

Constitutively Active Androgen Receptor in Hepatocellular Carcinoma

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
1.1. Sexual Dimorphism in Hepatocellular Carcinoma
1.2. Androgen Receptor Expression and Role in Disease Progression
1.3. AR and Epithelial–Mesenchymal Transition
1.4. Failed Therapeutic Approaches Targeting the AR
1.5. Reconciling the Role of AR in HCC with the Failure of Anti-Androgens
2. Alternative Mechanisms of AR Overexpression
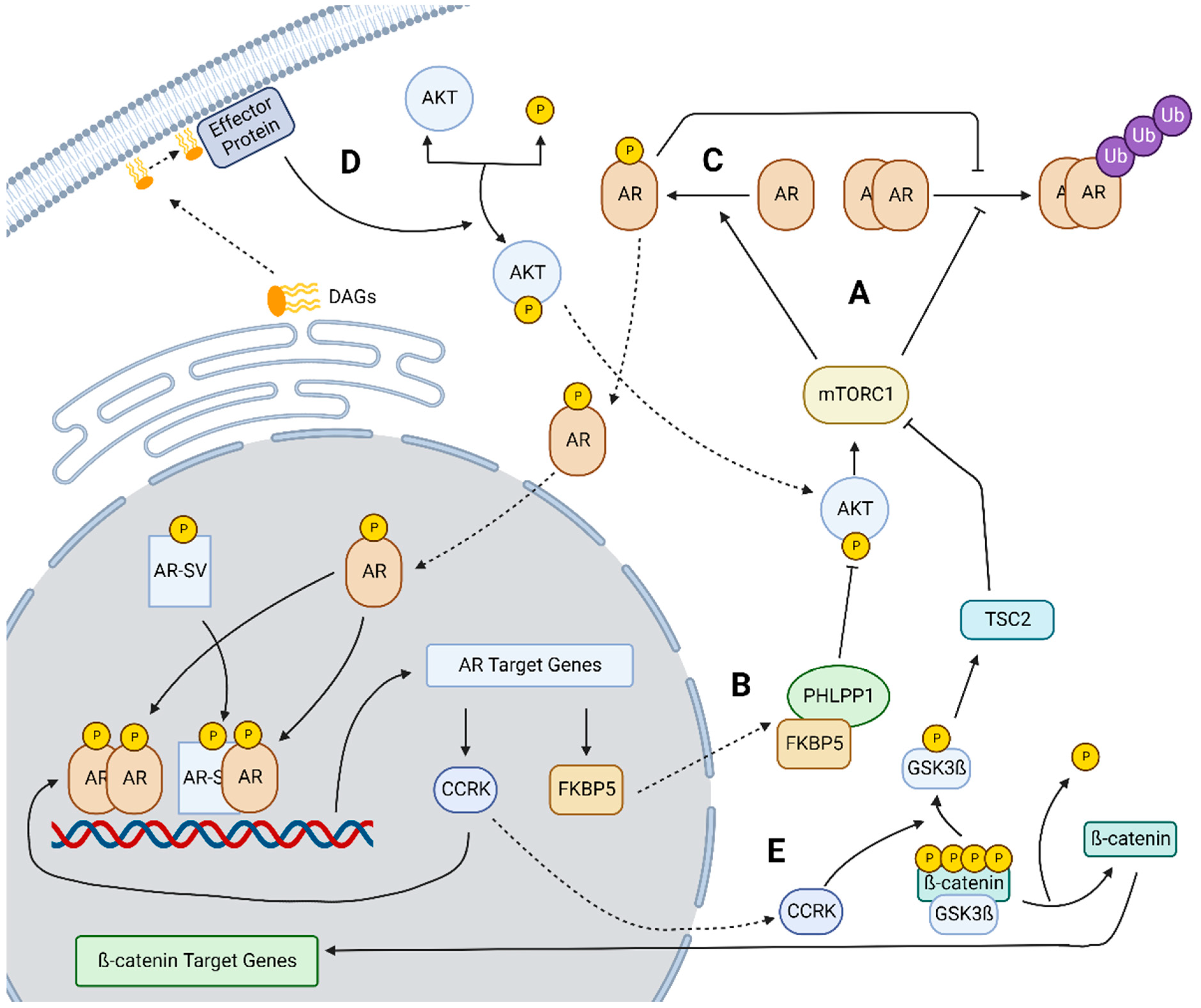
2.1. mTOR Overexpression and Signaling in HCC
2.2. AKT-mTOR and AR Crosstalk
2.3. Lipogenesis Driven Constitutive AR Activity
2.4. CCRK-AR-mTOR Pathway
2.5. FAK-Mediated Signaling
3. Difference between AR Expression and Activity
4. Alternative Splicing as a Means of Constitutive Activity
4.1. Decoupling of AR Activity from Androgen Binding
4.2. Variant Splicing in HCC
4.3. Variant AR Splicing Is a Function of AR Overexpression
4.4. Splicing Factor PRPF6 Associated with Increased AR Activity and Poor Prognosis
5. AR-Targeted Therapeutic Strategies for HCC
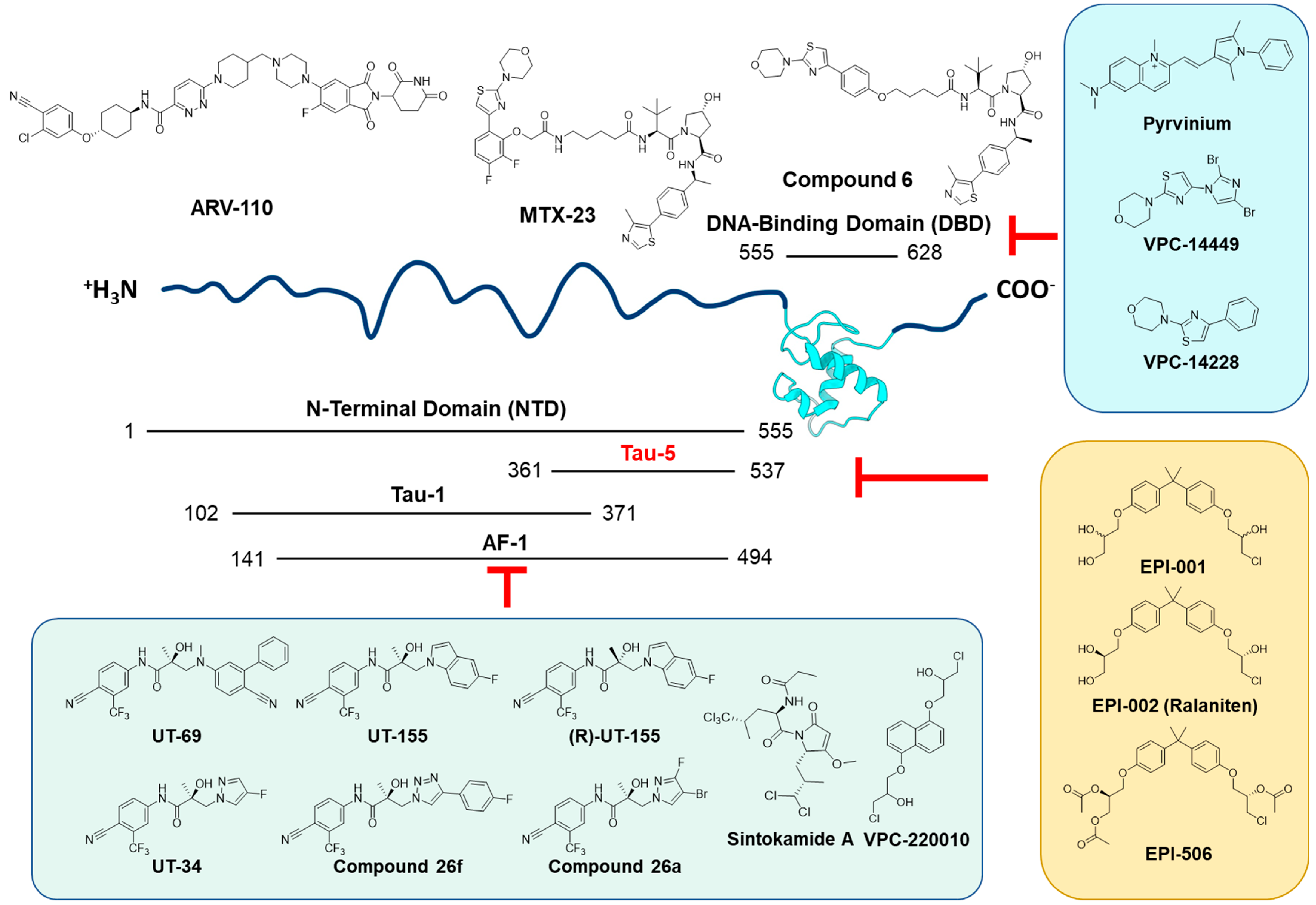
5.1. Effectively Targeting the Androgen Receptor in HCC
5.2. Novel AR-SV-Targeted Agents
5.2.1. DNA-Binding Domain-Targeted Agents
5.2.2. N-Terminal Domain-Targeted Agents
5.3. Repurposed
5.4. Combined mTOR and AR Inhibition
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Spencer, K.; Burley, S.K.; Zheng, X.F.S. Toward improving androgen receptor-targeted therapies in male-dominant hepatocellular carcinoma. Drug Discov. Today 2021, 26, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Burra, P.; Bizzaro, D.; Gonta, A.; Shalaby, S.; Gambato, M.; Morelli, M.C.; Trapani, S.; Floreani, A.; Marra, F.; Brunetto, M.R.; et al. Clinical impact of sexual dimorphism in non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Liver Int. 2021, 41, 1713–1733. [Google Scholar] [CrossRef] [PubMed]
- Bethesda, M.D. SEER Cancer Stat Facts: Liver and Intrahepatic Bile Duct Cancer. Available online: https://seer.cancer.gov/statfacts/html/livibd.html (accessed on 24 October 2022).
- Kanda, T.; Jiang, X.; Yokosuka, O. Androgen receptor signaling in hepatocellular carcinoma and pancreatic cancers. World J. Gastroenterol. 2014, 20, 9229–9236. [Google Scholar] [CrossRef]
- Ma, W.L.; Hsu, C.L.; Wu, M.H.; Wu, C.T.; Wu, C.C.; Lai, J.J.; Jou, Y.S.; Chen, C.W.; Yeh, S.; Chang, C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology 2008, 135, 947–955.e5. [Google Scholar] [CrossRef]
- Shi, M.W.; Zhang, N.A.; Shi, C.P.; Liu, C.J.; Luo, Z.H.; Wang, D.Y.; Guo, A.Y.; Chen, Z.X. SAGD: A comprehensive sex-associated gene database from transcriptomes. Nucleic Acids Res. 2019, 47, D835–D840. [Google Scholar] [CrossRef]
- Wu, M.H.; Ma, W.L.; Hsu, C.L.; Chen, Y.L.; Ou, J.H.; Ryan, C.K.; Hung, Y.C.; Yeh, S.; Chang, C. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci. Transl. Med. 2010, 2, 32ra35. [Google Scholar] [CrossRef]
- Liu, W.C.; Liu, Q.Y. Molecular mechanisms of gender disparity in hepatitis B virus-associated hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 6252–6261. [Google Scholar] [CrossRef]
- Wang, S.H.; Chen, P.J.; Yeh, S.H. Gender disparity in chronic hepatitis B: Mechanisms of sex hormones. J. Gastroenterol. Hepatol. 2015, 30, 1237–1245. [Google Scholar] [CrossRef]
- Kalra, M.; Mayes, J.; Assefa, S.; Kaul, A.K.; Kaul, R. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 5945–5961. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.X.; Yang, Y.; Zhang, Y.; Wang, H.Y.; Zheng, X.F.S. Significance and mechanism of androgen receptor overexpression and androgen receptor/mechanistic target of rapamycin cross-talk in hepatocellular carcinoma. Hepatology 2018, 67, 2271–2286. [Google Scholar] [CrossRef] [PubMed]
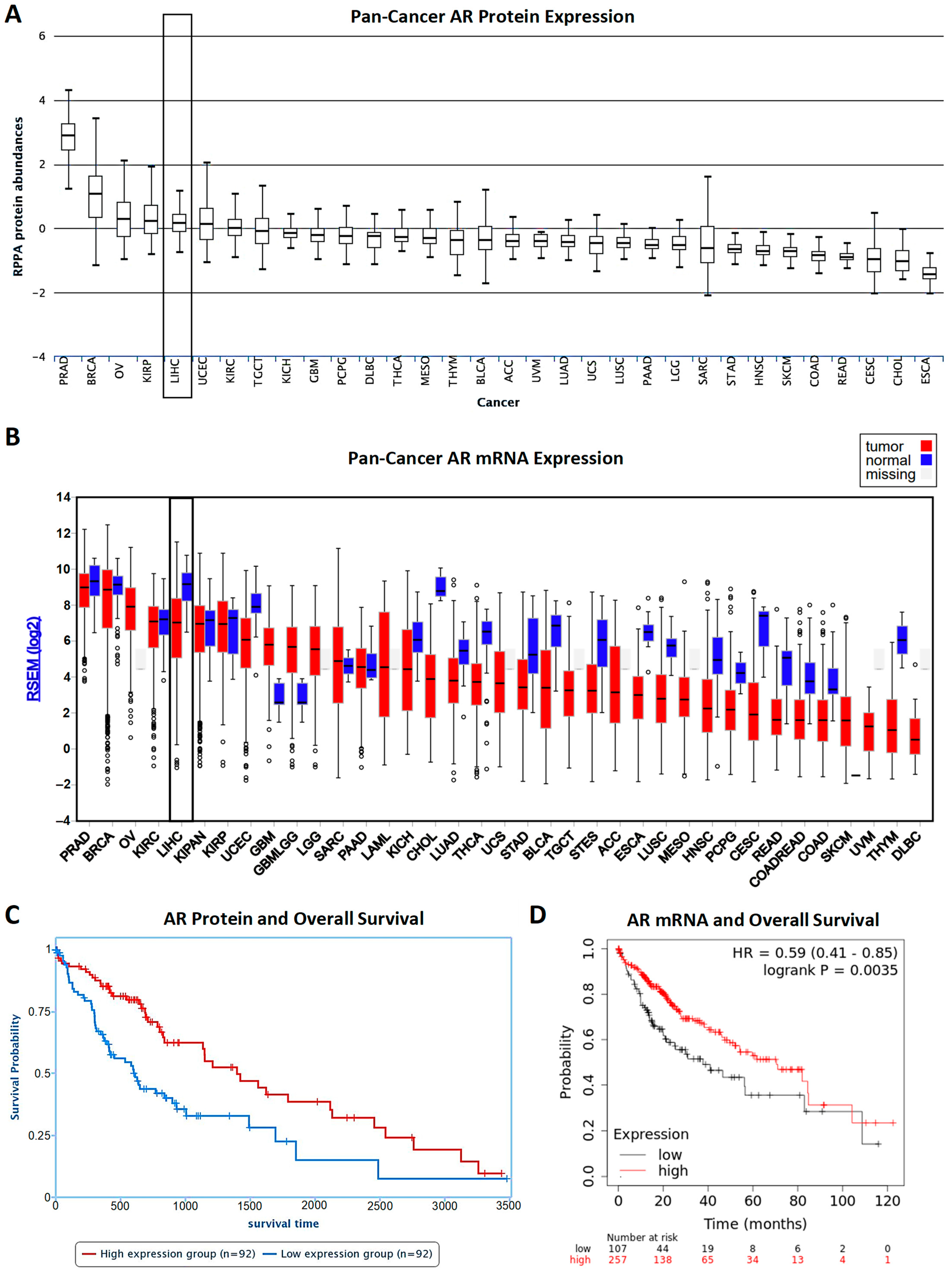
- Acosta-Lopez, S.; Diaz-Bethencourt, D.; Concepcion-Massip, T.; Martin-Fernandez de Basoa, M.C.; Plata-Bello, A.; Gonzalez-Rodriguez, A.; Perez-Hernandez, F.; Plata-Bello, J. The androgen receptor expression and its activity have different relationships with prognosis in hepatocellular carcinoma. Sci. Rep. 2020, 10, 22046. [Google Scholar] [CrossRef]
- Menyhart, O.; Nagy, A.; Gyorffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.Y.; Broom, B.M.; Verhaak, R.G.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef]
- Li, J.; Akbani, R.; Zhao, W.; Lu, Y.; Weinstein, J.N.; Mills, G.B.; Liang, H. Explore, Visualize, and Analyze Functional Cancer Proteomic Data Using the Cancer Proteome Atlas. Cancer Res. 2017, 77, e51–e54. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Liu, Y.N.; Liu, Y.; Lee, H.J.; Hsu, Y.H.; Chen, J.H. Activated androgen receptor downregulates E-cadherin gene expression and promotes tumor metastasis. Mol. Cell Biol. 2008, 28, 7096–7108. [Google Scholar] [CrossRef]
- Kong, D.; Sethi, S.; Li, Y.; Chen, W.; Sakr, W.A.; Heath, E.; Sarkar, F.H. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate 2015, 75, 161–174. [Google Scholar] [CrossRef]
- Dauki, A.M.; Blachly, J.S.; Kautto, E.A.; Ezzat, S.; Abdel-Rahman, M.H.; Coss, C.C. Transcriptionally Active Androgen Receptor Splice Variants Promote Hepatocellular Carcinoma Progression. Cancer Res. 2020, 80, 561–575. [Google Scholar] [CrossRef]
- Zhou, H.C.; Liu, C.X.; Pan, W.D.; Shang, L.R.; Zheng, J.L.; Huang, B.Y.; Chen, J.Y.; Zheng, L.; Fang, J.H.; Zhuang, S.M. Dual and opposing roles of the androgen receptor in VETC-dependent and invasion-dependent metastasis of hepatocellular carcinoma. J. Hepatol. 2021, 75, 900–911. [Google Scholar] [CrossRef]
- Rajaram, P.; Rivera, A.; Muthima, K.; Olveda, N.; Muchalski, H.; Chen, Q.H. Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer. Molecules 2020, 25, 2448. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.; Bleiberg, H.; Gay, F.; Messner, M.; Rougier, P.; Kok, T.C.; Cirera, L.; Cervantes, A.; De Greve, J.; Paillot, B.; et al. Evaluation of antiandrogen therapy in unresectable hepatocellular carcinoma: Results of a European Organization for Research and Treatment of Cancer multicentric double-blind trial. J. Clin. Oncol. 1998, 16, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Groupe d’Etude et de Traitement du Carcinome Hépatocellulaire. Randomized trial of leuprorelin and flutamide in male patients with hepatocellular carcinoma treated with tamoxifen. Hepatology 2004, 40, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Harding, J.J.; Kelley, R.K.; Tan, B.; Capanu, M.; Do, G.K.; Shia, J.; Chou, J.F.; Ferrer, C.S.; Boussayoud, C.; Muenkel, K.; et al. Phase Ib Study of Enzalutamide with or Without Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Oncologist 2020, 25, e1825–e1836. [Google Scholar] [CrossRef]
- Jacob, A.; Raj, R.; Allison, D.B.; Myint, Z.W. Androgen Receptor Signaling in Prostate Cancer and Therapeutic Strategies. Cancers 2021, 13, 5417. [Google Scholar] [CrossRef]
- Lee, D.K.; Chang, C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: Mechanism and clinical implication. J. Clin. Endocrinol. Metab. 2003, 88, 4043–4054. [Google Scholar] [CrossRef]
- Yu, L.; Nagasue, N.; Makino, Y.; Nakamura, T. Effect of androgens and their manipulation on cell growth and androgen receptor (AR) levels in AR-positive and -negative human hepatocellular carcinomas. J. Hepatol. 1995, 22, 295–302. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.K.; Lim, H.Y.; Poon, R.T.; Blanc, J.F.; Vogel, A.; Chen, C.L.; et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef]
- Ren, Q.N.; Zhang, H.; Sun, C.Y.; Zhou, Y.F.; Yang, X.F.; Long, J.W.; Li, X.X.; Mai, S.J.; Zhang, M.Y.; Zhang, H.Z.; et al. Phosphorylation of androgen receptor by mTORC1 promotes liver steatosis and tumorigenesis. Hepatology 2022, 75, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Chen, K.W.; Kuo, H.C.; Kuo, C.H.; Lin, W.H.; Chen, P.J.; Yeh, S.H. Specific diacylglycerols generated by hepatic lipogenesis stimulate the oncogenic androgen receptor activity in male hepatocytes. Int. J. Obes. 2019, 43, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Cheng, A.S.; Tsang, D.P.; Li, M.S.; Go, M.Y.; Cheung, Y.S.; Zhao, G.J.; Ng, S.S.; Lin, M.C.; Yu, J.; et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives beta-catenin/T cell factor-dependent hepatocarcinogenesis. J. Clin. Investig. 2011, 121, 3159–3175. [Google Scholar] [CrossRef]
- Sun, H.; Yang, W.; Tian, Y.; Zeng, X.; Zhou, J.; Mok, M.T.S.; Tang, W.; Feng, Y.; Xu, L.; Chan, A.W.H.; et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat. Commun. 2018, 9, 5214. [Google Scholar] [CrossRef]
- Shang, N.; Wang, H.; Bank, T.; Perera, A.; Joyce, C.; Kuffel, G.; Zilliox, M.J.; Cotler, S.J.; Ding, X.; Dhanarajan, A.; et al. Focal Adhesion Kinase and beta-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology 2019, 70, 1631–1645. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Lee, S.E.; Alcedo, K.P.; Kim, H.J.; Snider, N.T. Alternative Splicing in Hepatocellular Carcinoma. Cell Mol Gastroenterol. Hepatol. 2020, 10, 699–712. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Reviejo, M.; Soto, M.; Lozano, E.; Asensio, M.; Ortiz-Rivero, S.; Berasain, C.; Avila, M.A.; Herraez, E. Impact of Alternative Splicing Variants on Liver Cancer Biology. Cancers 2021, 14, 18. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, S.; Sowalsky, A.G.; Voznesensky, O.S.; Mostaghel, E.A.; Nelson, P.S.; Cai, C.; Balk, S.P. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res. 2014, 20, 1590–1600. [Google Scholar] [CrossRef]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H.; et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol. Cancer Res. 2017, 15, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lonergan, P.E.; Nacusi, L.P.; Wang, L.; Schmidt, L.J.; Sun, Z.; Van der Steen, T.; Boorjian, S.A.; Kosari, F.; Vasmatzis, G.; et al. The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. J. Urol. 2015, 193, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lan, T. Molecular Origin, Expression Regulation, and Biological Function of Androgen Receptor Splicing Variant 7 in Prostate Cancer. Urol. Int. 2021, 105, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. 2016, 21, 1084–1091. [Google Scholar] [CrossRef]
- Song, H.; Sun, N.; Lin, L.; Wei, S.; Zeng, K.; Liu, W.; Wang, C.; Zhong, X.; Wang, M.; Wang, S.; et al. Splicing factor PRPF6 upregulates oncogenic androgen receptor signaling pathway in hepatocellular carcinoma. Cancer Sci. 2020, 111, 3665–3678. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Gao, A.C. Current strategies for targeting the activity of androgen receptor variants. Asian J. Urol. 2019, 6, 42–49. [Google Scholar] [CrossRef]
- Xiang, W.; Wang, S. Therapeutic Strategies to Target the Androgen Receptor. J. Med. Chem. 2022, 65, 8772–8797. [Google Scholar] [CrossRef]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar] [PubMed]
- Lim, M.; Otto-Duessel, M.; He, M.; Su, L.; Nguyen, D.; Chin, E.; Alliston, T.; Jones, J.O. Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium. ACS Chem. Biol. 2014, 9, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Alen, P.; Devos, A.; Peeters, B.; Verhoeven, G.; Rombauts, W. The androgen-specific probasin response element 2 interacts differentially with androgen and glucocorticoid receptors. J. Biol. Chem. 1996, 271, 19013–19016. [Google Scholar] [CrossRef] [PubMed]
- Denayer, S.; Helsen, C.; Thorrez, L.; Haelens, A.; Claessens, F. The rules of DNA recognition by the androgen receptor. Mol. Endocrinol. 2010, 24, 898–913. [Google Scholar] [CrossRef]
- Zilliacus, J.; Wright, A.P.; Carlstedt-Duke, J.; Gustafsson, J.A. Structural determinants of DNA-binding specificity by steroid receptors. Mol. Endocrinol. 1995, 9, 389–400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dalal, K.; Roshan-Moniri, M.; Sharma, A.; Li, H.; Ban, F.; Hessein, M.; Hsing, M.; Singh, K.; LeBlanc, E.; Dehm, S.; et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J. Biol. Chem. 2014, 289, 26417–26429. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ban, F.; Dalal, K.; Leblanc, E.; Frewin, K.; Ma, D.; Adomat, H.; Rennie, P.S.; Cherkasov, A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 2014, 57, 6458–6467. [Google Scholar] [CrossRef]
- Dalal, K.; Ban, F.; Li, H.; Morin, H.; Roshan-Moniri, M.; Tam, K.J.; Shepherd, A.; Sharma, A.; Peacock, J.; Carlson, M.L.; et al. Selectively targeting the dimerization interface of human androgen receptor with small-molecules to treat castration-resistant prostate cancer. Cancer Lett. 2018, 437, 35–43. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.-J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Mol. Cancer Ther. 2021, 20, 490–499. [Google Scholar] [CrossRef]
- Bhumireddy, A.; Bandaru, N.V.M.R.; Raghurami Reddy, B.; Gore, S.T.; Mukherjee, S.; Balasubramanian, W.R.; Sumanth Kumar, V.; Alapati, K.S.; Venkata Gowri Chandra Sekhar, K.; Nellore, K.; et al. Design, synthesis, and biological evaluation of phenyl thiazole-based AR-V7 degraders. Bioorg. Med. Chem. Lett. 2022, 55, 128448. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; III, H.A.B.; Vuky, J.; Dreicer, R.; Sartor, A.O.; Sternberg, C.N.; Percent, I.J.; Hussain, M.H.A.; Kalebasty, A.R.; Shen, J.; et al. Phase 1/2 study of ARV-110, an androgen receptor (AR) PROTAC degrader, in metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 17. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016, 21, 1427–1435. [Google Scholar] [CrossRef]
- Sadar, M.D. Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin. Drug Discov. 2020, 15, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Betney, R.; Li, J.; Thompson, E.B.; McEwan, I.J. Induced alpha-helix structure in AF1 of the androgen receptor upon binding transcription factor TFIIF. Biochemistry 2004, 43, 3008–3013. [Google Scholar] [CrossRef]
- De Mol, E.; Szulc, E.; Di Sanza, C.; Martinez-Cristobal, P.; Bertoncini, C.W.; Fenwick, R.B.; Frigole-Vivas, M.; Masin, M.; Hunter, I.; Buzon, V.; et al. Regulation of Androgen Receptor Activity by Transient Interactions of Its Transactivation Domain with General Transcription Regulators. Structure 2018, 26, 145–152.e143. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. The human androgen receptor AF1 transactivation domain: Interactions with transcription factor IIF and molten-globule-like structural characteristics. Biochem. Soc. Trans. 2006, 34, 1054–1057. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. Functional characterization of the native NH2-terminal transactivation domain of the human androgen receptor: Binding kinetics for interactions with TFIIF and SRC-1a. Biochemistry 2008, 47, 3352–3359. [Google Scholar] [CrossRef]
- McEwan, I.J.; Gustafsson, J. Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF. Proc. Natl. Acad. Sci. USA 1997, 94, 8485–8490. [Google Scholar] [CrossRef]
- Banuelos, C.A.; Tavakoli, I.; Tien, A.H.; Caley, D.P.; Mawji, N.R.; Li, Z.; Wang, J.; Yang, Y.C.; Imamura, Y.; Yan, L.; et al. Sintokamide A Is a Novel Antagonist of Androgen Receptor That Uniquely Binds Activation Function-1 in Its Amino-terminal Domain. J. Biol. Chem. 2016, 291, 22231–22243. [Google Scholar] [CrossRef]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Soest, R.V.; Andersen, R.J. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org. Lett. 2008, 10, 4947–4950. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Banuelos, C.A.; Mawji, N.R.; Patrick, B.O.; Sadar, M.D.; Andersen, R.J. Structure-Activity Relationships for the Marine Natural Product Sintokamides: Androgen Receptor N-Terminus Antagonists of Interest for Treatment of Metastatic Castration-Resistant Prostate Cancer. J. Nat. Prod. 2021, 84, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Sadar, M.D. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res. 2011, 71, 1208–1213. [Google Scholar] [CrossRef]
- Andersen, R.J. Sponging off nature for new drug leads. Biochem. Pharmacol. 2017, 139, 3–14. [Google Scholar] [CrossRef]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef]
- De Mol, E.; Fenwick, R.B.; Phang, C.T.; Buzon, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; Garcia, J.; Bertoncini, C.W.; Estebanez-Perpina, E.; et al. EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Brand, L.J.; Olson, M.E.; Ravindranathan, P.; Guo, H.; Kempema, A.M.; Andrews, T.E.; Chen, X.; Raj, G.V.; Harki, D.A.; Dehm, S.M. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget 2015, 6, 3811–3824. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- Osbourne, E. EPI-002 and Enzalutamide Combination Therapy as a Potential Therapeutic Benefit for Castration-Resistant Prostate Cancer Patients; University of British Columbia: Vancouver, BC, Canada, 2014. [Google Scholar]
- Yang, Y.C.; Mawji, N.; Wang, J.; Sadar, M. Abstract 610: Preclinical evaluation of novel androgen receptor N-terminal domain inhibitor EPI-002 for the treatment of castration-resistant prostate cancer. Cancer Res. 2014, 74, 610. [Google Scholar] [CrossRef]
- Ding, R. EPI-002 Accelerates Ligand Dissociation from Androgen Receptor by Disrupting N-Terminus to C-Terminus Interaction; University of British Columbia: Vancouver, BC, Canada, 2013. [Google Scholar]
- Moigne, R.L.; Zhou, H.-J.; Obst, J.K.; Banuelos, C.A.; Jian, K.; Williams, D.; Virsik, P.; Andersen, R.J.; Sadar, M.; Perabo, F.; et al. Lessons learned from the metastatic castration-resistant prostate cancer phase I trial of EPI-506, a first-generation androgen receptor N-terminal domain inhibitor. J. Clin. Oncol. 2019, 37, 257. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Antonarakis, E.S.; Hussain, M.; Fizazi, K.; Joshua, A.M.; Attard, G.; Sadar, M.; Perabo, F.; Chi, K.N. A phase 1/2 open-label study of safety and antitumor activity of EPI-506, a novel AR N-terminal domain inhibitor, in men with metastatic castration-resistant prostate cancer (mCRPC) with progression after enzalutamide or abiraterone. J. Clin. Oncol. 2015, 33, TPS5072. [Google Scholar] [CrossRef]
- Maurice-Dror, C.; Le Moigne, R.; Vaishampayan, U.; Montgomery, R.B.; Gordon, M.S.; Hong, N.H.; DiMascio, L.; Perabo, F.; Chi, K.N. A phase 1 study to assess the safety, pharmacokinetics, and anti-tumor activity of the androgen receptor n-terminal domain inhibitor epi-506 in patients with metastatic castration-resistant prostate cancer. Investig. New Drugs 2022, 40, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Vaishampayan, U.N.; Gordon, M.S.; Smith, D.C.; Rudsinski, E.; Haas-Amatsaleh, A.D.; Thapar, N.; Perabo, F.; Montgomery, R.B. Efficacy, safety, tolerability, and pharmacokinetics of EPI-506 (ralaniten acetate), a novel androgen receptor (AR) N-terminal domain (NTD) inhibitor, in men with metastatic castration-resistant prostate cancer (mCRPC) progressing after enzalutamide and/or abiraterone. J. Clin. Oncol. 2017, 35, 5032. [Google Scholar] [CrossRef]
- Le Moigne, R.; Hong, N.; Pearson, P.; Lauriault, V.; Banuelos, C.; Mawji, N.; Tam, T.; Wang, J.; Virsik, P.; Andersen, R. 545P Preclinical profile of EPI-7386, a second-generation N-terminal domain androgen receptor inhibitor for the treatment of prostate cancer. J. Ann. Oncol. 2020, 31, S475. [Google Scholar] [CrossRef]
- Moigne, R.L.; Banuelos, C.A.; Mawji, N.R.; Tam, T.; Wang, J.; Jian, K.; Andersen, R.J.; Cesano, A.; Sadar, M.; Zhou, H.-J.; et al. IND candidate EPI-7386 as an N-terminal domain androgen receptor inhibitor in development for the treatment of prostate cancer. J. Clin. Oncol. 2020, 38, 142. [Google Scholar] [CrossRef]
- Ban, F.; Leblanc, E.; Cavga, A.D.; Huang, C.F.; Flory, M.R.; Zhang, F.; Chang, M.E.K.; Morin, H.; Lallous, N.; Singh, K.; et al. Development of an Androgen Receptor Inhibitor Targeting the N-Terminal Domain of Androgen Receptor for Treatment of Castration Resistant Prostate Cancer. Cancers 2021, 13, 3488. [Google Scholar] [CrossRef]
- Hwang, D.J.; He, Y.; Ponnusamy, S.; Mohler, M.L.; Thiyagarajan, T.; McEwan, I.J.; Narayanan, R.; Miller, D.D. New Generation of Selective Androgen Receptor Degraders: Our Initial Design, Synthesis, and Biological Evaluation of New Compounds with Enzalutamide-Resistant Prostate Cancer Activity. J. Med. Chem. 2019, 62, 491–511. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Coss, C.C.; Thiyagarajan, T.; Watts, K.; Hwang, D.J.; He, Y.; Selth, L.A.; McEwan, I.J.; Duke, C.B.; Pagadala, J.; et al. Novel Selective Agents for the Degradation of Androgen Receptor Variants to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 6282–6298. [Google Scholar] [CrossRef]
- Ponnusamy, S.; He, Y.; Hwang, D.J.; Thiyagarajan, T.; Houtman, R.; Bocharova, V.; Sumpter, B.G.; Fernandez, E.; Johnson, D.; Du, Z.; et al. Orally Bioavailable Androgen Receptor Degrader, Potential Next-Generation Therapeutic for Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 6764–6780. [Google Scholar] [CrossRef]
- He, Y.; Hwang, D.J.; Ponnusamy, S.; Thiyagarajan, T.; Mohler, M.L.; Narayanan, R.; Miller, D.D. Pyrazol-1-yl-propanamides as SARD and Pan-Antagonists for the Treatment of Enzalutamide-Resistant Prostate Cancer. J. Med. Chem. 2020, 63, 12642–12665. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hwang, D.J.; Ponnusamy, S.; Thiyagarajan, T.; Mohler, M.L.; Narayanan, R.; Miller, D.D. Exploration and Biological Evaluation of Basic Heteromonocyclic Propanamide Derivatives as SARDs for the Treatment of Enzalutamide-Resistant Prostate Cancer. J. Med. Chem. 2021, 64, 11045–11062. [Google Scholar] [CrossRef] [PubMed]
- Tummala, R.; Lou, W.; Gao, A.C.; Nadiminty, N. Quercetin Targets hnRNPA1 to Overcome Enzalutamide Resistance in Prostate Cancer Cells. Mol. Cancer Ther. 2017, 16, 2770–2779. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.M.; Ning, S.; Yang, J.C.; Lou, W.; Lombard, A.P.; Zhao, J.; Wu, C.Y.; Yu, A.; Evans, C.P.; et al. ARVib suppresses growth of advanced prostate cancer via inhibition of androgen receptor signaling. Oncogene 2021, 40, 5379–5392. [Google Scholar] [CrossRef]
- Ippolito, J.E.; Brandenburg, M.W.; Ge, X.; Crowley, J.R.; Kirmess, K.M.; Som, A.; D’Avignon, D.A.; Arbeit, J.M.; Achilefu, S.; Yarasheski, K.E.; et al. Extracellular pH Modulates Neuroendocrine Prostate Cancer Cell Metabolism and Susceptibility to the Mitochondrial Inhibitor Niclosamide. PLoS ONE 2016, 11, e0159675. [Google Scholar] [CrossRef]
- Chen, B.; Wei, W.; Ma, L.; Yang, B.; Gill, R.M.; Chua, M.S.; Butte, A.J.; So, S. Computational Discovery of Niclosamide Ethanolamine, a Repurposed Drug Candidate That Reduces Growth of Hepatocellular Carcinoma Cells In Vitro and in Mice by Inhibiting Cell Division Cycle 37 Signaling. Gastroenterology 2017, 152, 2022–2036. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S.; et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef]
- Barbosa, E.J.; Lobenberg, R.; de Araujo, G.L.B.; Bou-Chacra, N.A. Niclosamide repositioning for treating cancer: Challenges and nano-based drug delivery opportunities. Eur. J. Pharm. Biopharm. 2019, 141, 58–69. [Google Scholar] [CrossRef]
- Parikh, M.; Liu, C.; Wu, C.Y.; Evans, C.P.; Dall’Era, M.; Robles, D.; Lara, P.N.; Agarwal, N.; Gao, A.C.; Pan, C.X. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci. Rep. 2021, 11, 6377. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Mirshahi, F.; Aqbi, H.F.; Isbell, M.; Manjili, S.H.; Guo, C.; Saneshaw, M.; Bandyopadhyay, D.; Dozmorov, M.; Khosla, A.; Wack, K.; et al. Distinct hepatic immunological patterns are associated with the progression or inhibition of hepatocellular carcinoma. Cell Rep. 2022, 38, 110454. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montgomery, E.J.; Xing, E.; Campbell, M.J.; Li, P.-K.; Blachly, J.S.; Tsung, A.; Coss, C.C. Constitutively Active Androgen Receptor in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 13768. https://doi.org/10.3390/ijms232213768
Montgomery EJ, Xing E, Campbell MJ, Li P-K, Blachly JS, Tsung A, Coss CC. Constitutively Active Androgen Receptor in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(22):13768. https://doi.org/10.3390/ijms232213768
Chicago/Turabian StyleMontgomery, Emma J., Enming Xing, Moray J. Campbell, Pui-Kai Li, James S. Blachly, Allan Tsung, and Christopher C. Coss. 2022. "Constitutively Active Androgen Receptor in Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 22: 13768. https://doi.org/10.3390/ijms232213768
APA StyleMontgomery, E. J., Xing, E., Campbell, M. J., Li, P.-K., Blachly, J. S., Tsung, A., & Coss, C. C. (2022). Constitutively Active Androgen Receptor in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 23(22), 13768. https://doi.org/10.3390/ijms232213768