

Potential of Heterogeneous Compounds as Antidepressants: A Narrative Review

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neurological Alterations in Depression
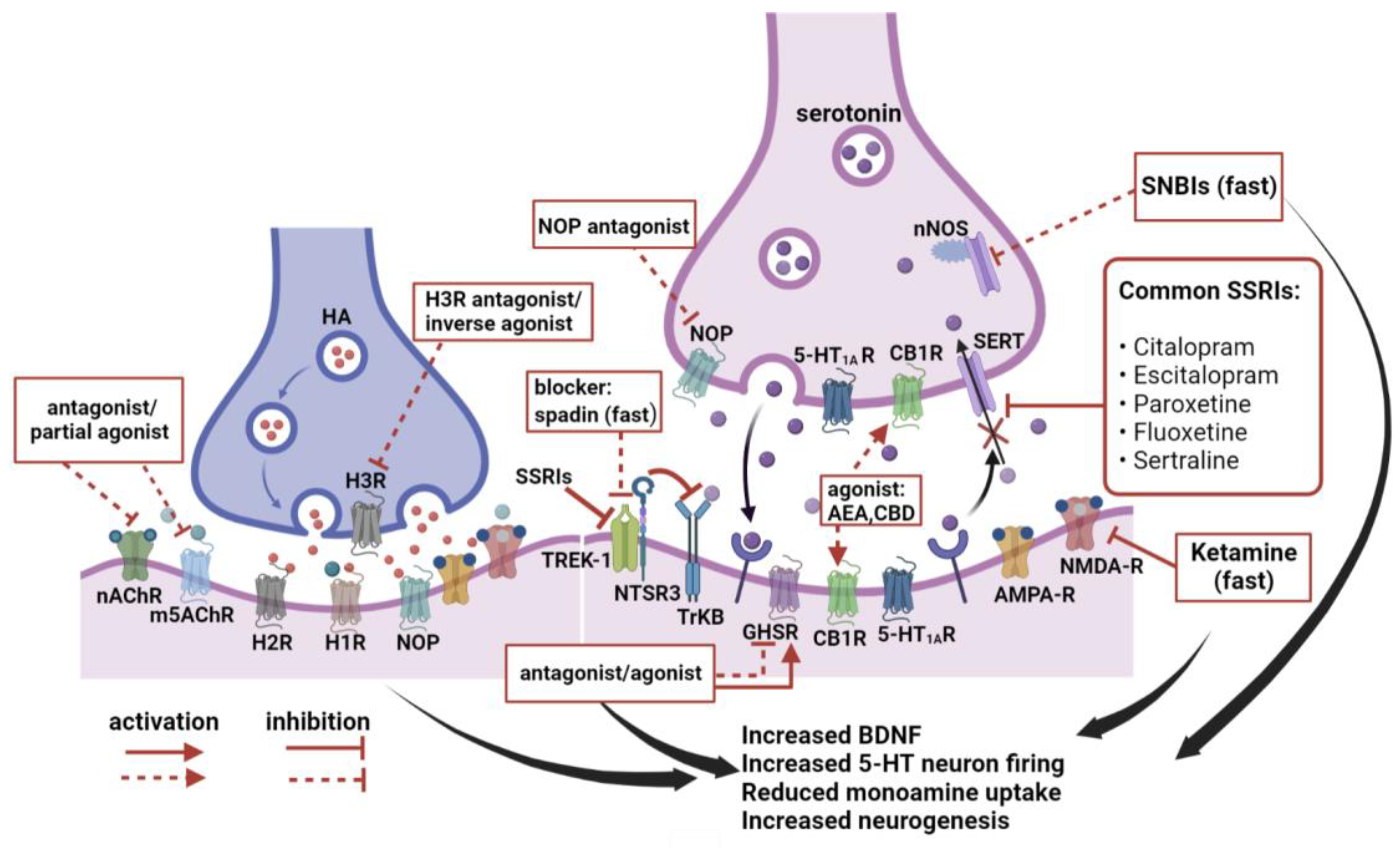
3. New Hotspots in Antidepressant Research
3.1. Histamine
3.2. Acetylcholine
3.3. Thyroid Hormones
3.4. Brain-Derived Peptides
3.4.1. Neurotrophic Factors
3.4.2. Sortilin and TWIK-Related K+ Channel 1 (TREK-1)
3.4.3. Sortilin-Derived Propeptide (PE) and Spadin
3.4.4. Opioid Peptides
3.4.5. Oxytocin and Arginine Vasopressin
3.5. Non-Brain-Derived Peptides
3.5.1. Adiponectin and Leptin
3.5.2. Ghrelin
3.6. Cannabinoids
3.7. Endogenous Digitalis-Like Compounds
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Witkin, J.M.; Martin, A.E.; Golani, L.K.; Xu, N.Z.; Smith, J.L. Rapid-acting antidepressants. Adv. Pharmacol. 2019, 86, 47–96. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Gardier, A.M. Fast-acting antidepressant activity of ketamine: Highlights on brain serotonin, glutamate, and GABA neurotransmission in preclinical studies. Pharmacol. Ther. 2019, 199, 58–90. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R.; Benkelfat, C.; Descarries, L. The neurobiology of depression--revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2378–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, R. Neurobiology of major depressive disorder. Neural Plast. 2013, 2013, 873278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, S. Oxidative stress and psychological disorders. Curr. Neuropharmacol. 2014, 12, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Hodes, G.E.; Menard, C.; Russo, S.J. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol. Stress 2016, 4, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]
- Salloum, N.C.; Fava, M.; Hock, R.S.; Freeman, M.P.; Flynn, M.; Hoeppner, B.; Cusin, C.; Iosifescu, D.V.; Trivedi, M.H.; Sanacora, G.; et al. Time to relapse after a single administration of intravenous ketamine augmentation in unipolar treatment-resistant depression. J. Affect. Disord. 2020, 260, 131–139. [Google Scholar] [CrossRef]
- Gold, P.W. The organization of the stress system and its dysregulation in depressive illness. Mol. Psychiatry 2015, 20, 32–47. [Google Scholar] [CrossRef]
- Gold, P.W. Endocrine Factors in Key Structural and Intracellular Changes in Depression. Trends Endocrinol. Metab. 2021, 32, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Price, R.B.; Duman, R. Neuroplasticity in cognitive and psychological mechanisms of depression: An integrative model. Mol. Psychiatry 2020, 25, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Scheinost, D.; Finnema, S.J.; Naganawa, M.; Davis, M.T.; DellaGioia, N.; Nabulsi, N.; Matuskey, D.; Angarita, G.A.; Pietrzak, R.H.; et al. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 2019, 10, 1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Irwin, M.R.; Eisenberger, N.I.; Lamkin, D.M.; Cole, S.W. Transcriptomic predictors of inflammation-induced depressed mood. Neuropsychopharmacology 2019, 44, 923–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyra, E.S.N.M.; Lam, M.P.; Soares, C.N.; Munoz, D.P.; Milev, R.; De Felice, F.G. Insulin Resistance as a Shared Pathogenic Mechanism Between Depression and Type 2 Diabetes. Front. Psychiatry 2019, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- de Kloet, E.R.; Otte, C.; Kumsta, R.; Kok, L.; Hillegers, M.H.; Hasselmann, H.; Kliegel, D.; Joels, M. Stress and Depression: A Crucial Role of the Mineralocorticoid Receptor. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef]
- Cooke, G.E.; Mullally, S.; Correia, N.; O’Mara, S.M.; Gibney, J. Hippocampal Volume Is Decreased in Adults with Hypothyroidism. Thyroid 2013, 24, 433–440. [Google Scholar] [CrossRef]
- Wright, C.; Shin, J.H.; Rajpurohit, A.; Deep-Soboslay, A.; Collado-Torres, L.; Brandon, N.J.; Hyde, T.M.; Kleinman, J.E.; Jaffe, A.E.; Cross, A.J.; et al. Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry 2017, 7, e1126. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Taguchi, Y.; Hayashi, H.; Tanaka, J.; Shiosaka, S.; Tohyama, M.; Kubota, H.; Terano, Y.; Wada, H. Evidence for the presence of a histaminergic neuron system in the rat brain: An immunohistochemical analysis. Neurosci. Lett. 1983, 39, 249–254. [Google Scholar] [CrossRef]
- Iida, T.; Yanai, K.; Yoshikawa, T. Histamine and Microglia. Curr. Top. Behav. Neurosci. 2022, 59, 241–259. [Google Scholar] [CrossRef]
- Carthy, E.; Ellender, T. Histamine, Neuroinflammation and Neurodevelopment: A Review. Front. Neurosci. 2021, 15, 680214. [Google Scholar] [CrossRef] [PubMed]
