
Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments
 , , ,
, , ,
Abstract
:1. Introduction
2. Astrocytic Role in AD Pathophysiology: Recent Developments
2.1. Astrocytes in Tau Protein and Amyloid Metabolism
2.2. Astrocytes, Neuroinflammation and Oxidative Stress
2.3. Astrocyte Role in Gliotransmission and Excitotoxicity
3. Astrocytes as a Therapeutic Target: Treatment and Neuroprotection Strategies Based on Astrocyte Modulation
3.1. Neuroinflammatory Control
3.2. Targeting Oxidative Stress
3.3. Modulation of Glutamatergic Activity
3.4. APOE and Lipid Metabolism
3.5. AGE Inhibitors
3.6. Neurovascular Unit and Blood–Brain Barrier Interventions
3.7. Interventions on Glymphatic System
3.8. Aβ Clearance
3.9. Calcium Signalling
3.10. Melatonin-Based Interventions
4. Non-Pharmacological Interventions That Impact Astrocyte Function
4.1. Physical Exercise
4.2. Dietary Approaches: Ketogenic Diet
4.3. Electromagnetic and Electric Stimulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Revi, M. Alzheimer’s Disease Therapeutic Approaches. Adv. Exp. Med. Biol. 2020, 1195, 105–116. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, C.; Sorbi, S. The Complexity of Alzheimer’s Disease: An Evolving Puzzle. Physiol. Rev. 2021, 101, 1047–1081. [Google Scholar] [CrossRef]
- Diociaiuti, M.; Bonanni, R.; Cariati, I.; Frank, C.; D’Arcangelo, G. Amyloid Prefibrillar Oligomers: The Surprising Commonalities in Their Structure and Activity. Int. J. Mol. Sci. 2021, 22, 6435. [Google Scholar] [CrossRef] [PubMed]
- Raskin, J.; Cummings, J.; Hardy, J.; Schuh, K.; Dean, R.A. Neurobiology of Alzheimer’s Disease: Integrated Molecular, Physiological, Anatomical, Biomarker, and Cognitive Dimensions. Curr. Alzheimer Res. 2015, 12, 712–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial Dysfunction, Oxidative Stress, Neuroinflammation, and Metabolic Alterations in the Progression of Alzheimer’s Disease: A Meta-Analysis of in Vivo Magnetic Resonance Spectroscopy Studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Armstrong, R.A. Risk Factors for Alzheimer’s Disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Setu, J.R.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Role of PSEN Mutations in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2020, 38, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2021, 12, 825816. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why Are Astrocytes Important? Neurochem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Murat, C.D.B.; García-Cáceres, C. Astrocyte Gliotransmission in the Regulation of Systemic Metabolism. Metabolites 2021, 11, 732. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte Function from Information Processing to Cognition and Cognitive Impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, M.; Magistretti, P.J. The Role of Astroglia in Neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [CrossRef]
- Liu, B.; Teschemacher, A.G.; Kasparov, S. Neuroprotective Potential of Astroglia. J. Neurosci. Res. 2017, 95, 2126–2139. [Google Scholar] [CrossRef] [Green Version]
- Valori, C.F.; Guidotti, G.; Brambilla, L.; Rossi, D. Astrocytes: Emerging Therapeutic Targets in Neurological Disorders. Trends Mol. Med. 2019, 25, 750–759. [Google Scholar] [CrossRef] [Green Version]
- McConnell, H.L.; Mishra, A. Cells of the Blood-Brain Barrier: An Overview of the Neurovascular Unit in Health and Disease. Methods Mol. Biol. 2022, 2492, 3–24. [Google Scholar] [CrossRef]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the Functions and Relationships of the Glymphatic System and Meningeal Lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s Disease and Other Age-Associated Dementias: A Supporting Player with a Central Role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer's disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, H.; Lee, C.J. Reactive Astrocytes in Alzheimer’s Disease: A Double-Edged Sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.R.; Li, Y.-M. The Role of Astrocytes in Amyloid Production and Alzheimer’s Disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.M.; Pottoo, F.H.; Dahiya, E.S.; Khan, F.A.; Kumar, J.B.S. Neuron-Glia Interactions: Molecular Basis of Alzheimer’s Disease and Applications of Neuroproteomics. Eur. J. Neurosci. 2020, 52, 2931–2943. [Google Scholar] [CrossRef]
- Nanclares, C.; Baraibar, A.M.; Araque, A.; Kofuji, P. Dysregulation of Astrocyte-Neuronal Communication in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7887. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte Dysfunction in Alzheimer Disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult Mouse Astrocytes Degrade Amyloid-Beta in Vitro and in Situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Pihlaja, R.; Koistinaho, J.; Malm, T.; Sikkilä, H.; Vainio, S.; Koistinaho, M. Transplanted Astrocytes Internalize Deposited Beta-Amyloid Peptides in a Transgenic Mouse Model of Alzheimer’s Disease. Glia 2008, 56, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, D. Astrocytic and Microglial Cells as the Modulators of Neuroinflammation in Alzheimer’s Disease. J. Neuroinflamm. 2022, 19, 206. [Google Scholar] [CrossRef] [PubMed]
- Saroja, S.R.; Gorbachev, K.; Julia, T.; Goate, A.M.; Pereira, A.C. Astrocyte-Secreted Glypican-4 Drives APOE4-Dependent Tau Hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2022, 119, e2108870119. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Uddin, M.S.; Lim, L.W. Glial Cells in Alzheimer’s Disease: From Neuropathological Changes to Therapeutic Implications. Ageing Res. Rev. 2022, 78, 101622. [Google Scholar] [CrossRef]
- Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600. [Google Scholar] [CrossRef]
- Pekny, M.; Nilsson, M. Astrocyte Activation and Reactive Gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Matthews, P.M.; Donat, C.K.; Livingston, N.R.; Femminella, G.D.; Guedes, S.S.; Myers, J.; Fan, Z.; Tyacke, R.J.; Venkataraman, A.V.; et al. Astrocyte Reactivity with Late-Onset Cognitive Impairment Assessed in Vivo Using 11C-BU99008 PET and Its Relationship with Amyloid Load. Mol. Psychiatry 2021, 26, 5848–5855. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Amarilla, P.; Arredondo, F.; Dapueto, R.; Boix, V.; Carvalho, D.; Santi, M.D.; Vasilskis, E.; Mesquita-Ribeiro, R.; Dajas-Bailador, F.; Abin-Carriquiry, J.A.; et al. Isolation and Characterization of Neurotoxic Astrocytes Derived from Adult Triple Transgenic Alzheimer’s Disease Mice. Neurochem. Int. 2022, 159, 105403. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Gsell, W.; Wahis, J.; Luckett, E.S.; Jamoulle, T.; Vermaercke, B.; Preman, P.; Moechars, D.; Hendrickx, V.; Jaspers, T.; et al. Astrocyte Calcium Dysfunction Causes Early Network Hyperactivity in Alzheimer’s Disease. Cell Rep. 2022, 40, 111280. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.-T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte Biomarker Signatures of Amyloid-β and Tau Pathologies in Alzheimer’s Disease. Mol. Psychiatry 2022. [Google Scholar] [CrossRef]
- Pillai, A.G.; Nadkarni, S. Amyloid Pathology Disrupts Gliotransmitter Release in Astrocytes. PLoS Comput. Biol. 2022, 18, e1010334. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient Astrocyte Metabolism Impairs Glutamine Synthesis and Neurotransmitter Homeostasis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef]
- Ong, W.-Y.; Tanaka, K.; Dawe, G.S.; Ittner, L.M.; Farooqui, A.A. Slow Excitotoxicity in Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 35, 643–668. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karran, E.; De Strooper, B. The Amyloid Hypothesis in Alzheimer Disease: New Insights from New Therapeutics. Nat. Rev. Drug. Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.-F.; Turk, J.; et al. Matrix Metalloproteinases Expressed by Astrocytes Mediate Extracellular Amyloid-Beta Peptide Catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoliu-Gaya, L.; Mulder, S.D.; Veerhuis, R.; Villegas, S. Effects of an Aβ-Antibody Fragment on Aβ Aggregation and Astrocytic Uptake Are Modulated by Apolipoprotein E and J Mimetic Peptides. PLoS ONE 2017, 12, e0188191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal Glial Activation Coincides with Increased BACE1 Activation and Precedes Amyloid Plaque Deposition in APP[V717I] Transgenic Mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; O’Connor, T.; Vassar, R. The Contribution of Activated Astrocytes to Aβ Production: Implications for Alzheimer’s Disease Pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol. Dis. 2014, 72, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Liao, F.; Yoon, H.; Kim, J. Apolipoprotein E Metabolism and Functions in Brain and Its Role in Alzheimer’s Disease. Curr. Opin. Lipidol. 2017, 28, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Lanfranco, M.F.; Sepulveda, J.; Kopetsky, G.; Rebeck, G.W. Expression and Secretion of ApoE Isoforms in Astrocytes and Microglia during Inflammation. Glia 2021, 69, 1478–1493. [Google Scholar] [CrossRef]
- Arnaud, L.; Benech, P.; Greetham, L.; Stephan, D.; Jimenez, A.; Jullien, N.; García-González, L.; Tsvetkov, P.O.; Devred, F.; Sancho-Martinez, I.; et al. APOE4 Drives Inflammation in Human Astrocytes via TAGLN3 Repression and NF-ΚB Activation. Cell Rep. 2022, 40, 111200. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L.; et al. Peripheral ApoE4 Enhances Alzheimer’s Pathology and Impairs Cognition by Compromising Cerebrovascular Function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Ba, M.; Kong, M.; Li, X.; Ng, K.P.; Rosa-Neto, P.; Gauthier, S. Is ApoE ε 4 a Good Biomarker for Amyloid Pathology in Late Onset Alzheimer’s Disease? Transl. Neurodegener. 2016, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisniewski, T.; Drummond, E. APOE-Amyloid Interaction: Therapeutic Targets. Neurobiol. Dis. 2020, 138, 104784. [Google Scholar] [CrossRef]
- Carter, D.B. The Interaction of Amyloid-Beta with ApoE. Subcell Biochem. 2005, 38, 255–272. [Google Scholar] [CrossRef]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective Removal of Astrocytic APOE4 Strongly Protects against Tau-Mediated Neurodegeneration and Decreases Synaptic Phagocytosis by Microglia. Neuron 2021, 109, 1657–1674.e7. [Google Scholar] [CrossRef]
- Wang, P.; Ye, Y. Astrocytes in Neurodegenerative Diseases: A Perspective from Tauopathy and α-Synucleinopathy. Life 2021, 11, 938. [Google Scholar] [CrossRef]
- Lebouvier, T.; Pasquier, F.; Buée, L. Update on Tauopathies. Curr. Opin. Neurol. 2017, 30, 589–598. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43–50. [Google Scholar] [CrossRef]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and Neuronal Tau Pathology in Tauopathies: Characterization of Disease-Specific Phenotypes and Tau Pathology Progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Holtzman, D.M. Glial Contributions to Neurodegeneration in Tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef] [Green Version]
- Amro, Z.; Yool, A.J.; Collins-Praino, L.E. The Potential Role of Glial Cells in Driving the Prion-like Transcellular Propagation of Tau in Tauopathies. Brain Behav. Immun. Health 2021, 14, 100242. [Google Scholar] [CrossRef]
- Smith, A.M.; Davey, K.; Tsartsalis, S.; Khozoie, C.; Fancy, N.; Tang, S.S.; Liaptsi, E.; Weinert, M.; McGarry, A.; Muirhead, R.C.J.; et al. Diverse Human Astrocyte and Microglial Transcriptional Responses to Alzheimer’s Pathology. Acta Neuropathol. 2022, 143, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Trinkaus, V.A.; Riera-Tur, I.; Imamoglu, R.; Schaller, T.F.; Wang, H.; Dudanova, I.; Hipp, M.S.; Bracher, A.; Hartl, F.U. The Extracellular Chaperone Clusterin Enhances Tau Aggregate Seeding in a Cellular Model. Nat. Commun. 2021, 12, 4863. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at CLU and CR1 Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.J.; Raissi, A.; Rajkovich, K.; Berto, S.; Kumar, J.; Molinaro, G.; Raduazzo, J.; Guo, Y.; Loerwald, K.; Konopka, G.; et al. MEF2C Regulates Cortical Inhibitory and Excitatory Synapses and Behaviors Relevant to Neurodevelopmental Disorders. eLife 2016, 5, e20059. [Google Scholar] [CrossRef]
- Adrião, A.; Santana, I.; Ribeiro, C.; Cancela, M.L.; Conceição, N.; Grazina, M. Identification of a Novel Mutation in MEF2C Gene in an Atypical Patient with Frontotemporal Lobar Degeneration. Neurol. Sci. 2022, 43, 319–326. [Google Scholar] [CrossRef]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.-P.; Younkin, S.G.; et al. Genome-Wide Association Meta-Analysis of Neuropathologic Features of Alzheimer’s Disease and Related Dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Wang, X.; Lopez, O.L.; Sweet, R.A.; Becker, J.T.; DeKosky, S.T.; Barmada, M.M.; Demirci, F.Y.; Kamboh, M.I. Genetic Determinants of Disease Progression in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 43, 649–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Devadoss, D.; Nair, M.; Chand, H.S.; Lakshmana, M.K. Novel Alzheimer Risk Factor IQ Motif Containing Protein K Is Abundantly Expressed in the Brain and Is Markedly Increased in Patients with Alzheimer’s Disease. Front. Cell Neurosci. 2022, 16, 954071. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.S.; De Muynck, L. Differentially Expressed Genes in Alzheimer’s Disease Highlighting the Roles of Microglia Genes Including OLR1 and Astrocyte Gene CDK2AP1. Brain Behav. Immun. Health 2021, 13, 100227. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ye, Y. Filamentous Recombinant Human Tau Activates Primary Astrocytes via an Integrin Receptor Complex. Nat. Commun. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic System, AQP4, and Their Implications in Alzheimer’s Disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Bégard, S.; Pythoud, C.; Rey, M.; et al. Tau Accumulation in Astrocytes of the Dentate Gyrus Induces Neuronal Dysfunction and Memory Deficits in Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced Gliotransmitter Release from Astrocytes Mediates Tau-Induced Synaptic Dysfunction in Cultured Hippocampal Neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef]
- Rostami, J.; Holmqvist, S.; Lindström, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergström, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [Green Version]
- Zaheer, S.; Thangavel, R.; Sahu, S.K.; Zaheer, A. Augmented Expression of Glia Maturation Factor in Alzheimer’s Disease. Neuroscience 2011, 194, 227–233. [Google Scholar] [CrossRef]
- Gimsa, U.; Mitchison, N.A.; Brunner-Weinzierl, M.C. Immune Privilege as an Intrinsic CNS Property: Astrocytes Protect the CNS against T-Cell-Mediated Neuroinflammation. Mediat. Inflamm. 2013, 2013, 320519. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, F.; Sgarlata, C.; Francis, M.; Maurizi, N.; Faragli, A.; Perna, S.; Rondanelli, M.; Rollone, M.; Ricevuti, G. Neuroinflammation, Immune System and Alzheimer Disease: Searching for the Missing Link. Aging Clin. Exp. Res. 2017, 29, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Rubiano, M.G. Astrocyte´s RAGE: More Than Just a Question of Mood. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, S.; Holsinger, R.M.D. Cyclical Amyloid Beta-Astrocyte Activity Induces Oxidative Stress in Alzheimer’s Disease. Biochimie 2020, 171–172, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, Y.; Feng, J. Cross Talk between Activation of Microglia and Astrocytes in Pathological Conditions in the Central Nervous System. Life Sci. 2011, 89, 141–146. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Park, J.; Griciuc, A.; Kim, E.; Choi, S.H.; Iwamoto, Y.; Kiss, M.G.; Christie, K.A.; Vinegoni, C.; Poller, W.C.; et al. Astrocytic Interleukin-3 Programs Microglia and Limits Alzheimer’s Disease. Nature 2021, 595, 701–706. [Google Scholar] [CrossRef]
- Britschgi, M.; Rufibach, K.; Huang, S.L.B.; Clark, C.M.; Kaye, J.A.; Li, G.; Peskind, E.R.; Quinn, J.F.; Galasko, D.R.; Wyss-Coray, T. Modeling of Pathological Traits in Alzheimer’s Disease Based on Systemic Extracellular Signaling Proteome. Mol. Cell Proteom. 2011, 10, M111.008862. [Google Scholar] [CrossRef] [Green Version]
- Kiddle, S.J.; Thambisetty, M.; Simmons, A.; Riddoch-Contreras, J.; Hye, A.; Westman, E.; Pike, I.; Ward, M.; Johnston, C.; Lupton, M.K.; et al. Plasma Based Markers of [11C] PiB-PET Brain Amyloid Burden. PLoS ONE 2012, 7, e44260. [Google Scholar] [CrossRef]
- Soares, H.D.; Potter, W.Z.; Pickering, E.; Kuhn, M.; Immermann, F.W.; Shera, D.M.; Ferm, M.; Dean, R.A.; Simon, A.J.; Swenson, F.; et al. Plasma Biomarkers Associated with the Apolipoprotein E Genotype and Alzheimer Disease. Arch. Neurol. 2012, 69, 1310–1317. [Google Scholar] [CrossRef] [Green Version]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Sama, D.M.; Norris, C.M. Calcium Dysregulation and Neuroinflammation: Discrete and Integrated Mechanisms for Age-Related Synaptic Dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative Stress in Alzheimer Disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kropf, E.; Fahnestock, M. Effects of Reactive Oxygen and Nitrogen Species on TrkA Expression and Signalling: Implications for ProNGF in Aging and Alzheimer’s Disease. Cells 2021, 10, 1983. [Google Scholar] [CrossRef]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the Antioxidant Glutathione in Neurons: Supply by Astrocytes of CysGly as Precursor for Neuronal Glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Cynader, M.S. Astrocytes Provide Cysteine to Neurons by Releasing Glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.-G.; Ransom, B.R.; Chen, X.-C.; Ye, Z.-C. Dual Pathways Mediate β-Amyloid Stimulated Glutathione Release from Astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef]
- Garg, S.K.; Vitvitsky, V.; Albin, R.; Banerjee, R. Astrocytic Redox Remodeling by Amyloid Beta Peptide. Antioxid. Redox Signal. 2011, 14, 2385–2397. [Google Scholar] [CrossRef] [Green Version]
- Zoufal, V.; Mairinger, S.; Krohn, M.; Wanek, T.; Filip, T.; Sauberer, M.; Stanek, J.; Kuntner, C.; Pahnke, J.; Langer, O. Measurement of Cerebral ABCC1 Transport Activity in Wild-Type and APP/PS1-21 Mice with Positron Emission Tomography. J. Cereb. Blood Flow Metab. 2020, 40, 954–965. [Google Scholar] [CrossRef]
- Allaman, I.; Gavillet, M.; Bélanger, M.; Laroche, T.; Viertl, D.; Lashuel, H.A.; Magistretti, P.J. Amyloid-Beta Aggregates Cause Alterations of Astrocytic Metabolic Phenotype: Impact on Neuronal Viability. J. Neurosci. 2010, 30, 3326–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akama, K.T.; Van Eldik, L.J. Beta-Amyloid Stimulation of Inducible Nitric-Oxide Synthase in Astrocytes Is Interleukin-1beta- and Tumor Necrosis Factor-Alpha (TNFalpha)-Dependent, and Involves a TNFalpha Receptor-Associated Factor- and NFkappaB-Inducing Kinase-Dependent Signaling Mechanism. J. Biol. Chem. 2000, 275, 7918–7924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallérac, G.; Rouach, N. Astrocytes as New Targets to Improve Cognitive Functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Huffels, C.F.M.; Middeldorp, J.; Hol, E.M. Aß Pathology and Neuron-Glia Interactions: A Synaptocentric View. Neurochem. Res. 2022; ahead of print. [Google Scholar] [CrossRef]
- Nava-Mesa, M.O.; Jiménez-Díaz, L.; Yajeya, J.; Navarro-Lopez, J.D. GABAergic Neurotransmission and New Strategies of Neuromodulation to Compensate Synaptic Dysfunction in Early Stages of Alzheimer’s Disease. Front. Cell Neurosci. 2014, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Garaschuk, O.; Verkhratsky, A. GABAergic Astrocytes in Alzheimer’s Disease. Aging 2019, 11, 1602–1604. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from Reactive Astrocytes Impairs Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; McGeer, P.L. Astrocytes Are GABAergic Cells That Modulate Microglial Activity. Glia 2011, 59, 152–165. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A.; Verkhratsky, A. Astrocyte Energy and Neurotransmitter Metabolism in Alzheimer’s Disease: Integration of the Glutamate/GABA-Glutamine Cycle. Prog. Neurobiol. 2022, 217, 102331. [Google Scholar] [CrossRef]
- Alfaro-Ruiz, R.; Martín-Belmonte, A.; Aguado, C.; Hernández, F.; Moreno-Martínez, A.E.; Ávila, J.; Luján, R. The Expression and Localisation of G-Protein-Coupled Inwardly Rectifying Potassium (GIRK) Channels Is Differentially Altered in the Hippocampus of Two Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11106. [Google Scholar] [CrossRef]
- Jeremic, D.; Sanchez-Rodriguez, I.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Therapeutic Potential of Targeting G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channels in the Central Nervous System. Pharmacol. Ther. 2021, 223, 107808. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodríguez, I.; Djebari, S.; Temprano-Carazo, S.; Vega-Avelaira, D.; Jiménez-Herrera, R.; Iborra-Lázaro, G.; Yajeya, J.; Jiménez-Díaz, L.; Navarro-López, J.D. Hippocampal Long-Term Synaptic Depression and Memory Deficits Induced in Early Amyloidopathy Are Prevented by Enhancing G-Protein-Gated Inwardly Rectifying Potassium Channel Activity. J. Neurochem. 2020, 153, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djebari, S.; Iborra-Lázaro, G.; Temprano-Carazo, S.; Sánchez-Rodríguez, I.; Nava-Mesa, M.O.; Múnera, A.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. G-Protein-Gated Inwardly Rectifying Potassium (Kir3/GIRK) Channels Govern Synaptic Plasticity That Supports Hippocampal-Dependent Cognitive Functions in Male Mice. J. Neurosci. 2021, 41, 7086–7102. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.; Binder, D.K. Astrocytes and Epilepsy; Academic Press: Cambridge, MA, USA, 2016; ISBN 0-12-802624-3. [Google Scholar]
- Danysz, W.; Parsons, C.G. The NMDA Receptor Antagonist Memantine as a Symptomatological and Neuroprotective Treatment for Alzheimer’s Disease: Preclinical Evidence. Int. J. Geriatr. Psychiatry 2003, 18, S23–S32. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of Glutamate Transporters Reveals a Major Role for Astroglial Transport in Excitotoxicity and Clearance of Glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The Role of Astrocytic Glutamate Transporters GLT-1 and GLAST in Neurological Disorders: Potential Targets for Neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Takahashi, K.; Kong, Q.; Lin, Y.; Stouffer, N.; Schulte, D.A.; Lai, L.; Liu, Q.; Chang, L.-C.; Dominguez, S.; Xing, X.; et al. Restored Glial Glutamate Transporter EAAT2 Function as a Potential Therapeutic Approach for Alzheimer’s Disease. J. Exp. Med. 2015, 212, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Terao, I.; Honyashiki, M.; Inoue, T. Comparative efficacy of lithium and aducanumab for cognitive decline in patients with mild cognitive impairment or Alzheimer’s disease: A systematic review and network meta-analysis. Ageing Res. Rev. 2022, 81, 101709. [Google Scholar] [CrossRef]
- Prillaman, M. Alzheimer’s Drug Slows Mental Decline in Trial—But Is It a Breakthrough? Nature 2022, 610, 15–16. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.-W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353.e5. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, K.; Ye, J.; Liu, Z.; Ren, Y.; He, W.; Xu, J.; He, Y.; Yuan, Y. Complement C3 Overexpression Activates JAK2/STAT3 Pathway and Correlates with Gastric Cancer Progression. J. Exp. Clin. Cancer Res. 2020, 39, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, M.; Kohro, Y.; Yano, T.; Tsujikawa, T.; Kitano, J.; Tozaki-Saitoh, H.; Koyanagi, S.; Ohdo, S.; Ji, R.-R.; Salter, M.W.; et al. JAK-STAT3 Pathway Regulates Spinal Astrocyte Proliferation and Neuropathic Pain Maintenance in Rats. Brain 2011, 134, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Toral-Rios, D.; Patiño-López, G.; Gómez-Lira, G.; Gutiérrez, R.; Becerril-Pérez, F.; Rosales-Córdova, A.; León-Contreras, J.C.; Hernández-Pando, R.; León-Rivera, I.; Soto-Cruz, I.; et al. Activation of STAT3 Regulates Reactive Astrogliosis and Neuronal Death Induced by AβO Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 7458. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Wang, Z.; Lei, M.; Che, J.; Zhang, S.; Zhang, T.; Hu, Y.; Shi, L.; Cui, L.; Liu, J.; et al. Daphnetin Ameliorates Aβ Pathogenesis via STAT3/GFAP Signaling in an APP/PS1 Double-Transgenic Mouse Model of Alzheimer’s Disease. Pharmacol. Res. 2022, 180, 106227. [Google Scholar] [CrossRef]
- Ito, K.; Noguchi, A.; Uosaki, Y.; Taga, T.; Arakawa, H.; Takizawa, T. Gfap and Osmr Regulation by BRG1 and STAT3 via Interchromosomal Gene Clustering in Astrocytes. Mol. Biol. Cell 2018, 29, 209–219. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-Mediated Astrogliosis Ameliorates Pathology in an Alzheimer’s Disease Model. EMBO Mol. Med. 2019, 11, e9665. [Google Scholar] [CrossRef]
- Babaei, P.; Eyvani, K.; Kouhestani, S. Sex-Independent Cognition Improvement in Response to Kaempferol in the Model of Sporadic Alzheimer’s Disease. Neurochem. Res. 2021, 46, 1480–1486. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Garcia-Martinez, V.; Gutierrez-Merino, C. Kaempferol Prevents the Activation of Complement C3 Protein and the Generation of Reactive A1 Astrocytes That Mediate Rat Brain Degeneration Induced by 3-Nitropropionic Acid. Food Chem. Toxicol. 2022, 164, 113017. [Google Scholar] [CrossRef]
- Yu, L.; Chen, C.; Wang, L.-F.; Kuang, X.; Liu, K.; Zhang, H.; Du, J.-R. Neuroprotective Effect of Kaempferol Glycosides against Brain Injury and Neuroinflammation by Inhibiting the Activation of NF-ΚB and STAT3 in Transient Focal Stroke. PLoS ONE 2013, 8, e55839. [Google Scholar] [CrossRef] [PubMed]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.-A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of Astrocyte Reactivity Improves Functional Deficits in Mouse Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Roy, A.; Prorok, T.; Patel, D.; Dasarathi, S.; Pahan, K. Aspirin Up-Regulates Suppressor of Cytokine Signaling 3 in Glial Cells via PPARα. J. Neurochem. 2019, 151, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes Are Important Mediators of Aβ-Induced Neurotoxicity and Tau Phosphorylation in Primary Culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [Green Version]
- Garwood, C.J.; Cooper, J.D.; Hanger, D.P.; Noble, W. Anti-Inflammatory Impact of Minocycline in a Mouse Model of Tauopathy. Front. Psychiatry 2010, 1, 136. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.-Q.; Gong, Z.-Y.; Lu, C.-D.; Wang, P. Prostaglandin I2 Is Responsible for Ameliorating Prostaglandin E2 Stress in Stimulating the Expression of Tumor Necrosis Factor α in a β-Amyloid Protein -Dependent Mechanism. Oncotarget 2017, 8, 102801–102819. [Google Scholar] [CrossRef] [Green Version]
- Mohri, I.; Kadoyama, K.; Kanekiyo, T.; Sato, Y.; Kagitani-Shimono, K.; Saito, Y.; Suzuki, K.; Kudo, T.; Takeda, M.; Urade, Y.; et al. Hematopoietic Prostaglandin D Synthase and DP1 Receptor Are Selectively Upregulated in Microglia and Astrocytes within Senile Plaques from Human Patients and in a Mouse Model of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 469–480. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Hand, T.; Andreasson, K. Prostaglandin D2 Mediates Neuronal Protection via the DP1 Receptor. J. Neurochem. 2005, 92, 477–486. [Google Scholar] [CrossRef]
- Mohan, S.; Ahmad, A.S.; Glushakov, A.V.; Chambers, C.; Doré, S. Putative Role of Prostaglandin Receptor in Intracerebral Hemorrhage. Front. Neurol. 2012, 3, 145. [Google Scholar] [CrossRef]
- Biringer, R.G. The Role of Eicosanoids in Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.S.; Ahmad, M.; Maruyama, T.; Narumiya, S.; Doré, S. Prostaglandin D2 DP1 Receptor Is Beneficial in Ischemic Stroke and in Acute Exicitotoxicity in Young and Old Mice. Age 2010, 32, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bate, C.; Kempster, S.; Williams, A. Prostaglandin D2 Mediates Neuronal Damage by Amyloid-Beta or Prions Which Activates Microglial Cells. Neuropharmacology 2006, 50, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An Epoxide Hydrolase Inhibitor Reduces Neuroinflammation in a Mouse Model of Alzheimer’s Disease. Sci. Transl. Med. 2020, 12, eabb1206. [Google Scholar] [CrossRef]
- Wu, Q.; Lin, M.; Wu, P.; Zhao, C.; Yang, S.; Yu, H.; Xian, W.; Song, J. TPPU Downregulates Oxidative Stress Damage and Induces BDNF Expression in PC-12 Cells. Comput. Math. Methods Med. 2022, 2022, 7083022. [Google Scholar] [CrossRef]
- Chen, W.; Wang, M.; Zhu, M.; Xiong, W.; Qin, X.; Zhu, X. 14,15-Epoxyeicosatrienoic Acid Alleviates Pathology in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2020, 40, 8188–8203. [Google Scholar] [CrossRef]
- Shi, Z.-M.; Han, Y.-W.; Han, X.-H.; Zhang, K.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Ting, C.; Zhen, Z.; Hong, W. Upstream Regulators and Downstream Effectors of NF-ΚB in Alzheimer’s Disease. J. Neurol. Sci. 2016, 366, 127–134. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Xu, Q.-Q.; Xian, Y.-F.; Lin, Z.-X. Sulforaphene Ameliorates Neuroinflammation and Hyperphosphorylated Tau Protein via Regulating the PI3K/Akt/GSK-3β Pathway in Experimental Models of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2020, 2020, 4754195. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-ΚB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef]
- Danilov, C.A.; Chandrasekaran, K.; Racz, J.; Soane, L.; Zielke, C.; Fiskum, G. Sulforaphane Protects Astrocytes against Oxidative Stress and Delayed Death Caused by Oxygen and Glucose Deprivation. Glia 2009, 57, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic Modification of Nrf2 by Sulforaphane Increases the Antioxidative and Anti-Inflammatory Capacity in a Cellular Model of Alzheimer’s Disease. Eur. J. Pharmacol. 2018, 824, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2929. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear Factor E2-Related Factor 2-Dependent Antioxidant Response Element Activation by Tert-Butylhydroquinone and Sulforaphane Occurring Preferentially in Astrocytes Conditions Neurons against Oxidative Insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 Activation in Astrocytes Protects against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-Z.; Zheng, X.-M.; Zhou, Y.-F.; Yun, L.-Y.; Luo, D.-M.; Hao, J.-J.; Liu, P.-F.; Zhang, W.-K.; Xu, J.-K.; Yan, Y.; et al. Cornuside Is a Potential Agent against Alzheimer’s Disease via Orchestration of Reactive Astrocytes. Nutrients 2022, 14, 3179. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Avila-Rodriguez, M.; Vega-Vela, N.E.; Echeverria, V.; González, J.; Hidalgo, O.A.; Santos, A.B.; Aliev, G.; Barreto, G.E. Growth Factors and Astrocytes Metabolism: Possible Roles for Platelet Derived Growth Factor. Med. Chem. 2016, 12, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Sycheva, M.; Sustarich, J.; Zhang, Y.; Selvaraju, V.; Geetha, T.; Gearing, M.; Babu, J.R. Pro-Nerve Growth Factor Induces Activation of RhoA Kinase and Neuronal Cell Death. Brain Sci. 2019, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Selles, M.C.; Fortuna, J.T.S.; Zappa-Villar, M.F.; de Faria, Y.P.R.; Souza, A.S.; Suemoto, C.K.; Leite, R.E.P.; Rodriguez, R.D.; Grinberg, L.T.; Reggiani, P.C.; et al. Adenovirus-Mediated Transduction of Insulin-Like Growth Factor 1 Protects Hippocampal Neurons from the Toxicity of Aβ Oligomers and Prevents Memory Loss in an Alzheimer Mouse Model. Mol. Neurobiol. 2020, 57, 1473–1483. [Google Scholar] [CrossRef]
- Albus, E.; Sinningen, K.; Winzer, M.; Thiele, S.; Baschant, U.; Hannemann, A.; Fantana, J.; Tausche, A.-K.; Wallaschofski, H.; Nauck, M.; et al. Milk Fat Globule-Epidermal Growth Factor 8 (MFG-E8) Is a Novel Anti-Inflammatory Factor in Rheumatoid Arthritis in Mice and Humans. J. Bone Miner. Res. 2016, 31, 596–605. [Google Scholar] [CrossRef]
- Kranich, J.; Krautler, N.J.; Falsig, J.; Ballmer, B.; Li, S.; Hutter, G.; Schwarz, P.; Moos, R.; Julius, C.; Miele, G.; et al. Engulfment of Cerebral Apoptotic Bodies Controls the Course of Prion Disease in a Mouse Strain-Dependent Manner. J. Exp. Med. 2010, 207, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, A.; Zhu, Y.; He, W.; Di, W.; Fang, Y.; Shi, X. MFG-E8 Reverses Microglial-Induced Neurotoxic Astrocyte (A1) via NF-ΚB and PI3K-Akt Pathways. J. Cell Physiol. 2018, 234, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, K.; Takano, K.; Moriyama, M.; Nakamura, Y. Microglia Endocytose Amyloid β Through the Binding of Transglutaminase 2 and Milk Fat Globule EGF Factor 8 Protein. Neurochem. Res. 2018, 43, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B. Strategies for Reducing or Preventing the Generation of Oxidative Stress. Oxid. Med. Cell Longev. 2011, 2011, 194586. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, M.A.; Beydoun, H.A.; Fanelli-Kuczmarski, M.T.; Weiss, J.; Hossain, S.; Canas, J.A.; Evans, M.K.; Zonderman, A.B. Association of Serum Antioxidant Vitamins and Carotenoids with Incident Alzheimer Disease and All-Cause Dementia Among US Adults. Neurology 2022, 98, e2150–e2162. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Geir Møller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.-S. Molecular Mechanisms behind Free Radical Scavengers Function against Oxidative Stress. Antioxidants 2017, 6, 51. [Google Scholar] [CrossRef]
- Dajas, F.; Andrés, A.-C.J.; Florencia, A.; Carolina, E.; Felicia, R.-M. Neuroprotective Actions of Flavones and Flavonols: Mechanisms and Relationship to Flavonoid Structural Features. Cent. Nerv. Syst. Agents Med. Chem. 2013, 13, 30–35. [Google Scholar] [CrossRef]
- Nakajima, A.; Ohizumi, Y. Potential Benefits of Nobiletin, A Citrus Flavonoid, against Alzheimer’s Disease and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 3380. [Google Scholar] [CrossRef] [Green Version]
- Yin, N.; Yao, X.; Zhou, Q.; Faiola, F.; Jiang, G. Vitamin E Attenuates Silver Nanoparticle-Induced Effects on Body Weight and Neurotoxicity in Rats. Biochem. Biophys. Res. Commun. 2015, 458, 405–410. [Google Scholar] [CrossRef]
- Behl, C. Vitamin E and Other Antioxidants in Neuroprotection. Int. J. Vitam. Nutr. Res. 1999, 69, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Abedi, Z.; Khaza’ai, H.; Vidyadaran, S.; Mutalib, M.S.A. The Modulation of NMDA and AMPA/Kainate Receptors by Tocotrienol-Rich Fraction and A-Tocopherol in Glutamate-Induced Injury of Primary Astrocytes. Biomedicines 2017, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of Vitamin E and Memantine on Functional Decline in Alzheimer Disease: The TEAM-AD VA Cooperative Randomized Trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef]
- Farina, N.; Llewellyn, D.; Isaac, M.G.E.K.N.; Tabet, N. Vitamin E for Alzheimer’s Dementia and Mild Cognitive Impairment. Cochrane Database Syst. Rev. 2017, 1, CD002854. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative Stress in Alzheimer’s Disease: Why Did Antioxidant Therapy Fail? Oxid. Med. Cell Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.-J.; Kim, H.; Kim, H.-S.; Chang, M.-J. Phloroglucinol Attenuates Oligomeric Amyloid Beta Peptide1-42-Induced Astrocytic Activation by Reducing Oxidative Stress. J. Pharmacol. Sci. 2021, 145, 308–312. [Google Scholar] [CrossRef]
- Wang, D.; Gao, F.; Hu, F.; Wu, J. Nobiletin Alleviates Astrocyte Activation and Oxidative Stress Induced by Hypoxia In Vitro. Molecules 2022, 27, 1962. [Google Scholar] [CrossRef]
- Quincozes-Santos, A.; Bobermin, L.D.; Latini, A.; Wajner, M.; Souza, D.O.; Gonçalves, C.-A.; Gottfried, C. Resveratrol Protects C6 Astrocyte Cell Line against Hydrogen Peroxide-Induced Oxidative Stress through Heme Oxygenase 1. PLoS ONE 2013, 8, e64372. [Google Scholar] [CrossRef]
- Yu, H.; Yamashita, T.; Hu, X.; Bian, Z.; Hu, X.; Feng, T.; Tadokoro, K.; Morihara, R.; Abe, K. Protective and anti-oxidative effects of curcumin and resveratrol on Aβ-oligomer-induced damage in the SH-SY5Y cell line. J. Neurol. Sci. 2022, 441, 120356. [Google Scholar] [CrossRef] [PubMed]
- Daverey, A.; Agrawal, S.K. Curcumin Alleviates Oxidative Stress and Mitochondrial Dysfunction in Astrocytes. Neuroscience 2016, 333, 92–103. [Google Scholar] [CrossRef] [PubMed]
- López, S.; Martá, M.; Sequeda, L.G.; Celis, C.; Sutachan, J.J.; Albarracín, S.L. Cytoprotective Action against Oxidative Stress in Astrocytes and Neurons by Bactris Guineensis (L.) H.E. Moore (Corozo) Fruit Extracts. Food Chem. Toxicol. 2017, 109, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Prah, J.; Winters, A.; Chaudhari, K.; Hersh, J.; Liu, R.; Yang, S.-H. Cholesterol Sulfate Alters Astrocyte Metabolism and Provides Protection against Oxidative Stress. Brain Res. 2019, 1723, 146378. [Google Scholar] [CrossRef]
- Lu, C.-Y.; Day, C.H.; Kuo, C.-H.; Wang, T.-F.; Ho, T.-J.; Lai, P.-F.; Chen, R.-J.; Yao, C.-H.; Viswanadha, V.P.; Kuo, W.-W.; et al. Calycosin Alleviates H2O2-Induced Astrocyte Injury by Restricting Oxidative Stress through the Akt/Nrf2/HO-1 Signaling Pathway. Environ. Toxicol. 2022, 37, 858–867. [Google Scholar] [CrossRef]
- Jeřábek, J.; Uliassi, E.; Guidotti, L.; Korábečný, J.; Soukup, O.; Sepsova, V.; Hrabinova, M.; Kuča, K.; Bartolini, M.; Peña-Altamira, L.E.; et al. Tacrine-Resveratrol Fused Hybrids as Multi-Target-Directed Ligands against Alzheimer’s Disease. Eur. J. Med. Chem. 2017, 127, 250–262. [Google Scholar] [CrossRef]
- Sun, J.; Xu, S.; Li, H.; Li, L.; Xu, Z.-Q.D. Galanin Protects Rat Cortical Astrocyte from Oxidative Stress: Involvement of GalR2 and PERK1/2 Signal Pathway. Mediat. Inflamm. 2019, 2019, 2716028. [Google Scholar] [CrossRef]
- Bordet, R.; Gelé, P.; Duriez, P.; Fruchart, J.-C. PPARs: A New Target for Neuroprotection. J. Neurol. Neurosurg. Psychiatry 2006, 77, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Giampietro, L.; Gallorini, M.; De Filippis, B.; Amoroso, R.; Cataldi, A.; di Giacomo, V. PPAR-γ Agonist GL516 Reduces Oxidative Stress and Apoptosis Occurrence in a Rat Astrocyte Cell Line. Neurochem. Int. 2019, 126, 239–245. [Google Scholar] [CrossRef]
- Fracassi, A.; Marcatti, M.; Zolochevska, O.; Tabor, N.; Woltjer, R.; Moreno, S.; Taglialatela, G. Oxidative Damage and Antioxidant Response in Frontal Cortex of Demented and Nondemented Individuals with Alzheimer’s Neuropathology. J. Neurosci. 2021, 41, 538–554. [Google Scholar] [CrossRef]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS Scavengers and ROS Producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, T.; Yamashita, T.; Sasaki, R.; Tadokoro, K.; Matsumoto, N.; Hishikawa, N.; Abe, K. Protective Effects of Edaravone on White Matter Pathology in a Novel Mouse Model of Alzheimer’s Disease with Chronic Cerebral Hypoperfusion. J. Cereb. Blood Flow Metab. 2021, 41, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.-S.; Yao, X.-Q.; Liu, Y.-H.; Wang, Q.-H.; Zeng, F.; Lu, J.-J.; Liu, J.; Zhu, C.; Shen, L.-L.; Liu, C.-H. Edaravone Alleviates Alzheimer’s Disease-Type Pathologies and Cognitive Deficits. Proc. Natl. Acad. Sci. USA 2015, 112, 5225–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Ma, L.; Gong, X.; Xu, C.; Zhang, Y.; Ma, M.; Watanabe, K.; Wen, J. Edaravone Exerts Brain Protective Function by Reducing the Expression of AQP4, APP and Aβ Proteins. Open Life Sci. 2019, 14, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanker, G.; Syversen, T.; Aschner, J.L.; Aschner, M. Modulatory Effect of Glutathione Status and Antioxidants on Methylmercury-Induced Free Radical Formation in Primary Cultures of Cerebral Astrocytes. Brain Res. Mol. Brain Res. 2005, 137, 11–22. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Machado, N.J.; dos Santos-Rodrigues, A.; Cunha, R.A.; Agostinho, P. Astrocytic Adenosine A2A Receptors Control the Amyloid-β Peptide-Induced Decrease of Glutamate Uptake. J. Alzheimer’s Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.-H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at Which Riluzole Treatment Prolongs Survival in Patients with Amyotrophic Lateral Sclerosis: A Retrospective Analysis of Data from a Dose-Ranging Study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-J.; Wang, K.-Y.; Wang, W.-C. Mechanisms Underlying the Riluzole Inhibition of Glutamate Release from Rat Cerebral Cortex Nerve Terminals (Synaptosomes). Neuroscience 2004, 125, 191–201. [Google Scholar] [CrossRef]
- Carbone, M.; Duty, S.; Rattray, M. Riluzole Elevates GLT-1 Activity and Levels in Striatal Astrocytes. Neurochem. Int. 2012, 60, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Lesuis, S.L.; Kaplick, P.M.; Lucassen, P.J.; Krugers, H.J. Treatment with the Glutamate Modulator Riluzole Prevents Early Life Stress-Induced Cognitive Deficits and Impairments in Synaptic Plasticity in APPswe/PS1dE9 Mice. Neuropharmacology 2019, 150, 175–183. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Dykes Hoberg, M.; Vidensky, S.; Chung, D.S.; et al. Beta-Lactam Antibiotics Offer Neuroprotection by Increasing Glutamate Transporter Expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Zumkehr, J.; Rodriguez-Ortiz, C.J.; Cheng, D.; Kieu, Z.; Wai, T.; Hawkins, C.; Kilian, J.; Lim, S.L.; Medeiros, R.; Kitazawa, M. Ceftriaxone Ameliorates Tau Pathology and Cognitive Decline via Restoration of Glial Glutamate Transporter in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chotibut, T.; Davis, R.W.; Arnold, J.C.; Frenchek, Z.; Gurwara, S.; Bondada, V.; Geddes, J.W.; Salvatore, M.F. Ceftriaxone Increases Glutamate Uptake and Reduces Striatal Tyrosine Hydroxylase Loss in 6-OHDA Parkinson’s Model. Mol. Neurobiol. 2014, 49, 1282–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.-C.; Hsu, C.-C.; Pawlak, C.R.; Tikhonova, M.A.; Lai, T.-J.; Amstislavskaya, T.G.; Ho, Y.-J. Effects of Ceftriaxone on the Behavioral and Neuronal Changes in an MPTP-Induced Parkinson’s Disease Rat Model. Behav. Brain Res. 2014, 268, 177–184. [Google Scholar] [CrossRef]
- Bisht, R.; Kaur, B.; Gupta, H.; Prakash, A. Ceftriaxone Mediated Rescue of Nigral Oxidative Damage and Motor Deficits in MPTP Model of Parkinson’s Disease in Rats. Neurotoxicology 2014, 44, 71–79. [Google Scholar] [CrossRef]
- Miller, B.R.; Dorner, J.L.; Shou, M.; Sari, Y.; Barton, S.J.; Sengelaub, D.R.; Kennedy, R.T.; Rebec, G.V. Up-Regulation of GLT1 Expression Increases Glutamate Uptake and Attenuates the Huntington’s Disease Phenotype in the R6/2 Mouse. Neuroscience 2008, 153, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Li, L.; Xian, X.; Liu, L.; Gao, J.; Li, W. Ceftriaxone Regulates Glutamate Production and Vesicular Assembly in Presynaptic Terminals through GLT-1 in APP/PS1 Mice. Neurobiol. Learn. Mem. 2021, 183, 107480. [Google Scholar] [CrossRef]
- Fan, S.; Xian, X.; Li, L.; Yao, X.; Hu, Y.; Zhang, M.; Li, W. Ceftriaxone Improves Cognitive Function and Upregulates GLT-1-Related Glutamate-Glutamine Cycle in APP/PS1 Mice. J. Alzheimer’s Dis. 2018, 66, 1731–1743. [Google Scholar] [CrossRef]
- Dzamba, D.; Honsa, P.; Anderova, M. NMDA Receptors in Glial Cells: Pending Questions. Curr. Neuropharmacol. 2013, 11, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional Synapse in Alzheimer’s Disease—A Focus on NMDA Receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef]
- Lee, M.-C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chang, L.; Song, Y.; Gao, X.; Roselli, F.; Liu, J.; Zhou, W.; Fang, Y.; Ling, W.; Li, H.; et al. Astrocytic GluN2A and GluN2B Oppose the Synaptotoxic Effects of Amyloid-Β1-40 in Hippocampal Cells. J. Alzheimer’s Dis. 2016, 54, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ Induces Astrocytic Glutamate Release, Extrasynaptic NMDA Receptor Activation, and Synaptic Loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, T.; Buisson, A. Reciprocal Disruption of Neuronal Signaling and Aβ Production Mediated by Extrasynaptic NMDA Receptors: A Downward Spiral. Cell Tissue Res. 2014, 356, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Palygin, O.; Lalo, U.; Verkhratsky, A.; Pankratov, Y. Ionotropic NMDA and P2X1/5 Receptors Mediate Synaptically Induced Ca2+ Signalling in Cortical Astrocytes. Cell Calcium 2010, 48, 225–231. [Google Scholar] [CrossRef]
- Ueda, Y.; Doi, T.; Nagatomo, K.; Tokumaru, J.; Takaki, M.; Willmore, L.J. Effect of Levetiracetam on Molecular Regulation of Hippocampal Glutamate and GABA Transporters in Rats with Chronic Seizures Induced by Amygdalar FeCl3 Injection. Brain Res. 2007, 1151, 55–61. [Google Scholar] [CrossRef]
- Sanz-Blasco, S.; Piña-Crespo, J.C.; Zhang, X.; McKercher, S.R.; Lipton, S.A. Levetiracetam Inhibits Oligomeric Aβ-Induced Glutamate Release from Human Astrocytes. Neuroreport 2016, 27, 705–709. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef] [Green Version]
- Cumbo, E.; Ligori, L.D. Levetiracetam, Lamotrigine, and Phenobarbital in Patients with Epileptic Seizures and Alzheimer’s Disease. Epilepsy Behav. 2010, 17, 461–466. [Google Scholar] [CrossRef]
- Kovacic, P.; Somanathan, R. Clinical Physiology and Mechanism of Dizocilpine (MK-801): Electron Transfer, Radicals, Redox Metabolites and Bioactivity. Oxid. Med. Cell Longev. 2010, 3, 13–22. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-Fatah, I.M.; Abdelrazek, H.M.A.; Ibrahim, S.M.; Abdallah, D.M.; El-Abhar, H.S. Dimethyl Fumarate Abridged Tauo-/Amyloidopathy in a D-Galactose/Ovariectomy-Induced Alzheimer’s-like Disease: Modulation of AMPK/SIRT-1, AKT/CREB/BDNF, AKT/GSK-3β, Adiponectin/Adipo1R, and NF-ΚB/IL-1β/ROS Trajectories. Neurochem. Int. 2021, 148, 105082. [Google Scholar] [CrossRef] [PubMed]
- Pao, P.-C.; Tsai, L.-H. Three Decades of Cdk5. J. Biomed. Sci. 2021, 28, 79. [Google Scholar] [CrossRef] [PubMed]
- Shupp, A.; Casimiro, M.C.; Pestell, R.G. Biological Functions of CDK5 and Potential CDK5 Targeted Clinical Treatments. Oncotarget 2017, 8, 17373–17382. [Google Scholar] [CrossRef] [Green Version]
- Posada-Duque, R.A.; Palacio-Castañeda, V.; Cardona-Gómez, G.P. CDK5 Knockdown in Astrocytes Provide Neuroprotection as a Trophic Source via Rac1. Mol. Cell. Neurosci. 2015, 68, 151–166. [Google Scholar] [CrossRef]
- Schaffer, S.; Kim, H.W. Effects and Mechanisms of Taurine as a Therapeutic Agent. Biomol. Ther. 2018, 26, 225–241. [Google Scholar] [CrossRef]
- Suárez, L.M.; Muñoz, M.-D.; Martín Del Río, R.; Solís, J.M. Taurine Content in Different Brain Structures during Ageing: Effect on Hippocampal Synaptic Plasticity. Amino Acids 2016, 48, 1199–1208. [Google Scholar] [CrossRef]
- Ripps, H.; Shen, W. Review: Taurine: A “Very Essential” Amino Acid. Mol. Vis. 2012, 18, 2673–2686. [Google Scholar]
- Vitvitsky, V.; Garg, S.K.; Banerjee, R. Taurine Biosynthesis by Neurons and Astrocytes. J. Biol. Chem. 2011, 286, 32002–32010. [Google Scholar] [CrossRef] [Green Version]
- Ochoa-de la Paz, L.; Zenteno, E.; Gulias-Cañizo, R.; Quiroz-Mercado, H. Taurine and GABA Neurotransmitter Receptors, a Relationship with Therapeutic Potential? Expert Rev. Neurother. 2019, 19, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, J.; Schousboe, A. Taurine Interaction with Neurotransmitter Receptors in the CNS: An Update. Neurochem. Res. 2005, 30, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Foos, T.M.; Wu, J.-Y. The Role of Taurine in the Central Nervous System and the Modulation of Intracellular Calcium Homeostasis. Neurochem. Res. 2002, 27, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Guerrero, S.; Guardo-Maya, S.; Medina-Rincón, G.J.; Orrego-González, E.E.; Cabezas-Pérez, R.; González-Reyes, R.E. Taurine and Astrocytes: A Homeostatic and Neuroprotective Relationship. Front. Mol. Neurosci. 2022, 15, 937789. [Google Scholar] [CrossRef] [PubMed]
- Louzada, P.R.; Paula Lima, A.C.; Mendonca-Silva, D.L.; Noël, F.; De Mello, F.G.; Ferreira, S.T. Taurine Prevents the Neurotoxicity of Beta-Amyloid and Glutamate Receptor Agonists: Activation of GABA Receptors and Possible Implications for Alzheimer’s Disease and Other Neurological Disorders. FASEB J. 2004, 18, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Lee, S.; Choi, S.L.; Kim, H.Y.; Baek, S.; Kim, Y. Taurine Directly Binds to Oligomeric Amyloid-β and Recovers Cognitive Deficits in Alzheimer Model Mice. Adv. Exp. Med. Biol. 2017, 975, 233–241. [Google Scholar] [CrossRef]
- Jahanshahi, M.; Nikmahzar, E.; Gorgani, S. Taurine Can Decrease Phosphorylated Tau Protein Levels in Alzheimer’s Model Rats’ Brains. Kathmandu Univ. Med. J. 2021, 19, 200–204. [Google Scholar]
- Reeta, K.H.; Singh, D.; Gupta, Y.K. Chronic Treatment with Taurine after Intracerebroventricular Streptozotocin Injection Improves Cognitive Dysfunction in Rats by Modulating Oxidative Stress, Cholinergic Functions and Neuroinflammation. Neurochem. Int. 2017, 108, 146–156. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, H.V.; Yoon, J.H.; Kang, B.R.; Cho, S.M.; Lee, S.; Kim, J.Y.; Kim, J.W.; Cho, Y.; Woo, J.; et al. Taurine in Drinking Water Recovers Learning and Memory in the Adult APP/PS1 Mouse Model of Alzheimer’s Disease. Sci. Rep. 2014, 4, 7467. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, Z.; García-Serrano, A.M.; Duarte, J.M. Taurine Supplementation as a Neuroprotective Strategy upon Brain Dysfunction in Metabolic Syndrome and Diabetes. Nutrients 2022, 14, 1292. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J. Lipid Metabolism in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Raha, S.; Ghosh, A.; Dutta, D.; Patel, D.R.; Pahan, K. Activation of PPARα Enhances Astroglial Uptake and Degradation of β-Amyloid. Sci. Signal. 2021, 14, eabg4747. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Konings, S.C.; Torres-Garcia, L.; Martinsson, I.; Gouras, G.K. Astrocytic and Neuronal Apolipoprotein E Isoforms Differentially Affect Neuronal Excitability. Front. Neurosci. 2021, 15, 734001. [Google Scholar] [CrossRef]
- Colton, C.A.; Brown, C.M.; Cook, D.; Needham, L.K.; Xu, Q.; Czapiga, M.; Saunders, A.M.; Schmechel, D.E.; Rasheed, K.; Vitek, M.P. APOE and the Regulation of Microglial Nitric Oxide Production: A Link between Genetic Risk and Oxidative Stress. Neurobiol. Aging 2002, 23, 777–785. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An Emerging Therapeutic Target for Alzheimer’s Disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A. Resistance to Autosomal Dominant Alzheimer’s Disease in an APOE3 Christchurch Homozygote: A Case Report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Mamun, A.A.; Uddin, M.; Bashar, B.; Fahim, M.; Zaman, S.; Begum, Y.; Bulbul, I.J.; Islam, M.; Sarwar, M.; Mathew, B. Molecular Insight into the Therapeutic Promise of Targeting APOE4 for Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 5086250. [Google Scholar] [CrossRef]
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Wang, C.; Gratuze, M.; Hoyle, R.; Bien-Ly, N.; Silverman, A.P.; Sullivan, P.M.; et al. APOE Immunotherapy Reduces Cerebral Amyloid Angiopathy and Amyloid Plaques While Improving Cerebrovascular Function. Sci. Transl. Med. 2021, 13, eabd7522. [Google Scholar] [CrossRef]
- Sadowski, M.; Pankiewicz, J.; Scholtzova, H.; Ripellino, J.A.; Li, Y.; Schmidt, S.D.; Mathews, P.M.; Fryer, J.D.; Holtzman, D.M.; Sigurdsson, E.M.; et al. A Synthetic Peptide Blocking the Apolipoprotein E/β-Amyloid Binding Mitigates β-Amyloid Toxicity and Fibril Formation in Vitro and Reduces β-Amyloid Plaques in Transgenic Mice. Am. J. Pathol. 2004, 165, 937–948. [Google Scholar] [CrossRef]
- Liu, S.; Breitbart, A.; Sun, Y.; Mehta, P.D.; Boutajangout, A.; Scholtzova, H.; Wisniewski, T. Blocking the Apolipoprotein E/Amyloid β Interaction in Triple Transgenic Mice Ameliorates Alzheimer’s Disease Related Amyloid β and Tau Pathology. J. Neurochem. 2014, 128, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwel, D.; Steffensen, K.R.; Verghese, P.B.; Kummer, M.P.; Gustafsson, J.-Å.; Holtzman, D.M.; Heneka, M.T. Critical Role of Astroglial Apolipoprotein E and Liver X Receptor-α Expression for Microglial Aβ Phagocytosis. J. Neurosci. 2011, 31, 7049–7059. [Google Scholar] [CrossRef] [PubMed]
- Skerrett, R.; Pellegrino, M.P.; Casali, B.T.; Taraboanta, L.; Landreth, G.E. Combined Liver X Receptor/Peroxisome Proliferator-Activated Receptor γ Agonist Treatment Reduces Amyloid β Levels and Improves Behavior in Amyloid Precursor Protein/Presenilin 1 Mice. J. Biol. Chem. 2015, 290, 21591–21602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Cabrera, J.M.; Sandoval-Hernández, A.G.; Niño, A.; Báez, T.; Bustos-Rangel, A.; Cardona-Gómez, G.P.; Múnera, A.; Arboleda, G. Bexarotene Therapy Ameliorates Behavioral Deficits and Induces Functional and Molecular Changes in Very-Old Triple Transgenic Mice Model of Alzheimer´s Disease. PLoS ONE 2019, 14, e0223578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Yamagishi, S. Possible Involvement of Advanced Glycation End-Products (AGEs) in the Pathogenesis of Alzheimer’s Disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced Glycation End Products (AGEs) and Other Adducts in Aging-Related Diseases and Alcohol-Mediated Tissue Injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Aliev, G.; Ávila-Rodrigues, M.; Barreto, G.E. Alterations in Glucose Metabolism on Cognition: A Possible Link Between Diabetes and Dementia. Curr. Pharm. Des. 2016, 22, 812–818. [Google Scholar] [CrossRef]
- Choi, B.-R.; Cho, W.-H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.-S. Increased Expression of the Receptor for Advanced Glycation End Products in Neurons and Astrocytes in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced Glycation Endproducts and Their Receptor RAGE in Alzheimer’s Disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Kamynina, A.; Esteras, N.; Koroev, D.O.; Angelova, P.R.; Volpina, O.M.; Abramov, A.Y. Activation of RAGE Leads to the Release of Glutamate from Astrocytes and Stimulates Calcium Signal in Neurons. J. Cell. Physiol. 2021, 236, 6496–6506. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, L.; Xu, Y.; Huang, Y.; Huang, J.; Zhu, J.; Wang, W.; Li, W.; Sun, A.; Li, X.; et al. Discovery of Novel Dual RAGE/SERT Inhibitors for the Potential Treatment of the Comorbidity of Alzheimer’s Disease and Depression. Eur. J. Med. Chem. 2022, 236, 114347. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Jia, P.; Zhang, D.; Yao, Z. TTP488 Ameliorates NLRP3-Associated Inflammation, Viability, Apoptosis, and ROS Production in an Alzheimer’s Disease Cell Model by Mediating the JAK1/STAT3/NFκB/IRF3 Pathway. Cell Biochem. Funct. 2021, 39, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Y.; Wang, Y.; Li, J.; Liu, N. Azeliragon Ameliorates Alzheimer’s Disease via the Janus Tyrosine Kinase and Signal Transducer and Activator of Transcription Signaling Pathway. Clinics 2021, 76, e2348. [Google Scholar] [CrossRef] [PubMed]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Yang, Y.; Ju, W.-N.; Wang, X.; Zhang, H.-L. Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse with Emphasis on Reactive Gliosis in the Context of Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The Role of Brain Vasculature in Neurodegenerative Disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef]
- Zapata-Acevedo, J.F.; García-Pérez, V.; Cabezas-Pérez, R.; Losada-Barragán, M.; Vargas-Sánchez, K.; González-Reyes, R.E. Laminin as a Biomarker of Blood-Brain Barrier Disruption under Neuroinflammation: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 6788. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular Dysfunction—The Disregarded Partner of Alzheimer’s Disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirabali, T.; Rust, R.; Rigotti, S.; Siccoli, A.; Nitsch, R.M.; Kulic, L. Distinct Changes in All Major Components of the Neurovascular Unit across Different Neuropathological Stages of Alzheimer’s Disease. Brain Pathol. 2020, 30, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Molina, L.A.; Villar-Vesga, J.; Henao-Restrepo, J.; Villegas, A.; Lopera, F.; Cardona-Gómez, G.P.; Posada-Duque, R. Extracellular Vesicles From 3xTg-AD Mouse and Alzheimer’s Disease Patient Astrocytes Impair Neuroglial and Vascular Components. Front. Aging Neurosci. 2021, 13, 593927. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E Controls Cerebrovascular Integrity via Cyclophilin, A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Natale, G.; Limanaqi, F.; Busceti, C.L.; Mastroiacovo, F.; Nicoletti, F.; Puglisi-Allegra, S.; Fornai, F. Glymphatic System as a Gateway to Connect Neurodegeneration from Periphery to CNS. Front. Neurosci. 2021, 15, 639140. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science 2013, 342, 1241224. [Google Scholar] [CrossRef] [Green Version]
- Reddy, O.C.; van der Werf, Y.D. The Sleeping Brain: Harnessing the Power of the Glymphatic System through Lifestyle Choices. Brain Sci. 2020, 10, 868. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Vinitsky, H.S.; Sun, Q.; Stæger, F.F.; Sigurdsson, B.; Mortensen, K.N.; Lilius, T.O.; Nedergaard, M. Increased Glymphatic Influx Is Correlated with High EEG Delta Power and Low Heart Rate in Mice under Anesthesia. Sci. Adv. 2019, 5, eaav5447. [Google Scholar] [CrossRef] [Green Version]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Shokri-Kojori, E.; Wang, G.-J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C. β-Amyloid Accumulation in the Human Brain after One Night of Sleep Deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Wang, M.X.; Ismail, O.; Braun, M.; Schindler, A.G.; Reemmer, J.; Wang, Z.; Haveliwala, M.A.; O’Boyle, R.P.; Han, W.Y.; et al. Loss of Perivascular Aquaporin-4 Localization Impairs Glymphatic Exchange and Promotes Amyloid β Plaque Formation in Mice. Alzheimer’s Res. Ther. 2022, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia Prevent Beta-Amyloid Plaque Formation in the Early Stage of an Alzheimer’s Disease Mouse Model with Suppression of Glymphatic Clearance. Alzheimer’s Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef]
- Ren, H.; Luo, C.; Feng, Y.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.-B.; Pei, Z.; Su, H. Omega-3 Polyunsaturated Fatty Acids Promote Amyloid-β Clearance from the Brain through Mediating the Function of the Glymphatic System. FASEB J. 2017, 31, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; O’Callaghan, P.; Li, H.; Tan, Y.; Zhang, G.; Barash, U.; Wang, X.; Lannfelt, L.; Vlodavsky, I.; Lindahl, U.; et al. Heparanase Overexpression Impedes Perivascular Clearance of Amyloid-β from Murine Brain: Relevance to Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 84. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, R.; Shi, C.; Mao, C.; Yang, Z.; Suo, Z.; Torp, R.; Xu, Y. AQP4 Association with Amyloid Deposition and Astrocyte Pathology in the Tg-ArcSwe Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, W.; Zhuo, Y.; Xiang, H.; Li, W.; Liu, H.; Xie, L.; Gao, Q.; Tan, S. L-3-n-Butylphthalide Effectively Improves the Glymphatic Clearance and Reduce Amyloid-β Deposition in Alzheimer’s Transgenic Mice. J. Mol. Neurosci. 2021, 71, 1266–1274. [Google Scholar] [CrossRef]
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood-Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell Neurosci. 2021, 15, 695479. [Google Scholar] [CrossRef]
- Arélin, K.; Kinoshita, A.; Whelan, C.M.; Irizarry, M.C.; Rebeck, G.W.; Strickland, D.K.; Hyman, B.T. LRP and Senile Plaques in Alzheimer’s Disease: Colocalization with Apolipoprotein E and with Activated Astrocytes. Brain Res. Mol. Brain Res. 2002, 104, 38–46. [Google Scholar] [CrossRef]
- Seok, H.; Lee, M.; Shin, E.; Yun, M.R.; Lee, Y.; Moon, J.H.; Kim, E.; Lee, P.H.; Lee, B.-W.; Kang, E.S.; et al. Low-Dose Pioglitazone Can Ameliorate Learning and Memory Impairment in a Mouse Model of Dementia by Increasing LRP1 Expression in the Hippocampus. Sci. Rep. 2019, 9, 4414. [Google Scholar] [CrossRef] [Green Version]
- Hellström-Lindahl, E.; Ravid, R.; Nordberg, A. Age-Dependent Decline of Neprilysin in Alzheimer’s Disease and Normal Brain: Inverse Correlation with Aβ Levels. Neurobiol. Aging 2008, 29, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Ach, K.; Schliebs, R. Aging-Related down-Regulation of Neprilysin, a Putative β-Amyloid-Degrading Enzyme, in Transgenic Tg2576 Alzheimer-like Mouse Brain Is Accompanied by an Astroglial Upregulation in the Vicinity of β-Amyloid Plaques. Neurosci. Lett. 2003, 339, 183–186. [Google Scholar] [CrossRef]
- Saito, T.; Iwata, N.; Tsubuki, S.; Takaki, Y.; Takano, J.; Huang, S.-M.; Suemoto, T.; Higuchi, M.; Saido, T.C. Somatostatin Regulates Brain Amyloid Beta Peptide Abeta42 through Modulation of Proteolytic Degradation. Nat. Med. 2005, 11, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Shibata, M.; Ishikuro, R.; Tanida, M.; Taniguchi, Y.; Ikeda-Matsuo, Y.; Sobue, K. Epigallocatechin Gallate Induces Extracellular Degradation of Amyloid β-Protein by Increasing Neprilysin Secretion from Astrocytes through Activation of ERK and PI3K Pathways. Neuroscience 2017, 362, 70–78. [Google Scholar] [CrossRef]
- Brezovakova, V.; Sykova, E.; Jadhav, S. Astrocytes Derived from Familial and Sporadic Alzheimer’s Disease IPSCs Show Altered Calcium Signaling and Respond Differently to Misfolded Protein Tau. Cells 2022, 11, 1429. [Google Scholar] [CrossRef]
- Li, M.-Z.; Zheng, L.-J.; Shen, J.; Li, X.-Y.; Zhang, Q.; Bai, X.; Wang, Q.-S.; Ji, J.-G. SIRT1 Facilitates Amyloid Beta Peptide Degradation by Upregulating Lysosome Number in Primary Astrocytes. Neural Regen. Res. 2018, 13, 2005–2013. [Google Scholar] [CrossRef]
- Lee, Y.F.; Lariviere, L.; Russ, A.N.; Choi, S.-Z.; Bacskai, B.J.; Kastanenka, K.V. Novel Botanical Therapeutic NB-02 Effectively Treats Alzheimer’s Neuropathophysiology in an APP/PS1 Mouse Model. eNeuro 2021, 8, ENEURO.0389-20.2021. [Google Scholar] [CrossRef]
- Pagnier, G.J.; Kastanenka, K.V.; Sohn, M.; Choi, S.; Choi, S.; Soh, H.; Bacskai, B.J. Novel Botanical Drug DA-9803 Prevents Deficits in Alzheimer’s Mouse Models. Alzheimer’s Res. Ther. 2018, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Gomes, S.; Sousa, N.; Pinto, L.; Oliveira, J.F. Functional Roles of Astrocyte Calcium Elevations: From Synapses to Behavior. Front. Cell Neurosci. 2017, 11, 427. [Google Scholar] [CrossRef] [Green Version]
- Semyanov, A.; Henneberger, C.; Agarwal, A. Making Sense of Astrocytic Calcium Signals—From Acquisition to Interpretation. Nat. Rev. Neurosci. 2020, 21, 551–564. [Google Scholar] [CrossRef]
- Zorec, R.; Araque, A.; Carmignoto, G.; Haydon, P.G.; Verkhratsky, A.; Parpura, V. Astroglial Excitability and Gliotransmission: An Appraisal of Ca2+ as a Signalling Route. ASN Neuro 2012, 4, e00080. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Perea, G.; Maglio, L.; Pastor, J.; García de Sola, R.; Araque, A. Astrocyte Calcium Signal and Gliotransmission in Human Brain Tissue. Cereb. Cortex 2013, 23, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Robinson, M.B. Regulation of Mitochondrial Dynamics in Astrocytes: Mechanisms, Consequences, and Unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y. Astrocytic Ca2+ Signaling Mediated by the Endoplasmic Reticulum in Health and Disease. J. Pharmacol. Sci. 2020, 144, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 Channels Regulate Astrocyte Resting Calcium Levels and Inhibitory Synapse Efficacy via GAT-3. Nat. Neurosci. 2011, 15, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Toescu, E.C.; Verkhratsky, A. The Importance of Being Subtle: Small Changes in Calcium Homeostasis Control Cognitive Decline in Normal Aging. Aging Cell 2007, 6, 267–273. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef]
- Parpura, V.; Grubišić, V.; Verkhratsky, A. Ca2+ Sources for the Exocytotic Release of Glutamate from Astrocytes. Biochim. Biophys. Acta 2011, 1813, 984–991. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptors of Human Neural Cells Play Crucial Roles in Alzheimer’s Disease. Front. Physiol. 2016, 7, 134. [Google Scholar] [CrossRef] [Green Version]
- Magi, S.; Castaldo, P.; Macrì, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular Calcium Dysregulation: Implications for Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef] [Green Version]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-Sensing Receptor Antagonist (Calcilytic) NPS 2143 Specifically Blocks the Increased Secretion of Endogenous Aβ42 Prompted by Exogenous Fibrillary or Soluble Aβ25-35 in Human Cortical Astrocytes and Neurons-Therapeutic Relevance to Alzheimer’s Disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143—Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells 2020, 9, 1386. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, M.; Lin, K.; Xiang, X.; Yang, J.; Zheng, Y.; Xiong, X.; Zhu, S. TRPC6 Attenuates Cortical Astrocytic Apoptosis and Inflammation in Cerebral Ischemic/Reperfusion Injury. Front. Cell Dev. Biol. 2020, 8, 594283. [Google Scholar] [CrossRef]
- Lu, R.; He, Q.; Wang, J. TRPC Channels and Alzheimer’s Disease. Adv. Exp. Med. Biol. 2017, 976, 73–83. [Google Scholar] [CrossRef]
- Griffith, T.N.; Varela-Nallar, L.; Dinamarca, M.C.; Inestrosa, N.C. Neurobiological Effects of Hyperforin and Its Potential in Alzheimer’s Disease Therapy. Curr. Med. Chem. 2010, 17, 391–406. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Fon Tacer, K.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Cheng, P.; Yu, K.; Han, Y.; Song, M.; Li, Y. Hyperforin Attenuates Aluminum-Induced Aβ Production and Tau Phosphorylation via Regulating Akt/GSK-3β Signaling Pathway in PC12 Cells. Biomed. Pharmacother. 2017, 96, 1–6. [Google Scholar] [CrossRef]
- Cerpa, W.; Hancke, J.L.; Morazzoni, P.; Bombardelli, E.; Riva, A.; Marin, P.P.; Inestrosa, N.C. The Hyperforin Derivative IDN5706 Occludes Spatial Memory Impairments and Neuropathological Changes in a Double Transgenic Alzheimer’s Mouse Model. Curr. Alzheimer Res. 2010, 7, 126–133. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Tapia-Rojas, C.; Griffith, T.N.; Carvajal, F.J.; Benito, M.J.; Rivera-Dictter, A.; Alvarez, A.R.; Serrano, F.G.; Hancke, J.L.; Burgos, P.V.; et al. Tetrahydrohyperforin Prevents Cognitive Deficit, Aβ Deposition, Tau Phosphorylation and Synaptotoxicity in the APPswe/PSEN1ΔE9 Model of Alzheimer’s Disease: A Possible Effect on APP Processing. Transl. Psychiatry 2011, 1, e20. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Chico, A.; Tepavcevic, V.; Manterola, A.; Utrilla, C.; Matute, C.; Mato, S. Endocannabinoid Signaling in Brain Diseases: Emerging Relevance of Glial Cells. Glia 2022. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Rodríguez, A.; Bonilla-Del Río, I.; Puente, N.; Gómez-Urquijo, S.M.; Fontaine, C.J.; Egaña-Huguet, J.; Elezgarai, I.; Ruehle, S.; Lutz, B.; Robin, L.M.; et al. Localization of the Cannabinoid Type-1 Receptor in Subcellular Astrocyte Compartments of Mutant Mouse Hippocampus. Glia 2018, 66, 1417–1431. [Google Scholar] [CrossRef] [PubMed]
- Achicallende, S.; Bonilla-Del Río, I.; Serrano, M.; Mimenza, A.; Lekunberri, L.; Anaut-Lusar, I.; Puente, N.; Gerrikagoitia, I.; Grandes, P. GLAST versus GFAP as Astroglial Marker for the Subcellular Study of Cannabinoid CB1 Receptors in Astrocytes. Histochem. Cell Biol. 2022; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vázquez, C.; Amores, M.; Ruiz-Pérez, G.; García-García, E.; et al. Cannabinoid CB2 Receptors in the Mouse Brain: Relevance for Alzheimer’s Disease. J. Neuroinflamm. 2018, 15, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Rueda, D.; Guerra-Ojeda, S.; Aldasoro, M.; Iradi, A.; Obrador, E.; Mauricio, M.D.; Vila, J.M.; Marchio, P.; Valles, S.L. WIN 55,212-2, Agonist of Cannabinoid Receptors, Prevents Amyloid Β1-42 Effects on Astrocytes in Primary Culture. PLoS ONE 2015, 10, e0122843. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in Vivo Blunts Beta-Amyloid Induced Neuroinflammation by Suppressing IL-1beta and INOS Expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.C.; Geiger, P.C.; Han, D.-H.; Jones, T.E.; Holloszy, J.O. Calcium Induces Increases in Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1alpha and Mitochondrial Biogenesis by a Pathway Leading to P38 Mitogen-Activated Protein Kinase Activation. J. Biol. Chem. 2007, 282, 18793–18799. [Google Scholar] [CrossRef] [Green Version]
- Rivera, A.; Vanzulli, I.; Butt, A.M. A Central Role for ATP Signalling in Glial Interactions in the CNS. Curr. Drug Targets 2016, 17, 1829–1833. [Google Scholar] [CrossRef]
- Erb, L.; Cao, C.; Ajit, D.; Weisman, G.A. P2Y Receptors in Alzheimer’s Disease. Biol. Cell 2015, 107, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor Signalling Mediates Astrocytic Hyperactivity in Vivo in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 Receptor Blockade Normalizes Network Dysfunction and Cognition in an Alzheimer’s Disease Model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-N.; Liu, R.-Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early Neuropathological Alzheimer’s Changes in Aged Individuals Are Accompanied by Decreased Cerebrospinal Fluid Melatonin Levels. J. Pineal Res. 2003, 35, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A. Antioxidants and Neuron-Astrocyte Interplay in Brain Physiology: Melatonin, a Neighbor to Rely On. Neurochem. Res. 2021, 46, 34–50. [Google Scholar] [CrossRef]
- Zhu, L.Q.; Wang, S.H.; Ling, Z.Q.; Wang, D.L.; Wang, J.-Z. Effect of Inhibiting Melatonin Biosynthesis on Spatial Memory Retention and Tau Phosphorylation in Rat. J. Pineal Res. 2004, 37, 71–77. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective Effect of Melatonin on Soluble Aβ1-42-Induced Memory Impairment, Astrogliosis, and Synaptic Dysfunction via the Musashi1/Notch1/Hes1 Signaling Pathway in the Rat Hippocampus. Alzheimer’s Res. Ther. 2016, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.-Y.; Han, S.-H. Melatonin Attenuates Kainic Acid-Induced Hippocampal Neurodegeneration and Oxidative Stress through Microglial Inhibition. J. Pineal Res. 2003, 34, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Olivier, P.; Fontaine, R.H.; Loron, G.; Van Steenwinckel, J.; Biran, V.; Massonneau, V.; Kaindl, A.; Dalous, J.; Charriaut-Marlangue, C.; Aigrot, M.-S.; et al. Melatonin Promotes Oligodendroglial Maturation of Injured White Matter in Neonatal Rats. PLoS ONE 2009, 4, e7128. [Google Scholar] [CrossRef]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.; Kaur, C.; Pandi-Perumal, S.; Brown, G.M.; Cardinali, D.P. Melatonin and Its Agonist Ramelteon in Alzheimer’s Disease: Possible Therapeutic Value. Int. J. Alzheimer’s Dis. 2010, 2011, 741974. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.; Zhu, W.; Yang, F.; Yu, Z.-H.; Cai, M.; Li, X.-T.; Zhang, J.-S.; Zhang, W.; Cai, D.-F. Melatonin-Induced ApoE Expression in Mouse Astrocytes Protects Endothelial Cells from OGD-R Induced Injuries. Transl. Psychiatry 2020, 10, 181. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Pei, J.; Zhan, Y.-J.; Cai, Y.-W. Overview of Meta-Analyses of Five Non-Pharmacological Interventions for Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 594432. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Lin, M.-S.; Tzeng, I.-S. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Jahangiri, Z.; Gholamnezhad, Z.; Hosseini, M. Neuroprotective Effects of Exercise in Rodent Models of Memory Deficit and Alzheimer’s. Metab. Brain Dis. 2019, 34, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Belaya, I.; Ivanova, M.; Sorvari, A.; Ilicic, M.; Loppi, S.; Koivisto, H.; Varricchio, A.; Tikkanen, H.; Walker, F.R.; Atalay, M.; et al. Astrocyte Remodeling in the Beneficial Effects of Long-Term Voluntary Exercise in Alzheimer’s Disease. J. Neuroinflamm. 2020, 17, 271. [Google Scholar] [CrossRef]
- Koppel, S.J.; Pei, D.; Wilkins, H.M.; Weidling, I.W.; Wang, X.; Menta, B.W.; Perez-Ortiz, J.; Kalani, A.; Manley, S.; Novikova, L.; et al. A Ketogenic Diet Differentially Affects Neuron and Astrocyte Transcription. J. Neurochem. 2021, 157, 1930–1945. [Google Scholar] [CrossRef]
- Horner, S.; Berger, L.; Gibas, K. Nutritional Ketosis and Photobiomodulation Remediate Mitochondria Warding off Alzheimer’s Disease in a Diabetic, ApoE4+ Patient with Mild Cognitive Impairment: A Case Report. Photodiagn. Photodyn. Ther. 2020, 30, 101777. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized Crossover Trial of a Modified Ketogenic Diet in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef]
- Nardone, R.; Höller, Y.; Tezzon, F.; Christova, M.; Schwenker, K.; Golaszewski, S.; Trinka, E.; Brigo, F. Neurostimulation in Alzheimer’s Disease: From Basic Research to Clinical Applications. Neurol. Sci. 2015, 36, 689–700. [Google Scholar] [CrossRef]
- Lin, Y.; Jin, J.; Lv, R.; Luo, Y.; Dai, W.; Li, W.; Tang, Y.; Wang, Y.; Ye, X.; Lin, W.-J. Repetitive Transcranial Magnetic Stimulation Increases the Brain’s Drainage Efficiency in a Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 102. [Google Scholar] [CrossRef]
- Tsoy, A.; Saliev, T.; Abzhanova, E.; Turgambayeva, A.; Kaiyrlykyzy, A.; Akishev, M.; Saparbayev, S.; Umbayev, B.; Askarova, S. The Effects of Mobile Phone Radiofrequency Electromagnetic Fields on β-Amyloid-Induced Oxidative Stress in Human and Rat Primary Astrocytes. Neuroscience 2019, 408, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Bonnì, S.; Pellicciari, M.C.; Casula, E.P.; Mancini, M.; Esposito, R.; Ponzo, V.; Picazio, S.; Di Lorenzo, F.; Serra, L.; et al. Transcranial Magnetic Stimulation of the Precuneus Enhances Memory and Neural Activity in Prodromal Alzheimer’s Disease. Neuroimage 2018, 169, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Darwish, E.S.; Khedr, E.M.; El Serogy, Y.M.; Ali, A.M. Effects of Low versus High Frequencies of Repetitive Transcranial Magnetic Stimulation on Cognitive Function and Cortical Excitability in Alzheimer’s Dementia. J. Neurol. 2012, 259, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-Y.; Xu, W.-T.; Zhang, X.-C.; Ni, G.-X. Mechanisms of Electroacupuncture on Alzheimer’s Disease: A Review of Animal Studies. Chin. J. Integr. Med. 2020, 26, 473–480. [Google Scholar] [CrossRef]
- Liang, P.-Z.; Li, L.; Zhang, Y.-N.; Shen, Y.; Zhang, L.-L.; Zhou, J.; Wang, Z.-J.; Wang, S.; Yang, S. Electroacupuncture Improves Clearance of Amyloid-β through the Glymphatic System in the SAMP8 Mouse Model of Alzheimer’s Disease. Neural Plast. 2021, 2021, 9960304. [Google Scholar] [CrossRef]
- Liu, P.-R.; Cao, F.; Zhang, Y.; Peng, S. Electroacupuncture Reduces Astrocyte Number and Oxidative Stress in Aged Rats with Surgery-Induced Cognitive Dysfunction. J. Int. Med. Res. 2019, 47, 3860–3873. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.-X.; Li, Q.-Q.; Yang, B.-F.; Liu, Y.; Guan, L.-P.; Wu, M.-M.; Wang, L.-P.; Liu, C.-Z. Acupuncture for Vascular Dementia: A Pragmatic Randomized Clinical Trial. Sci. World J. 2015, 2015, 161439. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Zhang, X.; Yu, J.; Han, J.; Yu, T.; Shi, J.; Zhao, L.; Nie, K. Acupuncture for Patients with Mild to Moderate Alzheimer’s Disease: A Randomized Controlled Trial. BMC Complement. Altern. Med. 2017, 17, 556. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Valla, J.; Sefidvash-Hockley, S.; Rogers, J.; Li, R. Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer's disease patients. J. Neurochem. 2002, 80, 807–814. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, Q.; Zhang, C.; Wang, M. Homoharringtonine Inhibits Alzheimer’s Disease Progression by Reducing Neuroinflammation via STAT3 Signaling in APP/PS1 Mice. Neurodegener. Dis. 2021, 21, 93–102. [Google Scholar] [CrossRef]
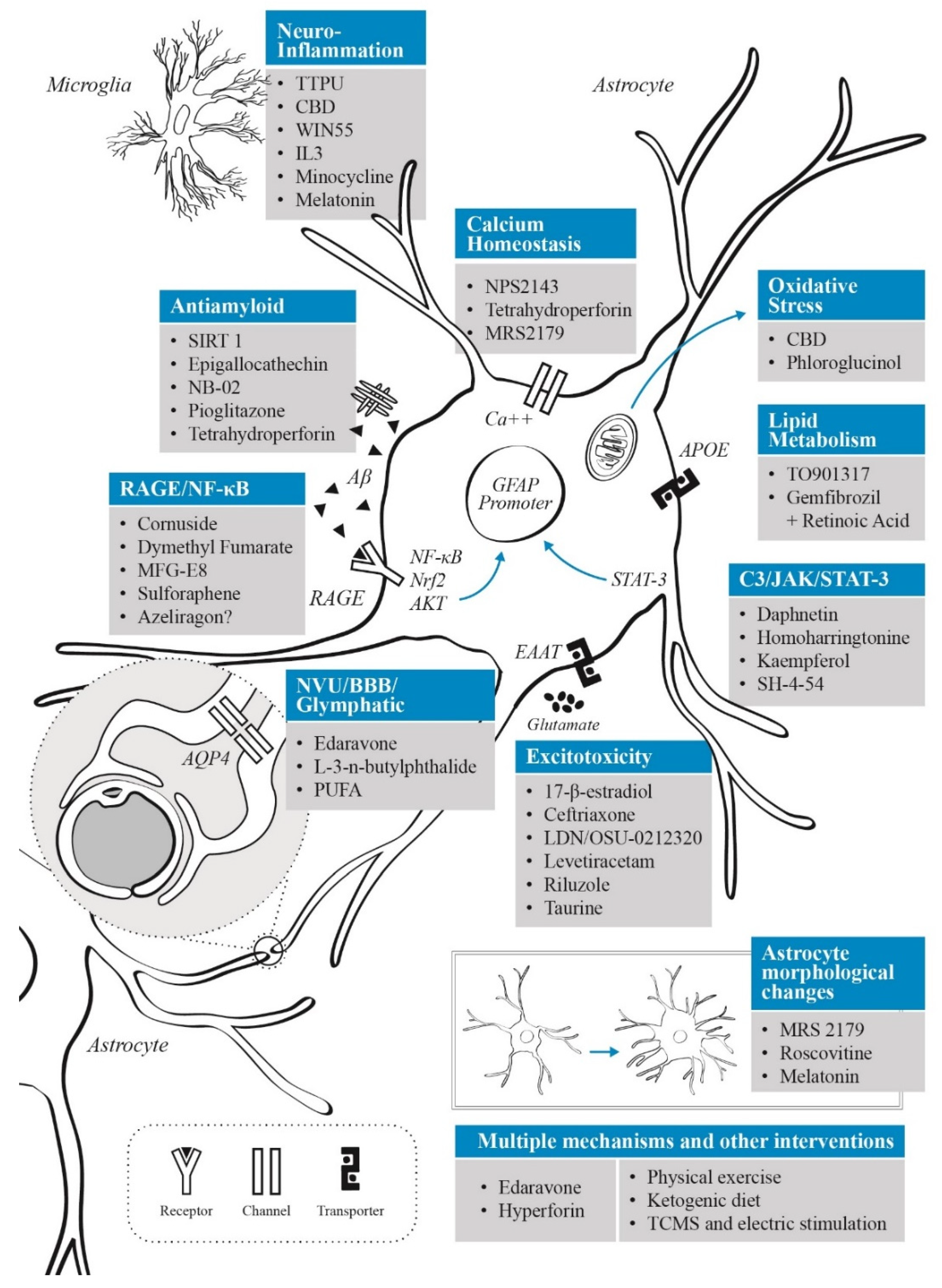

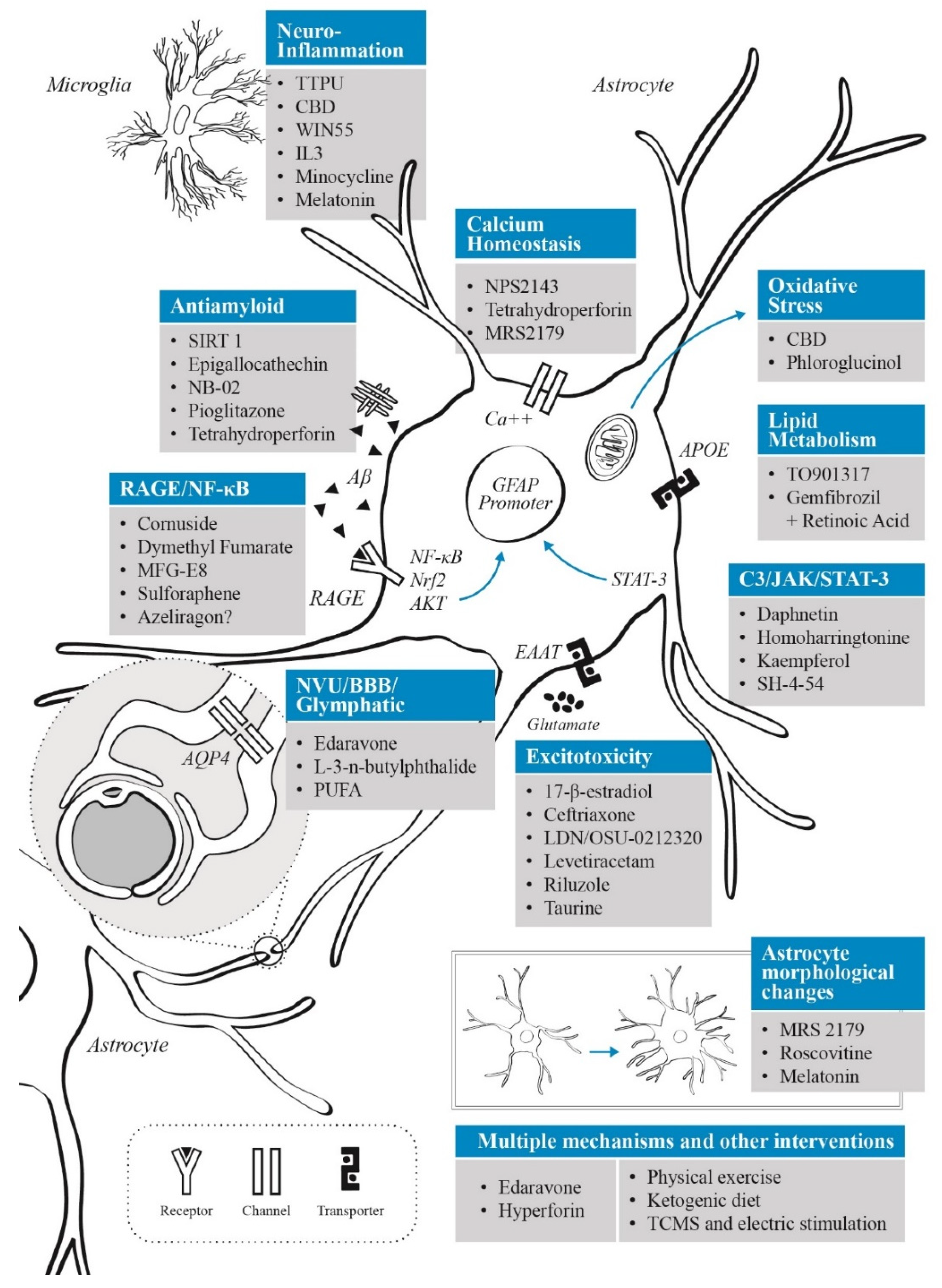{kind=link}
| Reference | Therapeutic Agent | Astrocytic Target and Mechanism | AD Model | Therapeutic Effects |
|---|---|---|---|---|
| [155] | 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) | Blocks astroglial sEH up-regulation, suppression of microglial reactivity through astrocyte-microglia | 5xFAD mice | Reduction in proinflammatory gene expression, reversion of immune pathways dysregulation, reduction in gliosis, reduction in number and size of Aβ plaques, cognitive improvement (object recognition test and fear conditioning paradigm) |
| [370] | 17-β-estradiol | Increases astrocytic glutamate uptake by an unknown mechanism that appears to be independent of estrogen receptors Increases expression of GLT-1 and GLAST in astrocytes | AD Human astrocytes | Increment glutamate reuptake in astrocytes |
| [337] | Cannabidiol (CBD) | Reduces GFAP, IL-1β and iNOS expression in mice exposed to Aβ1–42 Reduces Aβ-generated reactive gliosis | 3–5-month-old C57BL/6J mice | Reduces neuroinflammation. Delays onset and progression of Aβ neurotoxicity |
| [213,219] | Ceftriaxone | IncreaseGLT-1 expression, glutamine synthetase and N glutamine transporter 1 | APP/PS1 mice 3xTg-AD mouse model | Decreases cognitive impairment. Promotes the glutamate-glutamine cycle. Reduces tau pathology |
| [167] | Cornuside | AKT/Nrf2/NF-κB pathway and reduction in proinflammatory cytokines | 3xTg-AD mice | Prevention of cognitive impairment. Anti-amyloid. Reduction in tau phosphorylation Anti-inflammatory (reducing IL-1β, IL-6, TNF-α levels) Antioxidant |
| [137] | Daphnetin | Inhibits STAT3 phosphorylation at Ser727. Decreases astrogliosis (GFAP expression) | APP/PS1 mice | Reduces area and amount of Aβ deposition, decreases the soluble Aβ1–40 and Aβ1–42. |
| [233] | Dymethyl fumarate | Reduction in GFAP reactivity. Reduction in NF-κB-mediated inflammatory response. Activation of AMPK/SIRT-1 & AKT/CREB/BDNF hubs | D-galactose (D-Gal) administered to ovariectomized (OVX) female rats (postmenopausal AD model) | Ameliorated memory deficits. Anti-inflammatory. Antioxidant effects (via SOD and GSH). Reduction in tauo-/amyloidopathy |
| [203,204] | Edaravone | Attenuation of endothelium/astrocyte unit dysfunction. Reduction in astrocytosis (GFAP reactivity) | APP23 rodents associated with brain chronic hypoperfusion APPswe/PS1 mice | Reduction in oxidative stress and neuroinflammation. Improved damaged myelin (enhancing oligodendrocytes). Inhibition of Aβ aggregation and reduction in tau hyperphosphorylation |
| [305] | Epigallocatechin gallate | Increased expression of NEP | Cultured rat astrocytes treated with EGCG | Increased degradation of exogenous Aß |
| [252] | Gemfibrozil + Retinoic acid | PPARα-dependent pathway, low-density lipoprotein receptor, transcription factor EB | 5xFAD mice | RA switched to a neuroprotective state, lowered Aβ in brain, increased lysosomal astrocytic activity, increased autophagic flux, improved spatial learning and memory, enhancement in cognitive function, and reduction in Aβ plaques in hippocampus |
| [371] | Homoharringtonine | STAT3 signalling in glial cells Increases SOCS3 expression in the hippocampus. | APP/PS1 Mice | Alleviates cognitive deficits. Reduces accumulation of Aβ1–40 and Aβ1–42 in both soluble and insoluble forms. Attenuates synaptic function impairment. Suppresses STAT3 activation and reduces neuroinflammation |
| [328,329] | Hyperforin | TRPC6 agonist that stimulates activity of the nSOC pathway in the spines Regulates the Akt-GSK3β signaling pathway | APP knock-in and presenilin mouse models | Increases intracellular calcium levels, reduces astrogliosis, disaggregates Aβ aggregates, rescues mushroom spine loss Reduces tau phosphorylation and Aβ1–42 production in PC12 cells |
| [96] | IL-3 | Astrocytic IL-3 induces microglia activation and instructs it to clear aggregates of Aβ and tau. | 5xFAD mice | Allows microglia to clear aggregates of Aβ and tau. Reduced memory decline and Aβ load |
| [140,141] | Kaempferol | Complement C3 and STAT 3 pathways | AD model induced by ICV streptozotocin in Wistar rats | Prevents the activation of complement C3 protein and the generation of neurotoxic astrocytes. Antiamiloid, antioxidative (increased SOD and GSH levels) and anti-inflammatory mechanisms |
| [298] | L-3-n-butylphthalide | AQP4 | APP/PS1 mice | Reduce Aβ deposition and enhance perivascular localization of AQP4. |
| [129] | LDN/OSU-0212320 | Increases EAAT2 function | APPswe mice Primary neuron and astrocyte mixed culture | Reduce cognitive impairment and amyloid burden. Prevent Aβ25–35 oligomer–induced toxicity |
| [228] | Levetiracetam | Astrocytic SV2A | Primary cultures of human astrocytes exposed with oligomeric Aβ1–42 | Reduction in Aβ-induced glutamate release, reduction in gliotransmission-mediated excitation |
| [346] | Melatonin | Inhibition of Notch1 signalling pathway | Mice ICV injection with Aβ1–42 | Improved synaptic function, attenuation of astrogliosis and enhanced spatial memory performance, decrease in GFAP expression |
| [173,174] | MFG-E8 | Downregulation of NF-κB and upregulation of PI3K-Akt | Aβ1–42-activated microglia-conditioned medium to induce astrocytic activation Neuronal/glial mixed culture exposed to Aβ1–42 | Anti-inflammatory effects on astrocytes (modulation in IL-1α, TNF, and C1q) Increase microglial endocytosis of Aβ |
| [147] | Minocycline | Tetracycline antibiotic-protein synthesis inhibitor and increases expression of anti-inflammatory genes | htau mouse AD model | Decrease in astrocytic and caspase-3 activation. Reduction in neuronal loss. Reduce the number of RA, decreases the formation of abnormal tau species and attenuated the production of proinflammatory cytokines |
| [342] | MRS2179 | P2Y1R antagonist | Mice with human KM67/671NL mutation in APP and human L166P-mutated PS1 | Increase density and branches length of astrocytic process and points of terminal process close to senile plaques. Preserve structural synaptic integrity |
| [295] | n-3 polyunsaturated fatty acids (PUFA) | AQP4 | fat-1 mice C57BL/6 (ICV Aβ injection) | Promote interstitial Aβ clearance. Inhibit astrocyte activation and protect the AQP4 polarization |
| [308,309] | NB-02 (previously known as DA-9803) | Mechanism of action under study. Authors suggest that it could be acting by decreasing Aβ oligomerization and production while increasing clearance. | APP/PS1 mice, 5xFAD mice | Halts amyloid plaque deposition, preserves neuronal calcium homeostasis, restores spine density, alters astrocytic morphology, induces a phagocytic state in microglia |
| [322,323,324] | NPS2143 (CaSR negative allosteric modulator) | CaSR signaling secretion of proinflammatory agents Suppresses astrocytic Aβ1–42 by CaSR signaling. | Aβ25–35-exposed human cortical astrocytes Normal human adult astrocytes, fAβ25–35-treated astrocytes, HC-1A neurons in vitro | Reduces neuroinflammation and amyloidosis. Reduces intracellular accumulation and release of p-tau. Suppresses the release of IL-6, RANTES, ICAM-1 and MCP-2. |
| [189] | Phloroglucinol | Inhibits the generation of ROS | Oligomeric forms of Aβ1–42 in astrocyte cultures | Enhancing antioxidant enzymes expression such as SOD and GSH. Inhibits astrocytes activation induced by Aβ |
| [301] | Pioglitazone | Increased LRP1 expression | Senescence-accelerated mouse prone-8 (SAMP8) mice model | Reduction in Aß deposits and Aß1–40 levels improved performance in water maze test |
| [211] | Riluzole | Increase EAAT2 expression | APPswe/PS1dE9 mice | Prevent cognitive impairment and synaptic plasticity changes induced by early stress |
| [236] | Roscovitine | CDK inhibitor with affinity for CDK5 | SAMP8 mice | Reverses morphological changes produced by glutamate excitotoxicity |
| [132] | SH-4-54 (STAT3 phosphorylation site inhibitor) | C3aR1-STAT3 signaling mediated astrogliosis and astrocyte reactivity | PS19 mice | Reduction in neuroinflammation and partial rescue of tau pathology |
| [307] | Sirtuin 1 (SIRT 1) | Promotes oligomeric Aß degradation in astrocytes by deacetylation of lysosome-related proteins and upregulation of total lysosome number | Astrocyte culture of Sprague Dawley rats cortex samples. | Reduction in Aß deposits |
| [159,163,165] | Sulforaphene | Inhibition of NF-κB and stimulation of Nrf2 pathways | AD-Like pathology model induced by streptozotocin and N2a/APPswe cells | Anti-inflammatory and antioxidant effects. Inhibited the phosphorylation of tau protein and improved cognitive deficit in memory function. |
| [246,247] | Taurine | Produced and released as gliotransmitter by astrocytes. Activates GABAA and glycine receptors, and serves as GABAB agonist | APP/PS1 transgenic AD mouse | Homeostatic and neuroprotective effects. Regulates intracellular calcium levels, binds to oligomeric Aβ and helps to decrease tau phosphorylation |
| [263] | TO901317 | Astrocytic LXR agonist Increase ABCA1 and APOE expression in astrocytes | APP/PS1 mice | Increase phagocytosis of Aβ by microglial activity. Improve spatial learning |
| [326,330,331] | Tetrahydroperforin (IDN5607) | TRPC6 agonist that acts by modulating Aβ production, interacting with APP and C99, and blocking the cleavage of C99 by the γ-secretase | APPswe and PS-1dE9 mouse models | Prevents RA inflammatory response, decreases large Aβ deposits., reduces damage caused by oxidative stress, alleviates memory decline |
| [336] | WIN 55,212-2 | CB1 and CB2 receptors | Astrocytes exposed to Aβ1–42 | Prevented the elevation of TNF and IL-1β, p-65, COX-2 and iNOS proteins. Increased cell viability |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Giraldo, M.; González-Reyes, R.E.; Ramírez-Guerrero, S.; Bonilla-Trilleras, C.E.; Guardo-Maya, S.; Nava-Mesa, M.O. Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments. Int. J. Mol. Sci. 2022, 23, 13630. https://doi.org/10.3390/ijms232113630
Rodríguez-Giraldo M, González-Reyes RE, Ramírez-Guerrero S, Bonilla-Trilleras CE, Guardo-Maya S, Nava-Mesa MO. Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments. International Journal of Molecular Sciences. 2022; 23(21):13630. https://doi.org/10.3390/ijms232113630
Chicago/Turabian StyleRodríguez-Giraldo, Mateo, Rodrigo E. González-Reyes, Sofía Ramírez-Guerrero, Carlos E. Bonilla-Trilleras, Santiago Guardo-Maya, and Mauricio O. Nava-Mesa. 2022. "Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments" International Journal of Molecular Sciences 23, no. 21: 13630. https://doi.org/10.3390/ijms232113630
APA StyleRodríguez-Giraldo, M., González-Reyes, R. E., Ramírez-Guerrero, S., Bonilla-Trilleras, C. E., Guardo-Maya, S., & Nava-Mesa, M. O. (2022). Astrocytes as a Therapeutic Target in Alzheimer’s Disease–Comprehensive Review and Recent Developments. International Journal of Molecular Sciences, 23(21), 13630. https://doi.org/10.3390/ijms232113630