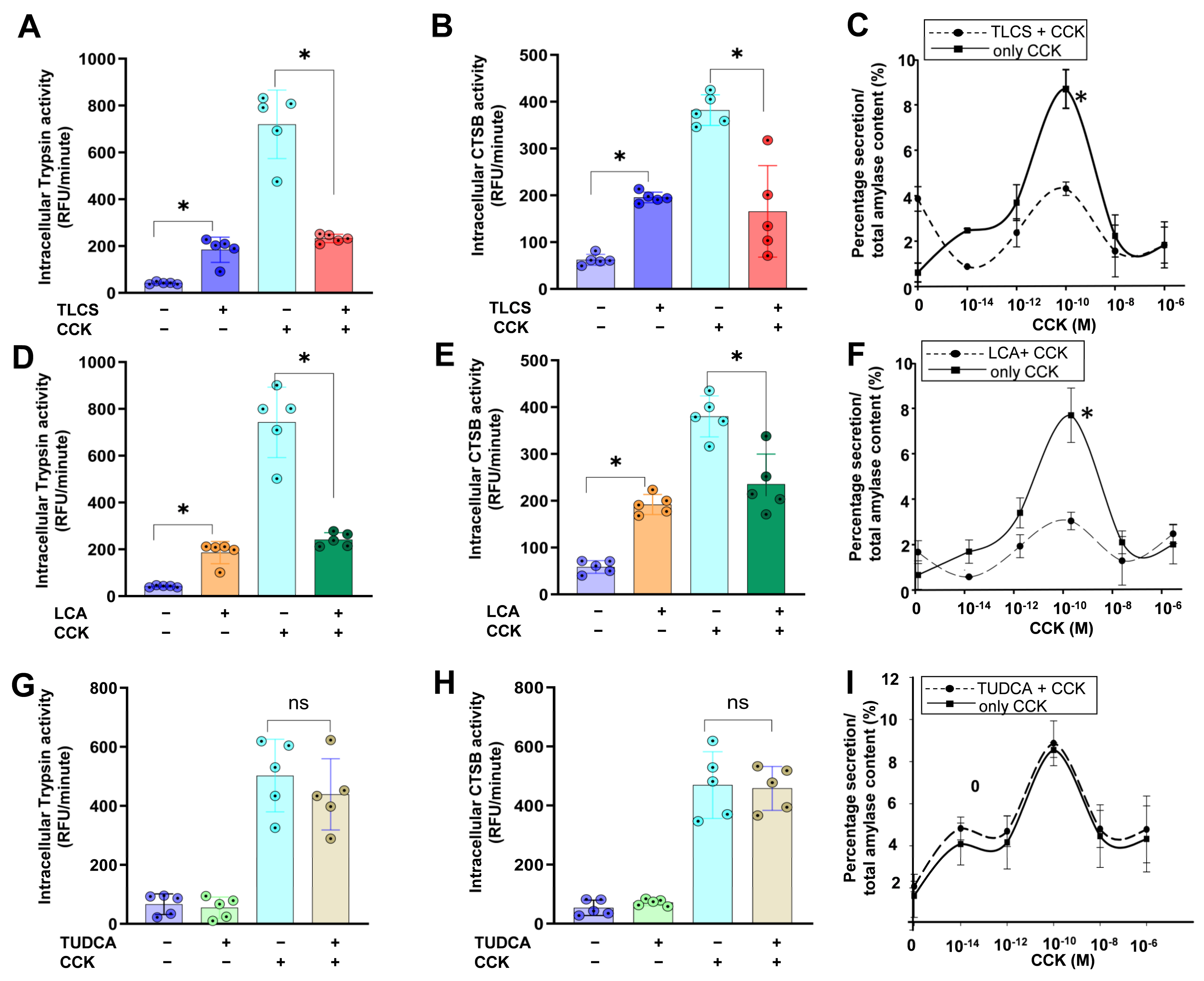
2.1. Impact of Hydrophobic and Hydrophilic BAs in Mouse Isolated Acini
2.2. BAs Were Elevated in the Serum and Reached the Pancreas after Intravenous or Intraperitoneal Administration in Mice
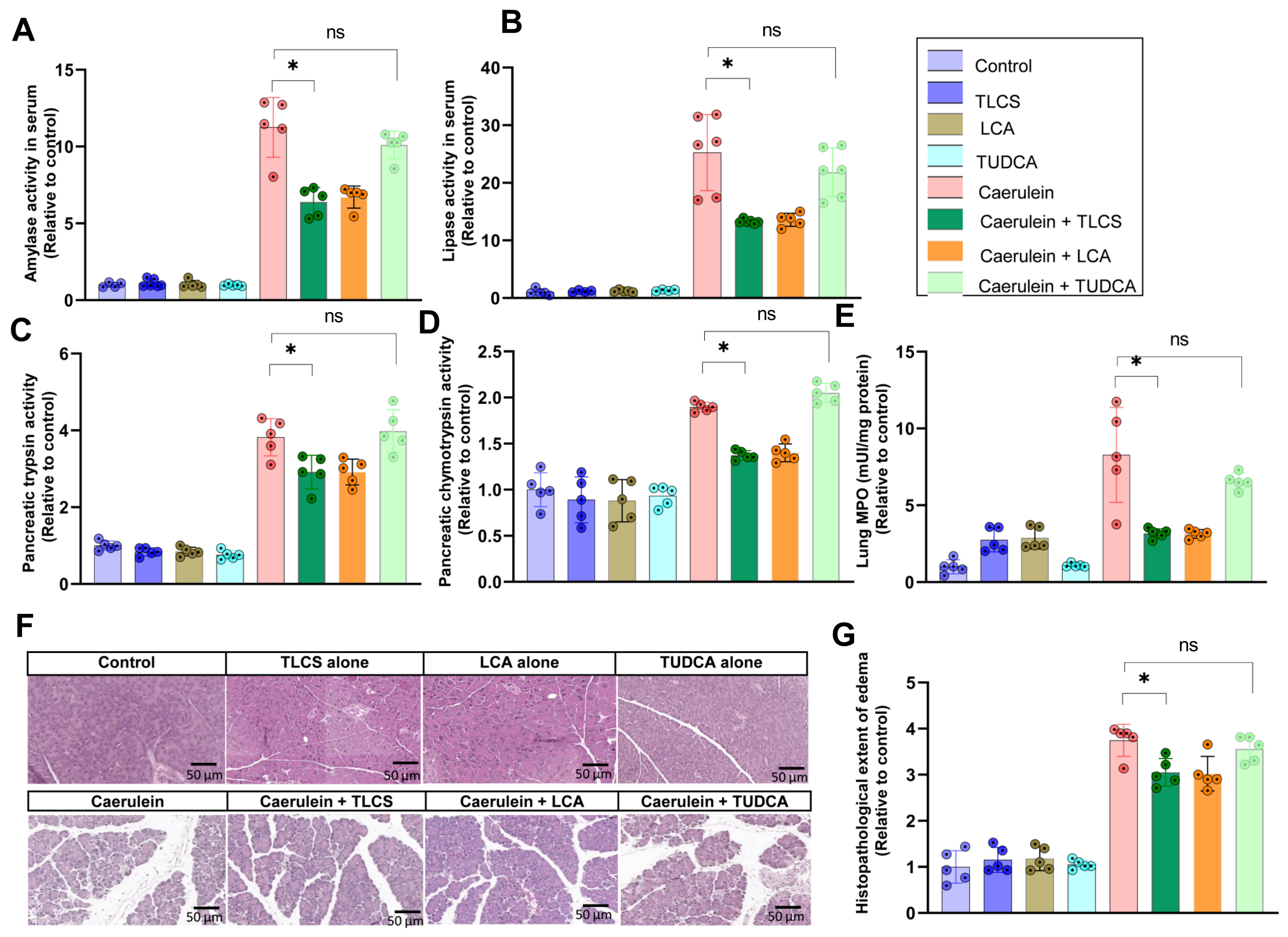
2.3. Hydrophobicity-Dependent Effects of BAs in CCK-Dependent Mouse AP Models
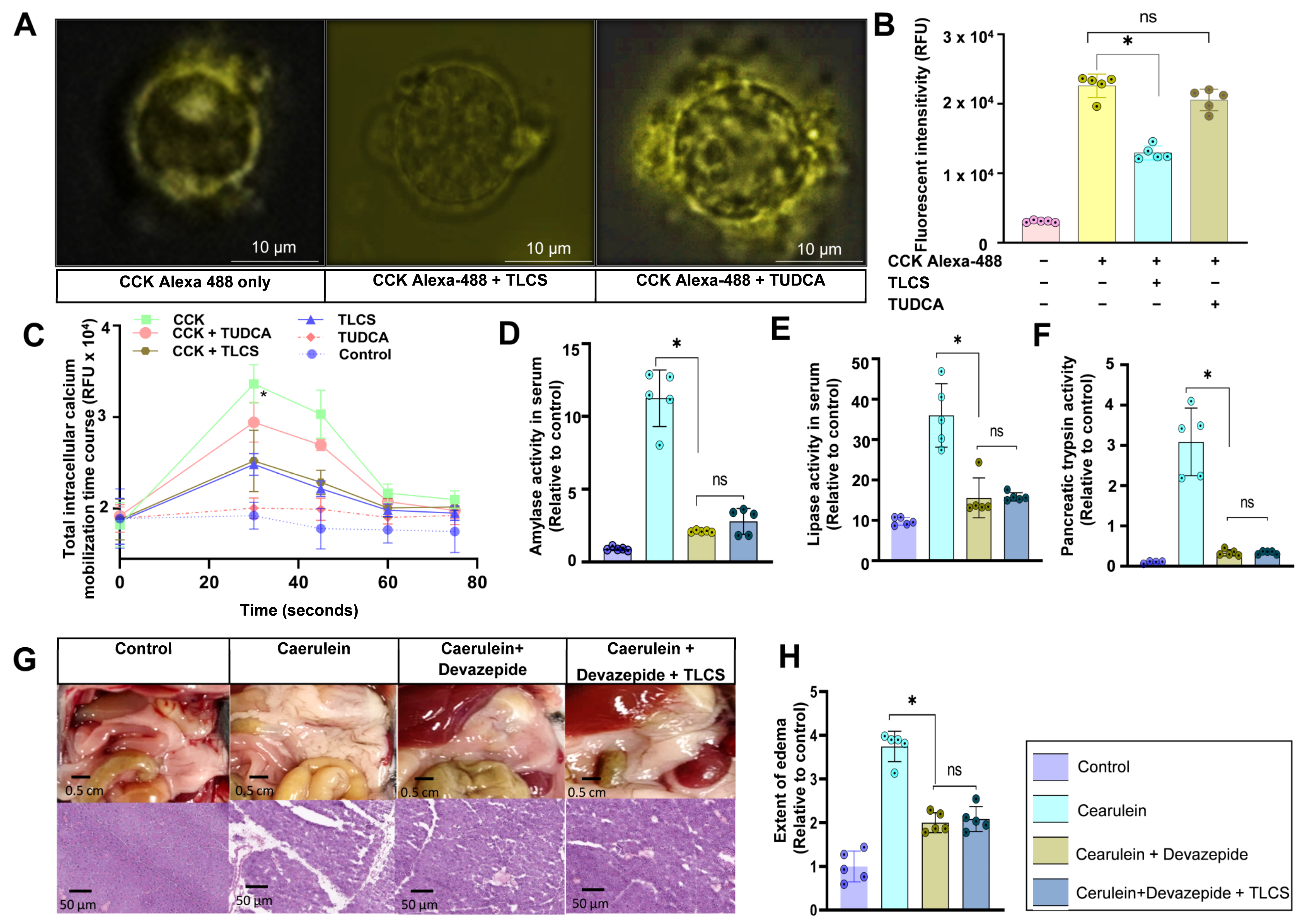
2.4. Aggravating Effects of Hydrophobic BAs in CCK-Independent AP Models in Mice
2.5. Interaction of Hydrophobic and Hydrophilic BAs with CCK1R on Mouse Isolated Acini
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Isolation and Stimulation of Mouse Pancreatic Acini
4.3. CCK1 Receptor-Binding Assay
4.4. Protease Activation Assays
4.5. Quantification of Intracellular Calcium Mobilization in Living Acinar Cells
4.6. Amylase and Lipase Measurement
4.7. Measurement of Propidium Iodide Exclusion and Release of LDH
4.8. Myeloperoxidase Measurement
4.9. Measurement of Total Bile Acid
4.10. Histopathological Examinations
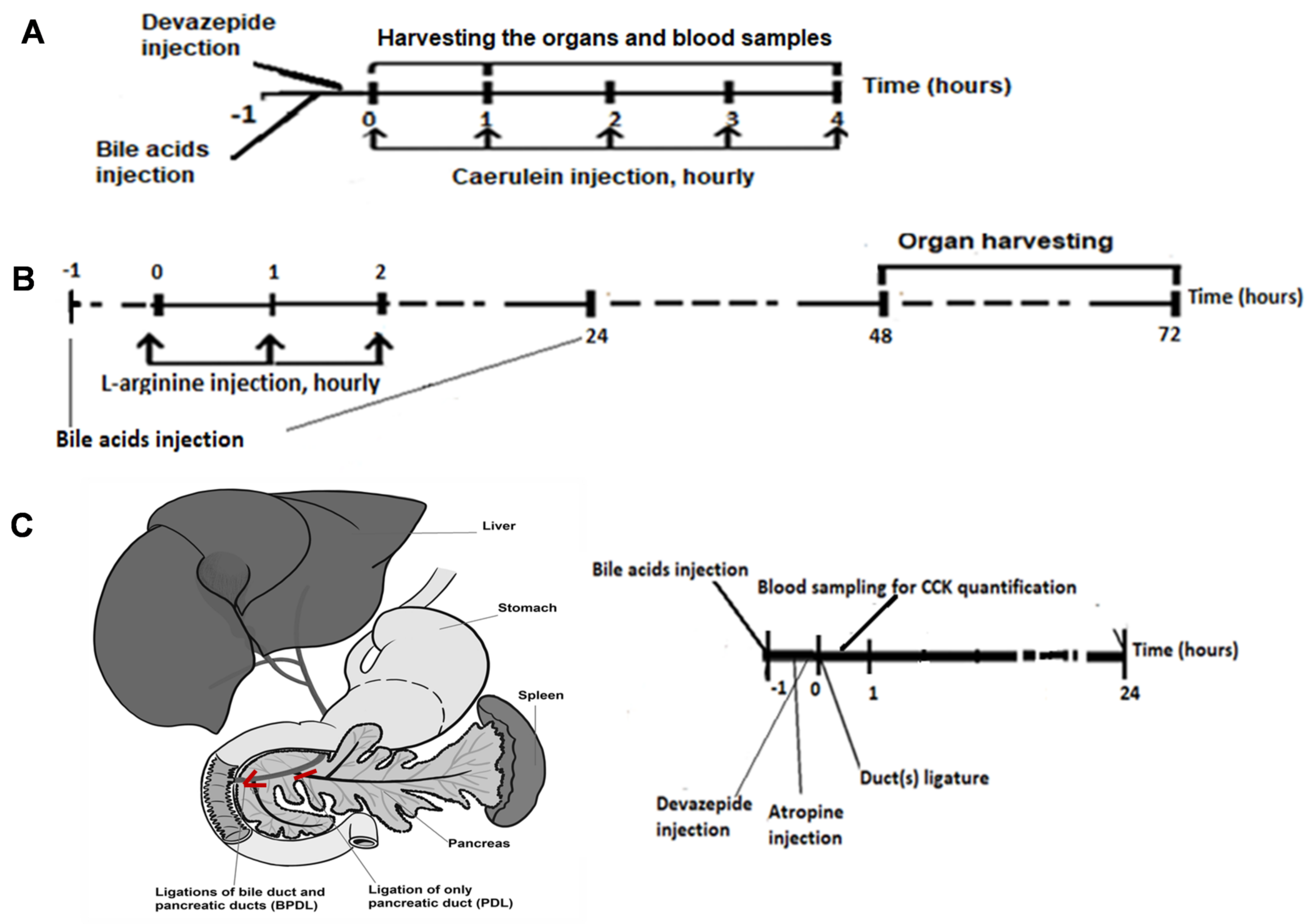
4.11. Animal Models
4.12. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Peery, A.F.; Crockett, S.D.; Barritt, A.S.; Dellon, E.S.; Eluri, S.; Gangarosa, L.M.; Jensen, E.T.; Lund, J.L.; Pasricha, S.; Runge, T.; et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology 2015, 149, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, J.P.; King, J.A.; Leong, J.H.; Quan, J.; Windsor, J.W.; Tanyingoh, D.; Coward, S.; Forbes, N.; Heitman, S.J.; Shaheen, A.A.; et al. Global Incidence of Acute Pancreatitis is Increasing Over Time: A systematic review and meta-analysis. Gastroenterology 2022, 162, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Petrov, M.S.; Shanbhag, S.; Chakraborty, M.; Phillips, A.R.; Windsor, J.A. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology 2010, 139, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D.; Wani, S.; Gardner, T.B.; Falck-Ytter, Y.; Barkun, A.N. American Gastroenterological Association Institute Guideline on initial management of acute pancreatitis. Gastroenterology 2018, 154, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Sendler, M.; Algül, H. Pathogenesis of acute pancreatitis. Internist 2021, 62, 1034–1043. [Google Scholar] [CrossRef]
- Halangk, W.; Krüger, B.; Ruthenbürger, M.; Stürzebecher, J.; Albrecht, E.; Lippert, H.; Lerch, M.M. Trypsin activity is not involved in premature, intrapancreatic trypsinogen activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G367–G374. [Google Scholar] [CrossRef]
- Wartmann, T.; Mayerle, J.; Kähne, T.; Sahin-Tóth, M.; Ruthenbürger, M.; Matthias, R.; Kruse, A.; Reinheckel, T.; Peters, C.; Weiss, F.U.; et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 2010, 138, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Lerch, M.M.; Aghdassi, A.A. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology 2010, 138, 429–433. [Google Scholar] [CrossRef]
- Perino, A.; Schoonjans, K. Metabolic Messengers: Bile acids. Nat. Metab. 2022, 4, 416–423. [Google Scholar] [CrossRef]
- Šarenac, T.M.; Mikov, M. Bile acid synthesis: From nature to the chemical modification and synthesis and their applications as drugs and dutrients. Front. Pharmacol. 2018, 9, 939. [Google Scholar] [CrossRef]
- Honda, A.; Ikegami, T.; Matsuzaki, Y. Intestinal digestion and absorption. In Bile Acids in Gastroenterology: Basic and Clinical; Tazuma, S., Takikawa, H., Eds.; Springer: Tokyo, Japan, 2017; pp. 27–41. [Google Scholar]
- Tran, Q.T.; Tran, V.H.; Sendler, M.; Doller, J.; Wiese, M.; Bolsmann, R.; Wilden, A.; Glaubitz, J.; Modenbach, J.M.; Thiel, F.G.; et al. Role of bile acids and bile salts in acute pancreatitis: From the experimental to clinical studies. Pancreas 2021, 50, 3–11. [Google Scholar] [CrossRef]
- Mori, H.; Svegliati Baroni, G.; Marzioni, M.; Di Nicola, F.; Santori, P.; Maroni, L.; Abenavoli, L.; Scarpellini, E. Farnesoid X receptor, bile acid metabolism, and gut microbiota. Metabolites 2022, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Carulli, N.; Bertolotti, M.; Carubbi, F.; Concari, M.; Martella, P.; Carulli, L.; Loria, P. Review article: Effect of bile salt pool composition on hepatic and biliary functions. Aliment. Pharmacol. Ther. 2000, 14 (Suppl. S2), 14–18. [Google Scholar] [CrossRef] [PubMed]
- Heuman, D.M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 1989, 30, 719–730. [Google Scholar] [CrossRef]
- Jakkampudi, A.; Jangala, R.; Reddy, R.; Mitnala, S.; Rao, G.V.; Pradeep, R.; Reddy, D.N.; Talukdar, R. Acinar injury and early cytokine response in human acute biliary pancreatitis. Sci. Rep. 2017, 7, 15276. [Google Scholar] [CrossRef] [Green Version]
- Muili, K.A.; Wang, D.; Orabi, A.I.; Sarwar, S.; Luo, Y.; Javed, T.A.; Eisses, J.F.; Mahmood, S.M.; Jin, S.; Singh, V.P.; et al. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J. Biol. Chem. 2013, 288, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Perides, G.; Laukkarinen, J.M.; Vassileva, G.; Steer, M.L. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 2010, 138, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Kotb, M.A. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects: Ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int. J. Mol. Sci. 2012, 13, 8882–8914. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Tazuma, S.; Kanno, K.; Igarashi, Y.; Inui, K.; Ohara, H.; Tsuyuguchi, T.; Ryozawa, S. Ursodeoxycholic acid after bile duct stone removal and risk factors for recurrence: A randomized trial. J. Hepato-Biliary-Pancreat. Sci. 2016, 23, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Seyhun, E.; Malo, A.; Schäfer, C.; Moskaluk, C.A.; Hoffmann, R.T.; Göke, B.; Kubisch, C.H. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G773–G782. [Google Scholar] [CrossRef]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes care 2009, 32 (Suppl. S2), S237–S245. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerch, M.M.; Aghdassi, A.A.; Sendler, M. Cell signaling of pancreatic duct pressure and its role in the onset of pancreatitis. Gastroenterology 2020, 159, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, S.; Sato, M.; Kozuka, S. Prevalence of pancreatitis in liver diseases of various etiologies: An analysis of 107,754 adult autopsies in Japan. Digestion 1992, 51, 86–94. [Google Scholar] [CrossRef]
- Williams, J.A.; Sans, M.D.; Tashiro, M.; Schäfer, C.; Bragado, M.J.; Dabrowski, A. Cholecystokinin activates a variety of intracellular signal transduction mechanisms in rodent pancreatic acinar cells. Pharmacol. Toxicol. 2002, 91, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yao, L.; Fu, X.; Mukherjee, R.; Xia, Q.; Jakubowska, M.A.; Ferdek, P.E.; Huang, W. Experimental acute pancreatitis models: History, current status, and role in translational research. Front. Physiol. 2020, 11, 614591. [Google Scholar] [CrossRef]
- Kremer, A.E.; Namer, B.; Bolier, R.; Fischer, M.J.; Oude Elferink, R.P.; Beuers, U. Pathogenesis and management of pruritus in PBC and PSC. Dig. Dis. 2015, 33 (Suppl. S2), 164–175. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cui, Z.J. NanoLuc Bioluminescence-Driven photodynamic activation of cholecystokinin 1 receptor with genetically-encoded protein photosensitizer MiniSOG. Int. J. Mol. Sci. 2020, 21, 3763. [Google Scholar] [CrossRef]
- Williams, J.A. Regulation of acinar cell function in the pancreas. Curr. Opin. Gastroenterol. 2010, 26, 478–483. [Google Scholar] [CrossRef] [Green Version]
- Gerasimenko, J.V.; Lur, G.; Sherwood, M.W.; Ebisui, E.; Tepikin, A.V.; Mikoshiba, K.; Gerasimenko, O.V.; Petersen, O.H. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc. Natl. Acad. Sci. USA 2009, 106, 10758–10763. [Google Scholar] [CrossRef]
- Pallagi, P.; Madácsy, T.; Varga, Á.; Maléth, J. Intracellular Ca2+ Signalling in the pathogenesis of acute pancreatitis: Recent advances and translational perspectives. Int. J. Mol. Sci. 2020, 21, 4005. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.J.; Dong, M.; Harikumar, K.G.; Miller, L.J. Impact of ursodeoxycholic acid on a CCK1R cholesterol-binding site may contribute to its positive effects in digestive function. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G377–G386. [Google Scholar] [CrossRef] [Green Version]
- Liddle, R.A. Regulation of cholecystokinin secretion by intraluminal releasing factors. Am. J. Physiol. 1995, 269, G319–G327. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, K.H.; Lee, J.A.; Namkung, W.; Sun, A.Q.; Ananthanarayanan, M.; Suchy, F.J.; Shin, D.M.; Muallem, S.; Lee, M.G. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology 2002, 122, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Cui, Z.J. Extracellular histones activate plasma membrane Toll-Like receptor 9 to trigger calcium oscillations in rat pancreatic acinar tumor cell AR4-2J. Cells 2018, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta Gene Regul. Mech. 2009, 1789, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.D.; Zhu, R.X.; Pan, X.T.; Sun, T.W. Bile acid supplementation improves murine pancreatitis in association with the gut microbiota. Front. Physiol. 2020, 11, 650. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 2017, 16, s15–s20. [Google Scholar] [CrossRef] [PubMed]
- Frost, F.; Weiss, F.U.; Lerch, M.M. The role of the microbiome in diseases of the pancreas. Internist 2022, 63, 372–378. [Google Scholar] [CrossRef]
- Arifin, W.N.; Zahiruddin, W.M. Sample size calculation in animal studies using resource equation approach. Malays. J. Med. Sci. 2017, 24, 101–105. [Google Scholar] [CrossRef]
- Mead, R.; Gilmour, S.; Mead, A. Designing useful experiments. In Statistical Principles for the Design of Experiments: Applications to Real Experiments; Cambridge Series in Statistical and Probabilistic Mathematics; Cambridge University Press: Cambridge, UK, 2012; pp. 538–564. [Google Scholar] [CrossRef]
- Paolini, M.; Pozzetti, L.; Piazza, F.; Cantelli-Forti, G.; Roda, A. Bile acid structure and selective modulation of murine hepatic cytochrome P450-linked enzymes. Hepatology 1999, 30, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Ferrell, L.D.; Grendell, J.H. Caerulein-induced acute necrotizing pancreatitis in mice: Protective effects of proglumide, benzotript, and secretin. Gastroenterology 1985, 88, 1192–1204. [Google Scholar] [CrossRef]
- Sendler, M.; Beyer, G.; Mahajan, U.M.; Kauschke, V.; Maertin, S.; Schurmann, C.; Homuth, G.; Völker, U.; Völzke, H.; Halangk, W.; et al. Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology 2015, 149, 765–776.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.J.; Wan, R.; Shen, J.Q.; Shen, J.; Wang, X.P. Effect of L-cysteine on remote organ injury in rats with severe acute pancreatitis induced by bile-pancreatic duct obstruction. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 428–435. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, Q.T.; Sendler, M.; Wiese, M.L.; Doller, J.; Zierke, L.; Gischke, M.; Glaubitz, J.; Tran, V.H.; Lalk, M.; Bornscheuer, U.T.; et al. Systemic Bile Acids Affect the Severity of Acute Pancreatitis in Mice Depending on Their Hydrophobicity and the Disease Pathogenesis. Int. J. Mol. Sci. 2022, 23, 13592. https://doi.org/10.3390/ijms232113592
Tran QT, Sendler M, Wiese ML, Doller J, Zierke L, Gischke M, Glaubitz J, Tran VH, Lalk M, Bornscheuer UT, et al. Systemic Bile Acids Affect the Severity of Acute Pancreatitis in Mice Depending on Their Hydrophobicity and the Disease Pathogenesis. International Journal of Molecular Sciences. 2022; 23(21):13592. https://doi.org/10.3390/ijms232113592
Chicago/Turabian StyleTran, Quang Trung, Matthias Sendler, Mats L. Wiese, Julia Doller, Lukas Zierke, Marcel Gischke, Juliane Glaubitz, Van Huy Tran, Michael Lalk, Uwe T. Bornscheuer, and et al. 2022. "Systemic Bile Acids Affect the Severity of Acute Pancreatitis in Mice Depending on Their Hydrophobicity and the Disease Pathogenesis" International Journal of Molecular Sciences 23, no. 21: 13592. https://doi.org/10.3390/ijms232113592