- Jutel, M.; Akdis, M.; Akdis, C.A. Histamine, histamine receptors and their role in immune pathology. Clin. Exp. Allergy 2009, 39, 1786–1800. [Google Scholar] [CrossRef]
- Sadek, B.; Saad, A.; Sadeq, A.; Jalal, F.; Stark, H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav. Brain Res. 2016, 312, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Femenía, T.; Magara, S.; DuPont, C.M.; Lindskog, M. Hippocampal-Dependent Antidepressant Action of the H3 Receptor Antagonist Clobenpropit in a Rat Model of Depression. Int. J. Neuropsychopharmacol. 2015, 18, pyv032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, T.; Yoshikawa, T.; Kárpáti, A.; Matsuzawa, T.; Kitano, H.; Mogi, A.; Harada, R.; Naganuma, F.; Nakamura, T.; Yanai, K. JNJ10181457, a histamine H3 receptor inverse agonist, regulates in vivo microglial functions and improves depression-like behaviours in mice. Biochem. Biophys. Res. Commun. 2017, 488, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dogra, S.; Sona, C.; Umrao, D.; Rashid, M.; Singh, S.K.; Wahajuddin, M.; Yadav, P.N. Chronic histamine 3 receptor antagonism alleviates depression like conditions in mice via modulation of brain-derived neurotrophic factor and hypothalamus-pituitary adrenal axis. Psychoneuroendocrinology 2019, 101, 128–137. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Zhong, S.; Dubiel, M.; Satała, G.; Sadek, B.; Stark, H. The Novel Pimavanserin Derivative ST-2300 with Histamine H(3) Receptor Affinity Shows Reduced 5-HT(2A) Binding, but Maintains Antidepressant- and Anxiolytic-like Properties in Mice. Biomolecules 2022, 12, 683. [Google Scholar] [CrossRef]
- Su, W.J.; Zhang, T.; Jiang, C.L.; Wang, W. Clemastine Alleviates Depressive-Like Behavior Through Reversing the Imbalance of Microglia-Related Pro-inflammatory State in Mouse Hippocampus. Front. Cell. Neurosci. 2018, 12, 412. [Google Scholar] [CrossRef] [Green Version]
- Yeni, Y.; Cakir, Z.; Hacimuftuoglu, A.; Taghizadehghalehjoughi, A.; Okkay, U.; Genc, S.; Yildirim, S.; Saglam, Y.S.; Calina, D.; Tsatsakis, A.; et al. A Selective Histamine H4 Receptor Antagonist, JNJ7777120, Role on glutamate Transporter Activity in Chronic Depression. J. Pers. Med. 2022, 12, 246. [Google Scholar] [CrossRef]
- Sanna, M.D.; Ghelardini, C.; Thurmond, R.L.; Masini, E.; Galeotti, N. Behavioural phenotype of histamine H(4) receptor knockout mice: Focus on central neuronal functions. Neuropharmacology 2017, 114, 48–57. [Google Scholar] [CrossRef]
- Zaborszky, L.; Hoemke, L.; Mohlberg, H.; Schleicher, A.; Amunts, K.; Zilles, K. Stereotaxic probabilistic maps of the magnocellular cell groups in human basal forebrain. NeuroImage 2008, 42, 1127–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apicella, P. Leading tonically active neurons of the striatum from reward detection to context recognition. Trends Neurosci. 2007, 30, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Risch, S.C.; Cohen, R.M.; Janowsky, D.S.; Kalin, N.H.; Murphy, D.L. Mood and behavioral effects of physostigmine on humans are accompanied by elevations in plasma beta-endorphin and cortisol. Science 1980, 209, 1545–1546. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Obayemi, A.; Wigestrand, M.B.; Fote, G.M.; Calarco, C.A.; Li, A.M.; Picciotto, M.R. Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and depression-like behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 3573–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetlin, V.I. Acetylcholine and Acetylcholine Receptors: Textbook Knowledge and New Data. Biomolecules 2020, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- al’Absi, M.; Carr, S.B.; Bongard, S. Anger and psychobiological changes during smoking abstinence and in response to acute stress: Prediction of smoking relapse. Int. J. Psychophysiol. 2007, 66, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Mineur, Y.S.; Eibl, C.; Young, G.; Kochevar, C.; Papke, R.L.; Gündisch, D.; Picciotto, M.R. Cytisine-based nicotinic partial agonists as novel antidepressant compounds. J. Pharmacol. Exp. Ther. 2009, 329, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Mineur, Y.S.; Einstein, E.B.; Seymour, P.A.; Coe, J.W.; O’Neill, B.T.; Rollema, H.; Picciotto, M.R. α4β2 nicotinic acetylcholine receptor partial agonists with low intrinsic efficacy have antidepressant-like properties. Behav. Pharmacol. 2011, 22, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Shytle, R.D.; Silver, A.A.; Lukas, R.J.; Newman, M.B.; Sheehan, D.V.; Sanberg, P.R. Nicotinic acetylcholine receptors as targets for antidepressants. Mol. Psychiatry 2002, 7, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Mineur, Y.S.; Cahuzac, E.L.; Mose, T.N.; Bentham, M.P.; Plantenga, M.E.; Thompson, D.C.; Picciotto, M.R. Interaction between noradrenergic and cholinergic signaling in amygdala regulates anxiety- and depression-related behaviors in mice. Neuropsychopharmacology 2018, 43, 2118–2125. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Santamaría, A.; Escobar, L.; Arias, H.R. The β4 nicotinic receptor subunit modulates the chronic antidepressant effect mediated by bupropion. Neurosci. Lett. 2013, 555, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, J.T.; Redrobe, J.P.; Nielsen, E.Ø.; Christensen, J.K.; Olsen, G.M.; Peters, D. A combined α7 nicotinic acetylcholine receptor agonist and monoamine reuptake inhibitor, NS9775, represents a novel profile with potential benefits in emotional and cognitive disturbances. Neuropharmacology 2013, 73, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Friedman Allyson, K.; Walsh Jessica, J.; Juarez, B.; Ku Stacy, M.; Chaudhury, D.; Wang, J.; Li, X.; Dietz David, M.; Pan, N.; Vialou Vincent, F.; et al. Enhancing Depression Mechanisms in Midbrain Dopamine Neurons Achieves Homeostatic Resilience. Science 2014, 344, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, K.M.; Nunes, E.; Hughley, S.; Addy, N.A. Ventral tegmental area muscarinic receptors modulate depression and anxiety-related behaviors in rats. Neurosci. Lett. 2016, 616, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Nunes, E.J.; Rupprecht, L.E.; Foster, D.J.; Lindsley, C.W.; Conn, P.J.; Addy, N.A. Examining the role of muscarinic M5 receptors in VTA cholinergic modulation of depressive-like and anxiety-related behaviors in rats. Neuropharmacology 2020, 171, 108089. [Google Scholar] [CrossRef]
- Foster, D.J.; Gentry, P.R.; Lizardi-Ortiz, J.E.; Bridges, T.M.; Wood, M.R.; Niswender, C.M.; Sulzer, D.; Lindsley, C.W.; Xiang, Z.; Conn, P.J. M5 receptor activation produces opposing physiological outcomes in dopamine neurons depending on the receptor’s location. J. Neurosci. 2014, 34, 3253–3262. [Google Scholar] [CrossRef] [Green Version]
- Mineur, Y.S.; Fote, G.M.; Blakeman, S.; Cahuzac, E.L.; Newbold, S.A.; Picciotto, M.R. Multiple Nicotinic Acetylcholine Receptor Subtypes in the Mouse Amygdala Regulate Affective Behaviors and Response to Social Stress. Neuropsychopharmacology 2016, 41, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Warner-Schmidt, J.L.; Schmidt, E.F.; Marshall, J.J.; Rubin, A.J.; Arango-Lievano, M.; Kaplitt, M.G.; Ibañez-Tallon, I.; Heintz, N.; Greengard, P. Cholinergic interneurons in the nucleus accumbens regulate depression-like behavior. Proc. Natl. Acad. Sci. USA 2012, 109, 11360–11365. [Google Scholar] [CrossRef] [Green Version]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Noda, M. Thyroid Hormone in the CNS: Contribution of Neuron-Glia Interaction. Vitam. Horm. 2018, 106, 313–331. [Google Scholar] [CrossRef]
- Remaud, S.; Gothié, J.D.; Morvan-Dubois, G.; Demeneix, B.A. Thyroid hormone signaling and adult neurogenesis in mammals. Front. Endocrinol. 2014, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Desouza, L.A.; Ladiwala, U.; Daniel, S.M.; Agashe, S.; Vaidya, R.A.; Vaidya, V.A. Thyroid hormone regulates hippocampal neurogenesis in the adult rat brain. Mol. Cell. Neurosci. 2005, 29, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Westerholz, S.; de Lima, A.D.; Voigt, T. Thyroid hormone-dependent development of early cortical networks: Temporal specificity and the contribution of trkB and mTOR pathways. Front. Cell. Neurosci. 2013, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berbel, P.; Marco, P.; Cerezo, J.R.; DeFelipe, J. Distribution of parvalbumin immunoreactivity in the neocortex of hypothyroid adult rats. Neurosci. Lett. 1996, 204, 65–68. [Google Scholar] [CrossRef]
- Bauer, M.; London, E.D.; Silverman, D.H.; Rasgon, N.; Kirchheiner, J.; Whybrow, P.C. Thyroid, brain and mood modulation in affective disorder: Insights from molecular research and functional brain imaging. Pharmacopsychiatry 2003, 36 (Suppl. S3), S215–S221. [Google Scholar] [CrossRef] [PubMed]
- Montero-Pedrazuela, A.; Fernández-Lamo, I.; Alieva, M.; Pereda-Pérez, I.; Venero, C.; Guadaño-Ferraz, A. Adult-onset hypothyroidism enhances fear memory and upregulates mineralocorticoid and glucocorticoid receptors in the amygdala. PLoS ONE 2011, 6, e26582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, B. Postpartum depression and thyroid antibody status. Thyroid 1999, 9, 699–703. [Google Scholar] [CrossRef]
- Ittermann, T.; Völzke, H.; Baumeister, S.E.; Appel, K.; Grabe, H.J. Diagnosed thyroid disorders are associated with depression and anxiety. Soc. Psychiatry Psychiatr. Epidemiol. 2015, 50, 1417–1425. [Google Scholar] [CrossRef]
- Manzano, J.; Bernal, J.; Morte, B. Influence of thyroid hormones on maturation of rat cerebellar astrocytes. Int. J. Dev. Neurosci. 2007, 25, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Heinz, A.; Whybrow, P.C. Thyroid hormones, serotonin and mood: Of synergy and significance in the adult brain. Mol. Psychiatry 2002, 7, 140–156. [Google Scholar] [CrossRef]
- Zhao, T.; Chen, B.M.; Zhao, X.M.; Shan, Z.Y. Subclinical hypothyroidism and depression: A meta-analysis. Transl. Psychiatry 2018, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.; Wang, J.; Yang, L.; Ding, X.; Zhong, Y.; Pan, J.; Yang, H.; Mu, L.; Chen, X.; Chen, Z. Subclinical Hypothyroidism and Depression: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2019, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bothwell, M. NGF, BDNF, NT3, and NT4. Handb. Exp. Pharmacol. 2014, 220, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Pareek, S.; Chrétien, M.; Murphy, R.A. Cellular processing of the neurotrophin precursors of NT3 and BDNF by the mammalian proprotein convertases. FEBS Lett. 1996, 379, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, B.L. Deciphering proneurotrophin actions. Handb. Exp. Pharmacol. 2014, 220, 17–32. [Google Scholar] [CrossRef]
- Deinhardt, K.; Chao, M.V. Trk receptors. Handb. Exp. Pharmacol. 2014, 220, 103–119. [Google Scholar] [CrossRef]
- Kraemer, B.R.; Yoon, S.O.; Carter, B.D. The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb. Exp. Pharmacol. 2014, 220, 121–164. [Google Scholar] [CrossRef]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Zarate, C., Jr. Antidepressant actions of ketamine: From molecular mechanisms to clinical practice. Curr. Opin. Neurobiol. 2015, 30, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Rantamäki, T.; Hendolin, P.; Kankaanpää, A.; Mijatovic, J.; Piepponen, P.; Domenici, E.; Chao, M.V.; Männistö, P.T.; Castrén, E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology 2007, 32, 2152–2162. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. Ketamine and rapid-acting antidepressants: A window into a new neurobiology for mood disorder therapeutics. Annu. Rev. Med. 2015, 66, 509–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshaw, B.A.; Malberg, J.E.; Lucki, I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005, 1037, 204–208. [Google Scholar] [CrossRef]
- Lepack, A.E.; Fuchikami, M.; Dwyer, J.M.; Banasr, M.; Duman, R.S. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 2014, 18, pyu033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, J.S.; Autio, H.; Vesa, L.; Antila, H.; Lindemann, L.; Hoener, M.C.; Skolnick, P.; Rantamäki, T.; Castrén, E. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf⁺/⁻ heterozygous null mice. Neuropharmacology 2012, 62, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Eisch, A.J.; Bolaños, C.A.; de Wit, J.; Simonak, R.D.; Pudiak, C.M.; Barrot, M.; Verhaagen, J.; Nestler, E.J. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: A role in depression. Biol. Psychiatry 2003, 54, 994–1005. [Google Scholar] [CrossRef]
- Petryshen, T.L.; Sabeti, P.C.; Aldinger, K.A.; Fry, B.; Fan, J.B.; Schaffner, S.F.; Waggoner, S.G.; Tahl, A.R.; Sklar, P. Population genetic study of the brain-derived neurotrophic factor (BDNF) gene. Mol. Psychiatry 2010, 15, 810–815. [Google Scholar] [CrossRef] [Green Version]
- Covaceuszach, S.; Peche, L.Y.; Konarev, P.V.; Grdadolnik, J.; Cattaneo, A.; Lamba, D. Untangling the Conformational Plasticity of V66M Human proBDNF Polymorphism as a Modifier of Psychiatric Disorder Susceptibility. Int. J. Mol. Sci. 2022, 23, 6596. [Google Scholar] [CrossRef]
- Tripp, A.; Oh, H.; Guilloux, J.P.; Martinowich, K.; Lewis, D.A.; Sibille, E. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 2012, 169, 1194–1202. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Zhang, H.; Mondal, A.C.; Roberts, R.C.; Conley, R.R.; Pandey, G.N. Neurotrophin receptor activation and expression in human postmortem brain: Effect of suicide. Biol. Psychiatry 2009, 65, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinowich, K.; Schloesser, R.J.; Lu, Y.; Jimenez, D.V.; Paredes, D.; Greene, J.S.; Greig, N.H.; Manji, H.K.; Lu, B. Roles of p75(NTR), long-term depression, and cholinergic transmission in anxiety and acute stress coping. Biol. Psychiatry 2012, 71, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrén, E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry 2013, 70, 983–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrén, E.; Hen, R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013, 36, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Nykjaer, A.; Willnow, T.E. Sortilin: A receptor to regulate neuronal viability and function. Trends Neurosci. 2012, 35, 261–270. [Google Scholar] [CrossRef]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef]
- Massa, F.; Devader, C.; Béraud-Dufour, S.; Brau, F.; Coppola, T.; Mazella, J. Focal adhesion kinase dependent activation of the PI3 kinase pathway by the functional soluble form of neurotensin receptor-3 in HT29 cells. Int. J. Biochem. Cell Biol. 2013, 45, 952–959. [Google Scholar] [CrossRef]
- Buttenschøn, H.N.; Demontis, D.; Kaas, M.; Elfving, B.; Mølgaard, S.; Gustafsen, C.; Kaerlev, L.; Petersen, C.M.; Børglum, A.D.; Mors, O.; et al. Increased serum levels of sortilin are associated with depression and correlated with BDNF and VEGF. Transl. Psychiatry 2015, 5, e677. [Google Scholar] [CrossRef] [Green Version]
- Stelzhammer, V.; Guest, P.C.; Rothermundt, M.; Sondermann, C.; Michael, N.; Schwarz, E.; Rahmoune, H.; Bahn, S. Electroconvulsive therapy exerts mainly acute molecular changes in serum of major depressive disorder patients. Eur. Neuropsychopharmacol. 2013, 23, 1199–1207. [Google Scholar] [CrossRef]
- Buttenschøn, H.N.; Nielsen, M.; Glerup, S.; Mors, O. Investigation of serum levels of sortilin in response to antidepressant treatment. Acta Neuropsychiatr. 2018, 30, 111–116. [Google Scholar] [CrossRef]
- Hervieu, G.J.; Cluderay, J.E.; Gray, C.W.; Green, P.J.; Ranson, J.L.; Randall, A.D.; Meadows, H.J. Distribution and expression of TREK-1, a two-pore-domain potassium channel, in the adult rat CNS. Neuroscience 2001, 103, 899–919. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Rennie, G.; Chapman, C.G.; Meadows, H.; Duckworth, M.D.; Kelsell, R.E.; Gloger, I.I.; Pangalos, M.N. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res. Mol. Brain Res. 2001, 86, 101–114. [Google Scholar] [CrossRef]
- Kennard, L.E.; Chumbley, J.R.; Ranatunga, K.M.; Armstrong, S.J.; Veale, E.L.; Mathie, A. Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br. J. Pharmacol. 2005, 144, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Lee, D.K.; Hong, S.G.; Han, J.; Kang, D. Activation of TREK-1, but Not TREK-2, Channel by Mood Stabilizers. Int. J. Mol. Sci. 2017, 18, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallee, N.; Lambrechts, K.; De Maistre, S.; Royal, P.; Mazella, J.; Borsotto, M.; Heurteaux, C.; Abraini, J.; Risso, J.J.; Blatteau, J.E. Fluoxetine Protection in Decompression Sickness in Mice is Enhanced by Blocking TREK-1 Potassium Channel with the “spadin” Antidepressant. Front. Physiol. 2016, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Giannoni-Guzman, M.A.; Kamitakahara, A.; Magalong, V.; Levitt, P.; McMahon, D.G. Circadian photoperiod alters TREK-1 channel function and expression in dorsal raphe serotonergic neurons via melatonin receptor 1 signaling. J. Pineal Res. 2021, 70, e12705. [Google Scholar] [CrossRef]
- Djillani, A.; Pietri, M.; Moreno, S.; Heurteaux, C.; Mazella, J.; Borsotto, M. Shortened Spadin Analogs Display Better TREK-1 Inhibition, In Vivo Stability and Antidepressant Activity. Front. Pharmacol. 2017, 8, 643. [Google Scholar] [CrossRef] [Green Version]
- Dillon, D.G.; Bogdan, R.; Fagerness, J.; Holmes, A.J.; Perlis, R.H.; Pizzagalli, D.A. Variation in TREK1 gene linked to depression-resistant phenotype is associated with potentiated neural responses to rewards in humans. Hum. Brain Mapp. 2010, 31, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Moreno, S.; Devader, C.M.; Pietri, M.; Borsotto, M.; Heurteaux, C.; Mazella, J. Altered Trek-1 Function in Sortilin Deficient Mice Results in Decreased Depressive-Like Behavior. Front. Pharmacol. 2018, 9, 863. [Google Scholar] [CrossRef]
- Munck Petersen, C.; Nielsen, M.S.; Jacobsen, C.; Tauris, J.; Jacobsen, L.; Gliemann, J.; Moestrup, S.K.; Madsen, P. Propeptide cleavage conditions sortilin/neurotensin receptor-3 for ligand binding. Embo J. 1999, 18, 595–604. [Google Scholar] [CrossRef]
- Mazella, J.; Pétrault, O.; Lucas, G.; Deval, E.; Béraud-Dufour, S.; Gandin, C.; El-Yacoubi, M.; Widmann, C.; Guyon, A.; Chevet, E.; et al. Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: A new concept in the antidepressant drug design. PLoS Biol. 2010, 8, e1000355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsotto, M.; Veyssiere, J.; Moha Ou Maati, H.; Devader, C.; Mazella, J.; Heurteaux, C. Targeting two-pore domain K(+) channels TREK-1 and TASK-3 for the treatment of depression: A new therapeutic concept. Br. J. Pharmacol. 2015, 172, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djillani, A.; Pietri, M.; Mazella, J.; Heurteaux, C.; Borsotto, M. Fighting against depression with TREK-1 blockers: Past and future. A focus on spadin. Pharmacol. Ther. 2019, 194, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Heurteaux, C.; Lucas, G.; Guy, N.; El Yacoubi, M.; Thümmler, S.; Peng, X.D.; Noble, F.; Blondeau, N.; Widmann, C.; Borsotto, M.; et al. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat. Neurosci. 2006, 9, 1134–1141. [Google Scholar] [CrossRef]
- Devader, C.; Khayachi, A.; Veyssière, J.; Moha Ou Maati, H.; Roulot, M.; Moreno, S.; Borsotto, M.; Martin, S.; Heurteaux, C.; Mazella, J. In vitro and in vivo regulation of synaptogenesis by the novel antidepressant spadin. Br. J. Pharmacol. 2015, 172, 2604–2617. [Google Scholar] [CrossRef] [Green Version]
- Moha Ou Maati, H.; Veyssiere, J.; Labbal, F.; Coppola, T.; Gandin, C.; Widmann, C.; Mazella, J.; Heurteaux, C.; Borsotto, M. Spadin as a new antidepressant: Absence of TREK-1-related side effects. Neuropharmacology 2012, 62, 278–288. [Google Scholar] [CrossRef]
- Devader, C.; Roulot, M.; Moréno, S.; Minelli, A.; Bortolomasi, M.; Congiu, C.; Gennarelli, M.; Borsotto, M.; Heurteaux, C.; Mazella, J. Serum sortilin-derived propeptides concentrations are decreased in major depressive disorder patients. J. Affect. Disord. 2017, 208, 443–447. [Google Scholar] [CrossRef]
- Pietri, M.; Djillani, A.; Mazella, J.; Borsotto, M.; Heurteaux, C. First evidence of protective effects on stroke recovery and post-stroke depression induced by sortilin-derived peptides. Neuropharmacology 2019, 158, 107715. [Google Scholar] [CrossRef]
- Crowley, N.A.; Kash, T.L. Kappa opioid receptor signaling in the brain: Circuitry and implications for treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 62, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Carlezon, W.A., Jr.; Krystal, A.D. Kappa-Opioid Antagonists for Psychiatric Disorders: From Bench to Clinical Trials. Depress. Anxiety 2016, 33, 895–906. [Google Scholar] [CrossRef]
- Spetea, M.; Schmidhammer, H. Kappa Opioid Receptor Ligands and Pharmacology: Diphenethylamines, a Class of Structurally Distinct, Selective Kappa Opioid Ligands. Handb. Exp. Pharmacol. 2022, 271, 163–195. [Google Scholar] [CrossRef] [PubMed]
- Toll, L.; Bruchas, M.R.; Calo, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavioli, E.C.; Calo, G. Nociceptin/orphanin FQ receptor antagonists as innovative antidepressant drugs. Pharmacol. Ther. 2013, 140, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Holanda, V.A.D.; Pacifico, S.; Azevedo Neto, J.; Finetti, L.; Lobão-Soares, B.; Calo, G.; Gavioli, E.C.; Ruzza, C. Modulation of the NOP receptor signaling affects resilience to acute stress. J. Psychopharmacol. 2019, 33, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Ma, Z.; Thakkar, M.M.; McCarley, R.W.; Auerbach, S.B. Nociceptin/orphanin FQ decreases serotonin efflux in the rat brain but in contrast to a kappa-opioid has no antagonistic effect on mu-opioid-induced increases in serotonin efflux. Neuroscience 2007, 147, 106–116. [Google Scholar] [CrossRef]
- Holanda, V.A.D.; Santos, W.B.; Asth, L.; Guerrini, R.; Calo, G.; Ruzza, C.; Gavioli, E.C. NOP agonists prevent the antidepressant-like effects of nortriptyline and fluoxetine but not R-ketamine. Psychopharmacology 2018, 235, 3093–3102. [Google Scholar] [CrossRef]
- Wang, L.N.; Liu, L.F.; Zhang, J.X.; Zhao, G.F. Plasma levels of nociceptin/orphanin FQ in patients with bipolar disorders and health adults. Zhonghua Yi Xue Za Zhi 2009, 89, 916–918. [Google Scholar]
- Post, A.; Smart, T.S.; Krikke-Workel, J.; Dawson, G.R.; Harmer, C.J.; Browning, M.; Jackson, K.; Kakar, R.; Mohs, R.; Statnick, M.; et al. A Selective Nociceptin Receptor Antagonist to Treat Depression: Evidence from Preclinical and Clinical Studies. Neuropsychopharmacology 2016, 41, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, R.; Neumann, I.D. Vasopressin and oxytocin release within the brain: A dynamic concept of multiple and variable modes of neuropeptide communication. Front. Neuroendocrinol. 2004, 25, 150–176. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A.; Domes, G.; Kirsch, P.; Heinrichs, M. Oxytocin and vasopressin in the human brain: Social neuropeptides for translational medicine. Nat. Rev. Neurosci. 2011, 12, 524–538. [Google Scholar] [CrossRef]
- Ring, R.H.; Schechter, L.E.; Leonard, S.K.; Dwyer, J.M.; Platt, B.J.; Graf, R.; Grauer, S.; Pulicicchio, C.; Resnick, L.; Rahman, Z.; et al. Receptor and behavioral pharmacology of WAY-267464, a non-peptide oxytocin receptor agonist. Neuropharmacology 2010, 58, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Thul, T.A.; Corwin, E.J.; Carlson, N.S.; Brennan, P.A.; Young, L.J. Oxytocin and postpartum depression: A systematic review. Psychoneuroendocrinology 2020, 120, 104793. [Google Scholar] [CrossRef] [PubMed]
- Emiliano, A.B.; Cruz, T.; Pannoni, V.; Fudge, J.L. The interface of oxytocin-labeled cells and serotonin transporter-containing fibers in the primate hypothalamus: A substrate for SSRIs therapeutic effects? Neuropsychopharmacology 2007, 32, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Keck, M.E.; Welt, T.; Müller, M.B.; Uhr, M.; Ohl, F.; Wigger, A.; Toschi, N.; Holsboer, F.; Landgraf, R. Reduction of hypothalamic vasopressinergic hyperdrive contributes to clinically relevant behavioral and neuroendocrine effects of chronic paroxetine treatment in a psychopathological rat model. Neuropsychopharmacology 2003, 28, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Bao, A.M.; Swaab, D.F. Corticotropin-releasing hormone and arginine vasopressin in depression focus on the human postmortem hypothalamus. Vitam. Horm. 2010, 82, 339–365. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Liu, F. Transcriptional and post-translational regulation of adiponectin. Biochem. J. 2009, 425, 41–52. [Google Scholar] [CrossRef]
- Yau, S.Y.; Li, A.; Hoo, R.L.; Ching, Y.P.; Christie, B.R.; Lee, T.M.; Xu, A.; So, K.F. Physical exercise-induced hippocampal neurogenesis and antidepressant effects are mediated by the adipocyte hormone adiponectin. Proc. Natl. Acad. Sci. USA 2014, 111, 15810–15815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, X.; Wang, B.; Garza, J.C.; Fang, X.; Wang, J.; Scherer, P.E.; Brenner, R.; Zhang, W.; Lu, X.Y. Adiponectin regulates contextual fear extinction and intrinsic excitability of dentate gyrus granule neurons through AdipoR2 receptors. Mol. Psychiatry 2017, 22, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, X.; Lu, X.Y. Adiponectin Exerts Neurotrophic Effects on Dendritic Arborization, Spinogenesis, and Neurogenesis of the Dentate Gyrus of Male Mice. Endocrinology 2016, 157, 2853–2869. [Google Scholar] [CrossRef] [Green Version]
- Narita, K.; Murata, T.; Takahashi, T.; Kosaka, H.; Omata, N.; Wada, Y. Plasma levels of adiponectin and tumor necrosis factor-alpha in patients with remitted major depression receiving long-term maintenance antidepressant therapy. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Li, C.; Lei, Y.; Xu, S.; Zhao, D.; Lu, X.Y. Role of the adipose PPARγ-adiponectin axis in susceptibility to stress and depression/anxiety-related behaviors. Mol. Psychiatry 2017, 22, 1056–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, J.; Sun, L.; Wang, J.; Sun, F.; Wang, W.; Wang, D.; Fan, X.; Liu, D.; Xu, Z.; Qiu, C.; et al. Role of Adiponectin-Notch pathway in cognitive dysfunction associated with depression and in the therapeutic effect of physical exercise. Aging Cell 2021, 20, e13387. [Google Scholar] [CrossRef] [PubMed]
- Farr, O.M.; Tsoukas, M.A.; Mantzoros, C.S. Leptin and the brain: Influences on brain development, cognitive functioning and psychiatric disorders. Metabolism 2015, 64, 114–130. [Google Scholar] [CrossRef]
- Bouret, S.G. Neurodevelopmental actions of leptin. Brain Res. 2010, 1350, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Milaneschi, Y.; Lamers, F.; Bot, M.; Drent, M.L.; Penninx, B.W. Leptin Dysregulation Is Specifically Associated With Major Depression With Atypical Features: Evidence for a Mechanism Connecting Obesity and Depression. Biol. Psychiatry 2017, 81, 807–814. [Google Scholar] [CrossRef]
- Ambrus, L.; Westling, S. Leptin, Anxiety Symptoms, and Hypothalamic-Pituitary-Adrenal Axis Activity among Drug-Free, Female Suicide Attempters. Neuropsychobiology 2019, 78, 145–152. [Google Scholar] [CrossRef]
- Andrews, Z.B. Ghrelin: What’s the function? J. Neuroendocrinol. 2019, 31, e12772. [Google Scholar] [CrossRef]
- Perello, M.; Cabral, A.; Cornejo, M.P.; De Francesco, P.N.; Fernandez, G.; Uriarte, M. Brain accessibility delineates the central effects of circulating ghrelin. J. Neuroendocrinol. 2019, 31, e12677. [Google Scholar] [CrossRef]
- Li, N.; Xiao, K.; Mi, X.; Li, N.; Guo, L.; Wang, X.; Sun, Y.; Li, G.D.; Zhou, Y. Ghrelin signaling in dCA1 suppresses neuronal excitability and impairs memory acquisition via PI3K/Akt/GSK-3beta cascades. Neuropharmacology 2022, 203, 108871. [Google Scholar] [CrossRef]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.J.; Emmerzaal, T.L.; Kozicz, T.; Andrews, Z.B. Ghrelin’s Role in the Hypothalamic-Pituitary-Adrenal Axis Stress Response: Implications for Mood Disorders. Biol. Psychiatry 2015, 78, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.J.; Xu, L.; Clarke, M.A.; Lemus, M.; Reichenbach, A.; Geenen, B.; Kozicz, T.; Andrews, Z.B. Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol. Psychiatry 2012, 72, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.; Ratner, C.; Rudenko, O.; Christiansen, S.H.; Skov, L.J.; Hundahl, C.; Woldbye, D.P.; Holst, B. Anxiolytic-Like Effects of Increased Ghrelin Receptor Signaling in the Amygdala. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.J.; Zhu, X.C.; Han, Q.Q.; Wang, Y.L.; Yue, N.; Wang, J.; Yu, R.; Li, B.; Wu, G.C.; Liu, Q.; et al. Ghrelin alleviates anxiety- and depression-like behaviors induced by chronic unpredictable mild stress in rodents. Behav. Brain Res. 2017, 326, 33–43. [Google Scholar] [CrossRef]
- Edvardsson, C.E.; Vestlund, J.; Jerlhag, E. A ghrelin receptor antagonist reduces the ability of ghrelin, alcohol or amphetamine to induce a dopamine release in the ventral tegmental area and in nucleus accumbens shell in rats. Eur. J. Pharmacol. 2021, 899, 174039. [Google Scholar] [CrossRef]
- Wang, J.Q.; Mao, L. The ERK Pathway: Molecular Mechanisms and Treatment of Depression. Mol. Neurobiol. 2019, 56, 6197–6205. [Google Scholar] [CrossRef]
- Skibicka, K.P.; Hansson, C.; Egecioglu, E.; Dickson, S.L. Role of ghrelin in food reward: Impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict. Biol. 2012, 17, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Stoyanova, I.; Lutz, D. Ghrelin-Mediated Regeneration and Plasticity After Nervous System Injury. Front. Cell Dev. Biol. 2021, 9, 595914. [Google Scholar] [CrossRef]
- Perea Vega, M.L.; Sanchez, M.S.; Fernández, G.; Paglini, M.G.; Martin, M.; de Barioglio, S.R. Ghrelin treatment leads to dendritic spine remodeling in hippocampal neurons and increases the expression of specific BDNF-mRNA species. Neurobiol. Learn. Mem. 2021, 179, 107409. [Google Scholar] [CrossRef]
- Serrenho, D.; Santos, S.D.; Carvalho, A.L. The Role of Ghrelin in Regulating Synaptic Function and Plasticity of Feeding-Associated Circuits. Front. Cell. Neurosci. 2019, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Niu, M.; Yang, J.; Li, L.; Liu, S.; Sun, Y.; Zhou, Z.; Zhou, Y. GHS-R1a Deficiency Alleviates Depression-Related Behaviors After Chronic Social Defeat Stress. Front. Neurosci. 2019, 13, 364. [Google Scholar] [CrossRef] [PubMed]
- van Andel, M.; van Schoor, N.M.; Korten, N.C.; Heijboer, A.C.; Drent, M.L. Ghrelin, leptin and high-molecular-weight adiponectin in relation to depressive symptoms in older adults: Results from the Longitudinal Aging Study Amsterdam. J. Affect. Disord. 2022, 296, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Homan, P.; Grob, S.; Milos, G.; Schnyder, U.; Hasler, G. Reduction in total plasma ghrelin levels following catecholamine depletion: Relation to bulimic and depressive symptoms. Psychoneuroendocrinology 2013, 38, 1545–1552. [Google Scholar] [CrossRef]
- Hansson, C.; Haage, D.; Taube, M.; Egecioglu, E.; Salome, N.; Dickson, S.L. Central administration of ghrelin alters emotional responses in rats: Behavioural, electrophysiological and molecular evidence. Neuroscience 2011, 180, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Currie, P.J.; Khelemsky, R.; Rigsbee, E.M.; Dono, L.M.; Coiro, C.D.; Chapman, C.D.; Hinchcliff, K. Ghrelin is an orexigenic peptide and elicits anxiety-like behaviors following administration into discrete regions of the hypothalamus. Behav. Brain Res. 2012, 226, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.M.; Burgos-Robles, A.; Liu, E.; Correia, S.S.; Goosens, K.A. A ghrelin-growth hormone axis drives stress-induced vulnerability to enhanced fear. Mol. Psychiatry 2014, 19, 1284–1294. [Google Scholar] [CrossRef] [Green Version]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Effect of electroconvulsive therapy on brain-derived neurotrophic factor levels in patients with major depressive disorder. Brain Behav. 2018, 8, e01101. [Google Scholar] [CrossRef] [Green Version]
- Yousufzai, M.; Harmatz, E.S.; Shah, M.; Malik, M.O.; Goosens, K.A. Ghrelin is a persistent biomarker for chronic stress exposure in adolescent rats and humans. Transl. Psychiatry 2018, 8, 74. [Google Scholar] [CrossRef]
- Lutter, M.; Sakata, I.; Osborne-Lawrence, S.; Rovinsky, S.A.; Anderson, J.G.; Jung, S.; Birnbaum, S.; Yanagisawa, M.; