

Pathology of Initial Axon Segments in Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Related Disorders
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Search Strategy
2.2. Data Extraction
2.3. Qualitative Analysis and Synthesis
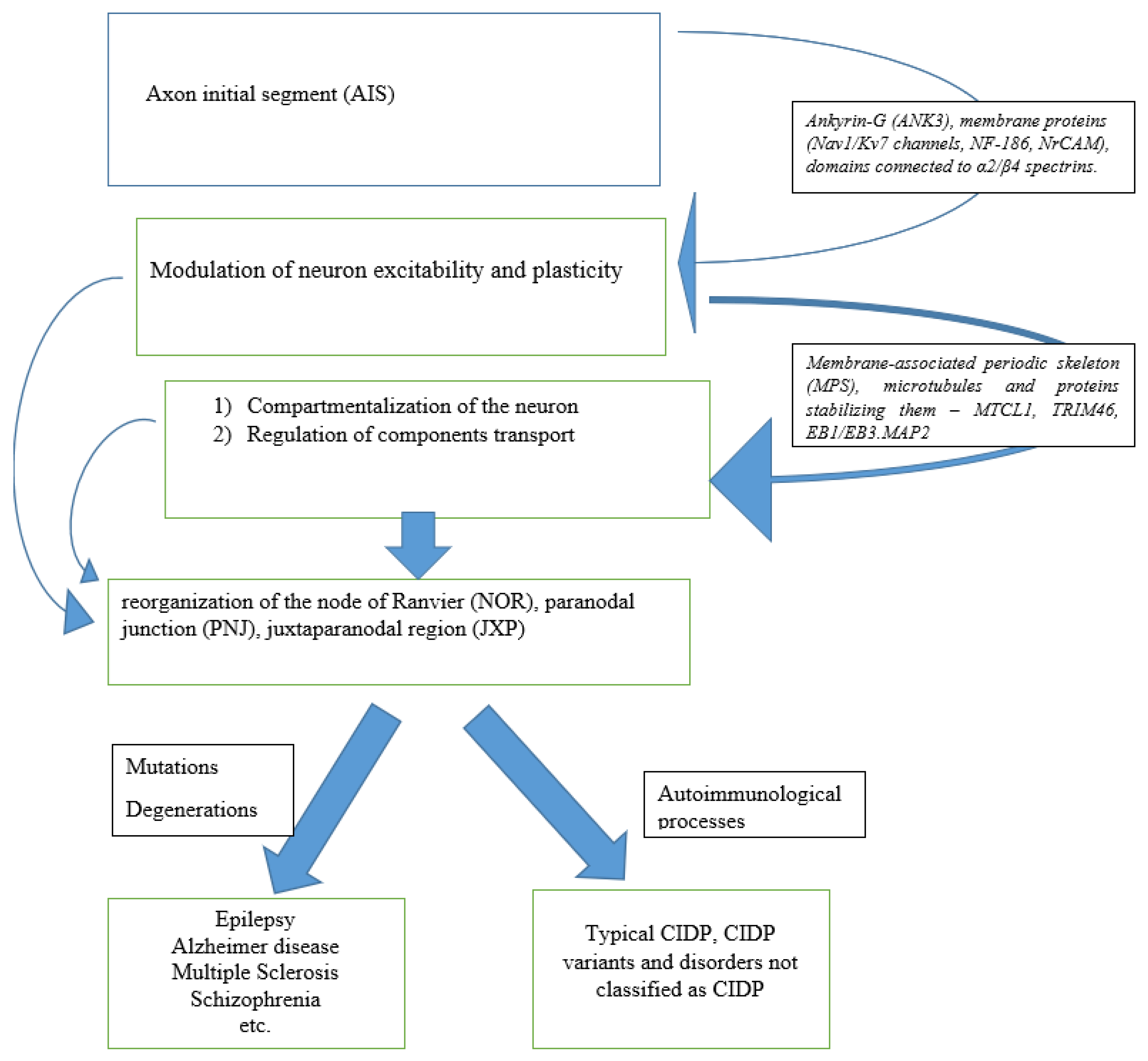
3. Anatomy
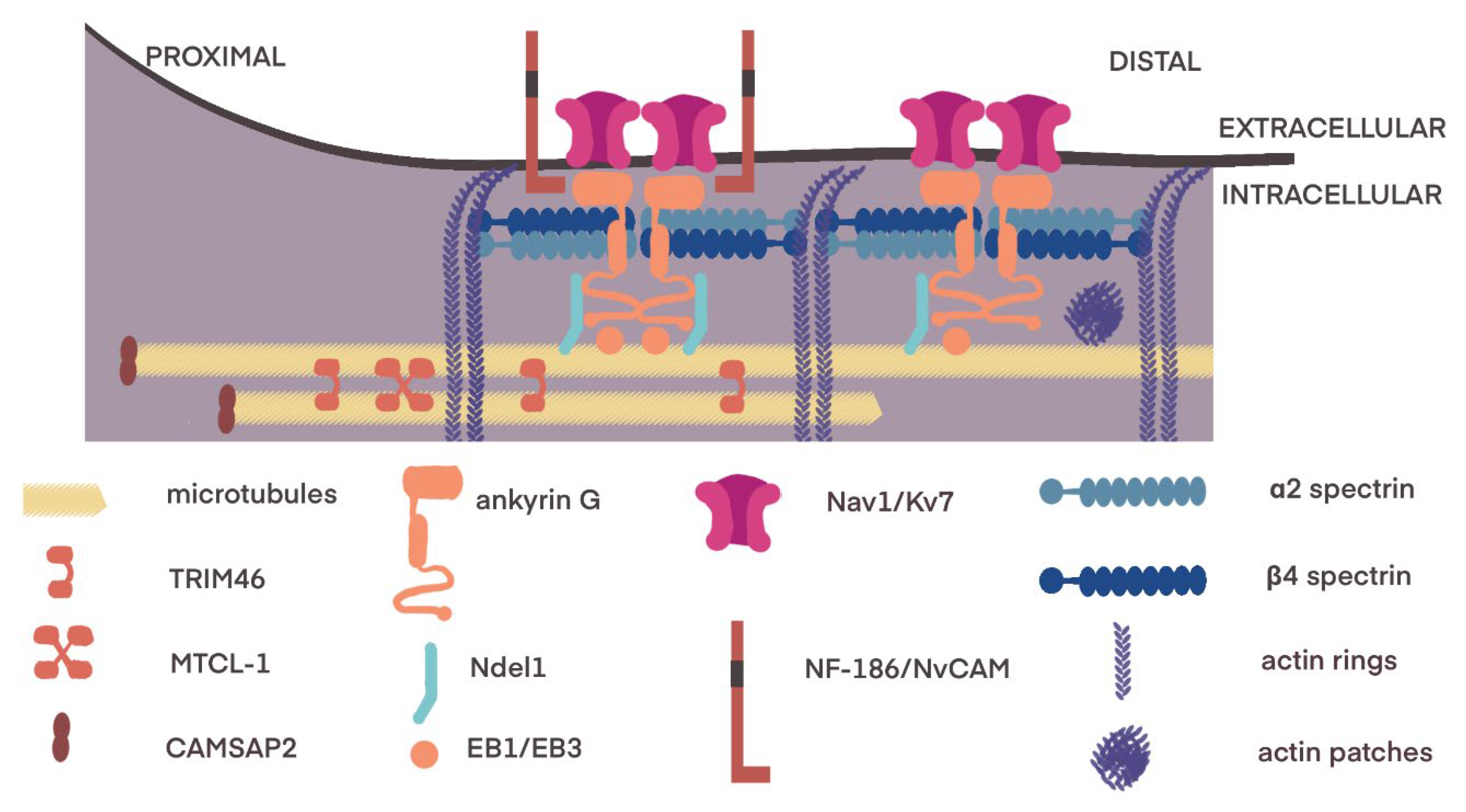
3.1. Axon Initial Segment
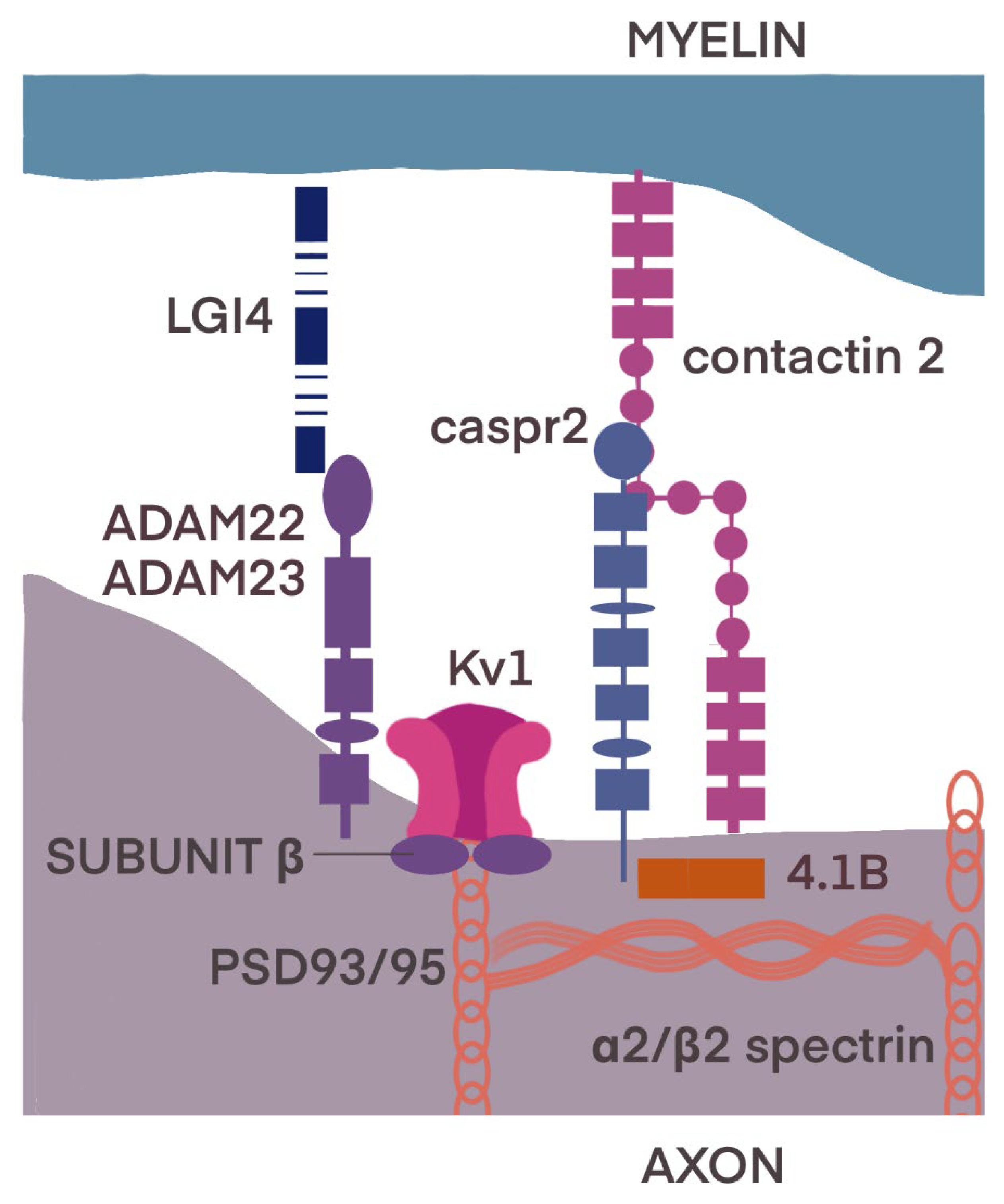
3.2. Juxtaparanode Region
3.3. Transport of Components
4. Pathology of Initial Axon Segments
4.1. Pathology of Initial Axon Segments in CIDP
4.2. Pathology of Initial Axon Segments in Related Disorders
5. Conclusions
6. Future Directions
- The anatomy of the axon initial segment: the node of Ranvier, the paranodal junction and the juxtaparanodal region, together with understanding the principles of axonal transport of various factors important in the physiology and pathology of peripheral nerves.
- The disclosure of specific pathomechanisms important in the peripheral nerve damage, including novel target antigens and their mutual dependence and dependence on the presence of other pro and anti-inflammatory factors.
- The reference of autoimmune disturbances to clinical findings, which may lead to the creation of a new classification of inflammatory neuropathies.
- All of the above-mentioned scientific challenges should result in new therapeutic options tailored to the individual patient.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIS | axon initial segment |
| AnkG | ankyrin G |
| BL | basal lamina |
| CCI | chronic constriction injury |
| CIAP | chronic inflammatory axonal polyneuropathy |
| CIDP | chronic inflammatory demyelinating polyradiculoneuropathy |
| CIP | chronic inflammatory polyradiculoneuropathy |
| CNS | central nervous system |
| EA-1 | episodic ataxia type 1 |
| JXP | juxtaparanodal region |
| LGI | leucine-rich glioma inactivated |
| MAGUK | membrane-associated guanylate kinase |
| MTCL1 | microtubule cross-linking factor 1 |
| MGUS | monoclonal gammopathies of undetermined significance |
| NOR | node of Ranvier |
| NAWM | normal-appearing white matter |
| PNJ | paranodal junction |
| SCLC | small-cell lung carcinoma |
| TRIM46 | tripartite-motif containing 46 |
| TNFα | tumor necrosis factor α |
References
- Van den Bergh, P.Y.K.; van Doorn, P.A.; Hadden, R.D.M.; Avau, B.; Vankrunkelsven, P.; Allen, J.A.; Attarian, S.; Blomkwist-Markens, P.H.; Cornblath, D.R.; Eftimov, F.; et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force—Second revision. Eur. J. Neurol. 2021, 28, 3556–3583. [Google Scholar] [CrossRef] [PubMed]
- Uncini, A.; Mathis, S.; Vallat, J.M. New classification of autoimmune neuropathies based on target antigens and involved domains of myelinated fibres. J. Neurol. Neurosurg. Psychiatry 2022, 93, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Vallat, J.M.; Deschamps, N.; Corcia, P.; Magy, L.; Mathis, S. Chronic Inflammatory or Chronic Inflammatory Demyelinating Polyradiculoneuropathy? Front. Neurol. 2022, 13, 862335. [Google Scholar] [CrossRef] [PubMed]
- Susuki, K.; Yuki, N.; Schafer, D.P.; Hirata, K.; Zhang, G.; Funakoshi, K.; Rasband, M.N. Dysfunction of nodes of Ranvier: A mechanism for anti-ganglioside antibody-mediated neuropathies. Exp. Neurol. 2012, 233, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Uncini, A.; Susuki, K.; Yuki, N. Nodo-paranodopathy: Beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2013, 124, 1928–1934. [Google Scholar] [CrossRef]
- Oh, S.J.; Lu, L.; Alsharabati, M.; Morgan, M.B.; King, P. Chronic inflammatory axonal polyneuropathy. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1175–1180. [Google Scholar] [CrossRef]
- Rasband, M.N.; Peles, E. Mechanisms of node of Ranvier assembly. Nat. Rev. Neurosci. 2021, 22, 7–20. [Google Scholar] [CrossRef]
- Dziadkowiak, E.; Waliszewska-Prosół, M.; Nowakowska-Kotas, M.; Budrewicz, S.; Koszewicz, Z.; Koszewicz, M. Pathophysiology of the different clinical phenotypes of chronic inflammatory demyelinating polyradiculoneuropathy (Cidp). Int. J. Mol. Sci. 2022, 23, 179. [Google Scholar] [CrossRef]
- Palay, S.L.; Sotelo, C.; Peters, A.; Orkand, P.M. The axon hillock and the initial segment. J. Cell Biol. 1968, 38, 193–201. [Google Scholar] [CrossRef]
- Peters, A.; Proskauer, C.C.; Kaiserman-Abramof, I.R. The small pyramidal neuron of the rat cerebral cortex. The axon hillock and initial segment. J. Cell Biol. 1968, 39, 604–619. [Google Scholar] [CrossRef]
- Debanne, D.; Campanac, E.; Bialowas, A.; Carlier, E.; Alcaraz, G. Axon physiology. Physiol. Rev. 2011, 91, 555–602. [Google Scholar] [CrossRef]
- Ogawa, Y.; Rasband, M.N. The functional organization and assembly of the axon initial segment. Curr. Opin. Neurobiol. 2008, 18, 307–313. [Google Scholar] [CrossRef]
- Leterrier, C. The axon initial segment: An updated viewpoint. J. Neurosci. 2018, 38, 2135–2145. [Google Scholar] [CrossRef]
- Fujitani, M.; Otani, Y.; Miyajima, H. Pathophysiological roles of abnormal axon initial segments in neurodevelopmental disorders. Cells 2021, 10, 2110. [Google Scholar] [CrossRef]
- Kordeli, E.; Bennett, V. Distinct ankyrin isoforms at neuron cell bodies and nodes of Ranvier resolved using erythrocyte ankyrin-deficient mice. J. Cell Biol. 1991, 114, 1243–1259. [Google Scholar] [CrossRef]
- Pinatel, D.; Faivre-Sarrailh, C. Assembly and Function of the Juxtaparanodal Kv1 Complex in Health and Disease. Life 2020, 11, 8. [Google Scholar] [CrossRef]
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.-Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and Na V channels at electrically active domains of the axon. J. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef]
- Bennett, V.; Lorenzo, D.N. Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr. Top Membr. 2013, 72, 1–37. [Google Scholar] [CrossRef]
- Davis, J.Q.; Lambert, S.; Bennett, V. Molecular composition of the node of Ranvier: Identification of ankyrin- binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J. Cell Biol. 1996, 135, 1355–1367. [Google Scholar] [CrossRef]
- Leterrier, C.; Vacher, H.; Fache, M.-P.; D’Ortoli, S.A.; Castets, F.; Autillo-Touati, A.; Dargent, B. End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc. Natl. Acad. Sci. USA 2011, 108, 8826–8831. [Google Scholar] [CrossRef]
- Albrecht, D.; Winterflood, C.M.; Sadeghi, M.; Tschager, T.; Noé, F.; Ewers, H. Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J. Cell Biol. 2016, 215, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Biller, L.; Michel, U.; Bähr, M.; Koch, J.C. Cytoskeletal assembly in axonal outgrowth and regeneration analyzed on the nanoscale. Sci. Rep. 2022, 12, 14387. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, M.; Jorgensen, E.M.; Bastiani, M.J. Axons break in animals lacking β-spectrin. J. Cell Biol. 2007, 176, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, S.; Gibaud, S.; Jimenez, A.; Caillol, G.; Leterrier, C. Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. Nat. Commun. 2019, 10, 5803. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Hahn, I.; Webb, S.E.D.; Pearce, S.P.; Prokop, A. Periodic actin structures in neuronal axons are required to maintain microtubules. Mol. Biol. Cell 2017, 28, 296–308. [Google Scholar] [CrossRef]
- Komada, M.; Soriano, P. βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J. Cell Biol. 2002, 156, 337–348. [Google Scholar] [CrossRef]
- Lorenzo, D.N.; Badea, A.; Zhou, R.; Mohler, P.J.; Zhuang, X.; Bennett, V. βiI-spectrin promotes mouse brain connectivity through stabilizing axonal plasma membranes and enabling axonal organelle transport. Proc. Natl. Acad. Sci. USA 2019, 116, 15686–15695. [Google Scholar] [CrossRef]
- Satake, T.; Yamashita, K.; Hayashi, K.; Miyatake, S.; Tamura-Nakano, M.; Doi, H.; Furuta, Y.; Shioi, G.; Miura, E.; Takeo, Y.H.; et al. MTCL1 plays an essential role in maintaining Purkinje neuron axon initial segment. EMBO J. 2017, 36, 1227–1242. [Google Scholar] [CrossRef]
- Vuong, J.K.; Ergin, V.; Chen, L.; Zheng, S. Multilayered regulations of alternative splicing, NMD, and protein stability control temporal induction and tissue-specific expression of TRIM46 during axon formation. Nat. Commun. 2022, 13, 2081. [Google Scholar] [CrossRef]
- Dwenger, M.M.; Raph, S.M.; Baba, S.P.; Iv, J.B.M.; Nystoriak, M.A. Diversification of Potassium Currents in Excitable Cells via Kv β Proteins. Cells 2022, 11, 2230. [Google Scholar] [CrossRef]
- Wang, H.; Kunkel, D.D.; Martin, T.M.; Schwartzkroin, P.A.; Tempel, B.L. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature 1993, 365, 75–79. [Google Scholar] [CrossRef]
- Rasband, M.; Trimmer, J.S.; Schwarz, T.L.; Levinson, S.R.; Ellisman, M.H.; Schachner, M.; Shrager, P. Potassium channel distribution, clustering, and function in remyelinating rat axons. J. Neurosci. 1998, 18, 36–47. [Google Scholar] [CrossRef]
- Rhodes, K.J.; Strassle, B.W.; Monaghan, M.M.; Bekele-Arcuri, Z.; Matos, M.F.; Trimmer, J.S. Association and colocalization of the Kvβ1 and Kvβ2 β-subunits with Kv1 α-subunits in mammalian brain K+ channel complexes. J. Neurosci. 1997, 17, 8246–8258. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef]
- Koźmiński, W.; Pera, J. Involvement of the Peripheral Nervous System in Episodic Ataxias. Biomedicines 2020, 8, 448. [Google Scholar] [CrossRef]
- Laube, G.; Röper, J.; Pitt, J.C.; Sewing, S.; Kistner, U.; Garner, C.C.; Pongs, O.; Veh, R.W. Ultrastructural localization of Shaker-related potassium channel subunits and synapse-associated protein 90 to septate-like junctions in rat cerebellar Pinceaux. Brain Res. Mol. Brain Res. 1996, 42, 51–61. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, C.L.; Messing, A.; Chiu, S.Y. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: Role of potassium channels under the myelin sheath in young nerves. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 7200–7215. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef]
- Poliak, S.; Gollan, L.; Martinez, R.; Custer, A.; Einheber, S.; Salzer, J.L.; Trimmer, J.S.; Shrager, P.; Peles, E. Caspr2, a new member of the Neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999, 24, 1037–1047. [Google Scholar] [CrossRef]
- Sinha, S.; Shivaram, S.; Nagappa, M.; Seshagiri, D.; Mahadevan, A.; Gangadhar, Y.; Sathyaprabha, T.; Kumavat, V.; Bharath, R.; Taly, A. Clinical profile and treatment response in patients with CASPR2 antibody-associated neurological disease. Ann. Indian Acad. Neurol. 2021, 24, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Kannoth, S.; Nambiar, V.; Gopinath, S.; Anandakuttan, A.; Mathai, A.; Rajan, P.K. Expanding spectrum of contactin-associated protein 2 (CASPR2) autoimmunity-syndrome of parkinsonism and ataxia. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2018, 39, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.; Santos, S.; Coutinho, E.; Whitt, J.L.; Beltrão, N.; Rondão, T.; Leite, M.I.; Buckley, C.; Lee, H.-K.; Carvalho, A.L. Disrupted AMPA Receptor Function upon Genetic- or Antibody-Mediated Loss of Autism-Associated CASPR2. Cereb. Cortex. 2019, 29, 4919–4931. [Google Scholar] [CrossRef] [PubMed]
- Vural, A.; Doppler, K.; Meinl, E. Autoantibodies against the node of ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: Diagnostic, pathogenic, and therapeutic relevance. Front. Immunol. 2018, 9, 1029. [Google Scholar] [CrossRef] [PubMed]
- Horresh, I.; Bar, V.; Kissil, J.L.; Peles, E. Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J. Neurosci. 2010, 30, 2480–2489. [Google Scholar] [CrossRef]
- Labasque, M.; Hivert, B.; Nogales-Gadea, G.; Querol, L.; Illa, I.; Faivre-Sarrailh, C. Specific contactin N-glycans are implicated in neurofascin binding and autoimmune targeting in peripheral neuropathies. J. Biol. Chem. 2014, 289, 7907–7918. [Google Scholar] [CrossRef]
- Xu, D.-E.; Zou, Y.; Zhang, W.-F.; Liu, H.-Y.; Li, X.; Zhang, X.; Ma, X.-F.; Sun, Y.; Jiang, S.-Y.; Ma, Q.-H. Structure and function of the contactin-associated protein family in myelinated axons and their relationship with nerve diseases. Neural. Regen. Res. 2017, 12, 1551–1558. [Google Scholar] [CrossRef]
- Traka, M.; Dupree, J.L.; Popko, B.; Karagogeos, D. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J. Neurosci. 2002, 22, 3016–3024. [Google Scholar] [CrossRef]
- Furley, A.J.; Morton, S.B.; Manalo, D.; Karagogeos, D.; Dodd, J.; Jessell, T.M. The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 1990, 61, 157–170. [Google Scholar] [CrossRef]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.-Y.; Shrager, P.; et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef]
- Ohara, R.; Yamakawa, H.; Nakayama, M.; Ohara, O. Type II brain 4.1 (4.1B/KIAA0987), a member of the protein 4.1 family, is localized to neuronal paranodes. Mol. Brain Res. 2000, 85, 41–52. [Google Scholar] [CrossRef]
- Arroyo, E.J.; Sirkowski, E.E.; Chitale, R.; Scherer, S.S. Acute demyelination disrupts the molecular organization of peripheral nervous system nodes. J. Comp. Neurol. 2004, 479, 424–434. [Google Scholar] [CrossRef]
- Ivanovic, A.; Horresh, I.; Golan, N.; Spiegel, I.; Sabanay, H.; Frechter, S.; Ohno, S.; Terada, N.; Möbius, W.; Rosenbluth, J.; et al. The cytoskeletal adapter protein 4.1G organizes the internodes in peripheral myelinated nerves. J. Cell Biol. 2012, 196, 337–344. [Google Scholar] [CrossRef]
- Ogawa, Y.; Oses-Prieto, J.; Kim, M.Y.; Horresh, I.; Peles, E.; Burlingame, A.L.; Trimmer, J.S.; Meijer, D.; Rasband, M.N. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J. Neurosci. 2010, 30, 1038–1048. [Google Scholar] [CrossRef]
- Özkaynak, E.; Abello, G.; Jaegle, M.; Van Berge, L.; Hamer, D.; Kegel, L.; Driegen, S.; Sagane, K.; Bermingham, J.R.; Meijer, D. Adam22 is a major neuronal receptor for Lgi4-mediated Schwann cell signaling. J. Neurosci. 2010, 30, 3857–3864. [Google Scholar] [CrossRef]
- Hivert, B.; Marien, L.; Agbam, K.N.; Faivre-Sarrailh, C. ADAM22 and ADAM23 modulate the targeting of the Kv1 channel-associated protein LGI1 to the axon initial segment. J. Cell Sci. 2019, 132, jcs.219774. [Google Scholar] [CrossRef]
- Zhang, Y.; Bekku, Y.; Dzhashiashvili, Y.; Armenti, S.; Meng, X.; Sasaki, Y.; Milbrandt, J.; Salzer, J.L. Assembly and maintenance of nodes of ranvier rely on distinct sources of proteins and targeting mechanisms. Neuron 2012, 73, 92–107. [Google Scholar] [CrossRef]
- Nelson, A.D.; Jenkins, P.M. Axonal Membranes and Their Domains: Assembly and Function of the Axon Initial Segment and Node of Ranvier. Front. Cell Neurosci. 2017, 11, 136. [Google Scholar] [CrossRef]
- Bréchet, A.; Fache, M.-P.; Brachet, A.; Ferracci, G.; Baude, A.; Irondelle, M.; Pereira, S.; Leterrier, C.; Dargent, B. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J. Cell Biol. 2008, 183, 1101–1114. [Google Scholar] [CrossRef]
- Gu, C.; Zhou, W.; Puthenveedu, M.A.; Xu, M.; Jan, Y.N.; Jan, L.Y. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron 2006, 52, 803–816. [Google Scholar] [CrossRef]
- Vacher, H.; Yang, J.W.; Cerda, O.; Autillo-Touati, A.; Dargent, B.; Trimmer, J.S. Cdk-mediated phosphorylation of the Kvβ2 auxiliary subunit regulates Kv1 channel axonal targeting. J. Cell Biol. 2011, 192, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Pinatel, D.; Hivert, B.; Saint-Martin, M.; Noraz, N.; Savvaki, M.; Karagogeos, D.; Faivre-Sarrailh, C. The Kv1-associated molecules TAG-1 and Caspr2 are selectively targeted to the axon initial segment in hippocampal neurons. J. Cell Sci. 2017, 130, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Bel, C.; Oguievetskaia, K.; Pitaval, C.; Goutebroze, L.; Faivre-Sarrailh, C. Axonal targeting of Caspr2 in hippocampal neurons via selective somatodendritic endocytosis. J. Cell Sci. 2009, 122, 3403–3413. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kato, K.; Yamaguchi, T.; Fukata, Y.; Ohno, S.; Kaibuchi, K. Role of the PAR-3-KIF3 complex in the establishment of neuronal polarity. Nat. Cell Biol. 2004, 6, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.R.; Holtmann, L.; Bobylev, I.; Schneider, C.; Ritter, C.; Weis, J.; Lehmann, H.C. Loss of Schwann cell plasticity in chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neuroinflamm. 2016, 13, 255. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Belfiore, L.; Chu, T.-H.; Fleming, T.; Midha, R.; Biernaskie, J.; Schuurmans, C. Insights Into the Role and Potential of Schwann Cells for Peripheral Nerve Repair From Studies of Development and Injury. Front. Mol. Neurosci. 2021, 13, 608442. [Google Scholar] [CrossRef]
- Panzer, K.V.; Burrell, J.C.; Helm, K.V.T.; Purvis, E.M.; Zhang, Q.; Le, A.D.; O’Donnell, J.C.; Cullen, D.K. Tissue Engineered Bands of Büngner for Accelerated Motor and Sensory Axonal Outgrowth. Front. Bioeng. Biotechnol. 2020, 8, 580654. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef]
- Parkinson, D.B.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.A.; Woodhoo, A.; Lloyd, A.C.; Feltri, M.L.; Wrabetz, L.; Behrens, A.; Mirsky, R.; et al. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef]
- Boerboom, A.; Dion, V.; Chariot, A.; Franzen, R. Molecular mechanisms involved in schwann cell plasticity. Front. Mol. Neurosci. 2017, 10, 38. [Google Scholar] [CrossRef]
- Tzekova, N.; Heinen, A.; Bunk, S.; Hermann, C.; Hartung, H.-P.; Reipert, B.; Küry, P. Immunoglobulins stimulate cultured Schwann cell maturation and promote their potential to induce axonal outgrowth. J. Neuroinflamm. 2015, 12, 107. [Google Scholar] [CrossRef][Green Version]
- Renthal, W.; Tochitsky, I.; Yang, L.; Cheng, Y.-C.; Li, E.; Kawaguchi, R.; Geschwind, D.H.; Woolf, C.J. Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 2020, 108, 128–144.e9. [Google Scholar] [CrossRef]
- Lee, N.; Neitzel, K.L.; Devlin, B.K.; MacLennan, A.J. STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: Potential role for STAT3 as a retrograde signaling transcription factor. J. Comp. Neurol. 2004, 474, 535–545. [Google Scholar] [CrossRef]
- Jankowski, M.P.; Cornuet, P.K.; McIlwrath, S.; Koerber, H.R.; Albers, K.M. SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience 2006, 143, 501–514. [Google Scholar] [CrossRef]
- Okuyama, N.; Kiryu-Seo, S.; Kiyama, H. Altered expression of Smad family members in injured motor neurons of rat. Brain Res. 2007, 1132, 36–41. [Google Scholar] [CrossRef]
- Wang, G.; Simon, D.J.; Wu, Z.; Belsky, D.M.; Heller, E.; O’Rourke, M.K.; Hertz, N.T.; Molina, H.; Zhong, G.; Tessier-Lavigne, M.; et al. Structural plasticity of actin-spectrin membrane skeleton and functional role of actin and spectrin in axon degeneration. Elife 2019, 8, e38730. [Google Scholar] [CrossRef]
- Bonetti, B.; Valdo, P.; Stegagno, C.; Tanel, R.; Zanusso, G.; Ramarli, D.; Fiorini, E.; Turazzi, S.; Carner, M.; Moretto, G. Tumor necrosis factor α and human Schwann cells: Signalling and phenotype modulation without cell death. J. Neuropathol. Exp. Neurol. 2000, 59, 74–84. [Google Scholar] [CrossRef][Green Version]
- Nikoloudaki, G.; Brooks, S.; Peidl, A.P.; Tinney, D.; Hamilton, D.W. JNK signaling as a key modulator of soft connective tissue physiology, pathology, and healing. Int. J. Mol. Sci. 2020, 21, 1015. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Moss, K.R.; Bopp, T.S.; Johnson, A.E.; Höke, A. New evidence for secondary axonal degeneration in demyelinating neuropathies. Neurosci. Lett. 2021, 744, 135595. [Google Scholar] [CrossRef]
- Iijima, M.; Koike, H.; Katsuno, M.; Sobue, G. Polymorphism of transient axonal glycoprotein-1 in chronic inflammatory demyelinating polyneuropathy. J. Peripher. Nerv. Syst. 2011, 16 (Suppl. 1), 52–55. [Google Scholar] [CrossRef] [PubMed]
- Doppler, K.; Schuster, Y.; Appeltshauser, L.; Biko, L.; Villmann, C.; Weishaupt, A.; Werner, C.; Sommer, C. Anti-CNTN1 IgG3 induces acute conduction block and motor deficits in a passive transfer rat model. J. Neuroinflamm. 2019, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, A.; Nusser, Z. Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci. 2008, 28, 14329–14340. [Google Scholar] [CrossRef] [PubMed]
- Vacher, H.; Trimmer, J.S. Trafficking mechanisms underlying neuronal voltage-gated ion channel localization at the axon initial segment. Epilepsia 2012, 53 (Suppl. 9), 21–31. [Google Scholar] [CrossRef]
- Huang, C.Y.M.; Rasband, M.N. Axon initial segments: Structure, function, and disease. Ann. N. Y. Acad. Sci. 2018, 1420, 46–61. [Google Scholar] [CrossRef]
- Clark, K.C.; Josephson, A.; Benusa, S.D.; Hartley, R.K.; Baer, M.; Thummala, S.; Joslyn, M.; Sword, B.A.; Elford, H.; Oh, U.; et al. Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia 2016, 64, 1190–1209. [Google Scholar] [CrossRef]
- Kastriti, M.E.; Sargiannidou, I.; Kleopa, K.A.; Karagogeos, D. Differential modulation of the juxtaparanodal complex in Multiple Sclerosis. Mol. Cell Neurosci. 2015, 67, 93–103. [Google Scholar] [CrossRef]
- Nip, K.; Kashiwagura, S.; Kim, J.H. Loss of β4-spectrin impairs Nav channel clustering at the heminode and temporal fidelity of presynaptic spikes in developing auditory brain. Sci. Rep. 2022, 12, 5854. [Google Scholar] [CrossRef]
- Buelow, M.; Süßmuth, D.; Smith, L.D.; Aryani, O.; Castiglioni, C.; Stenzel, W.; Bertini, E.; Schuelke, M.; Knierim, E. Novel bi-allelic variants expand the SPTBN4-related genetic and phenotypic spectrum. Eur. J. Hum. Genet. 2021, 29, 1121–1128. [Google Scholar] [CrossRef]
- Knierim, E.; Gill, E.; Seifert, F.; Morales-Gonzalez, S.; Unudurthi, S.D.; Hund, T.J.; Stenzel, W.; Schuelke, M. A recessive mutation in beta-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum. Genet. 2017, 136, 903–910. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, M.; Wang, H.; Li, Y.; Zhanghao, K.; Xi, P.; Zhang, Y. The largest isoform of Ankyrin-G is required for lattice structure of the axon initial segment. Biochem. Biophys. Res. Commun. 2021, 578, 28–34. [Google Scholar] [CrossRef]
- Sun, X.; Wu, Y.; Gu, M.; Liu, Z.; Ma, Y.; Li, J.; Zhang, Y. Selective filtering defect at the axon initial segment in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 2014, 111, 14271–14276. [Google Scholar] [CrossRef]
- Wimmer, V.; Reid, C.; Mitchell, S.; Richards, K.L.; Scaf, B.B.; Leaw, B.T.; Hill-Yardin, E.; Royeck, M.; Horstmann, M.-T.; Cromer, B.; et al. Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J. Clin. Investig. 2010, 120, 2661–2671. [Google Scholar] [CrossRef]
- Van Coevorden-Hameete, M.H.; Van Beuningen, S.F.; Perrenoud, M.; Will, L.M.; Hulsenboom, E.; Demonet, J.-F.; Sabater, L.; Kros, J.M.; Verschuuren, J.; Titulaer, M.J.; et al. Antibodies to TRIM46 are associated with paraneoplastic neurological syndromes. Ann. Clin. Transl. Neurol. 2017, 4, 680–686. [Google Scholar] [CrossRef]
- Manganas, L.N.; Akhtar, S.; Antonucci, D.E.; Campomanes, C.R.; Dolly, J.O.; Trimmer, J.S. Episodic Ataxia Type-1 Mutations in the Kv1.1 Potassium Channel Display Distinct Folding and Intracellular Trafficking Properties. J. Biol. Chem. 2001, 276, 49427–49434. [Google Scholar] [CrossRef]
- Berghs, S.; Ferracci, F.; Maksimova, E.; Gleason, S.; Leszczynski, N.; Butler, M.; De Camilli, P.; Solimena, M. Autoimmunity to βIV spectrin in paraneoplastic lower motor neuron syndrome. Proc. Natl. Acad. Sci. USA 2001, 98, 6945–6950. [Google Scholar] [CrossRef]
- Bartley, C.M.; Ngo, T.T.; Alvarenga, B.D.; Kung, A.F.; Teliska, L.H.; Sy, M.; DeRisi, J.L.; Rasband, M.N.; Pittock, S.J.; Dubey, D.; et al. βIV-Spectrin Autoantibodies in 2 Individuals With Neuropathy of Possible Paraneoplastic Origin. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1188. [Google Scholar] [CrossRef]
- Beijer, D.; Deconinck, T.; De Bleecker, J.L.; Dotti, M.T.; Malandrini, A.; Urtizberea, J.A.; Zulaica, M.; De Munain, A.L.; Asselbergh, B.; De Jonghe, P.; et al. Nonsense mutations in alpha-II spectrin in three families with juvenile onset hereditary motor neuropathy. Brain 2019, 142, 2605–2616. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziadkowiak, E.; Nowakowska-Kotas, M.; Budrewicz, S.; Koszewicz, M. Pathology of Initial Axon Segments in Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Related Disorders. Int. J. Mol. Sci. 2022, 23, 13621. https://doi.org/10.3390/ijms232113621
Dziadkowiak E, Nowakowska-Kotas M, Budrewicz S, Koszewicz M. Pathology of Initial Axon Segments in Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Related Disorders. International Journal of Molecular Sciences. 2022; 23(21):13621. https://doi.org/10.3390/ijms232113621
Chicago/Turabian StyleDziadkowiak, Edyta, Marta Nowakowska-Kotas, Sławomir Budrewicz, and Magdalena Koszewicz. 2022. "Pathology of Initial Axon Segments in Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Related Disorders" International Journal of Molecular Sciences 23, no. 21: 13621. https://doi.org/10.3390/ijms232113621
APA StyleDziadkowiak, E., Nowakowska-Kotas, M., Budrewicz, S., & Koszewicz, M. (2022). Pathology of Initial Axon Segments in Chronic Inflammatory Demyelinating Polyradiculoneuropathy and Related Disorders. International Journal of Molecular Sciences, 23(21), 13621. https://doi.org/10.3390/ijms232113621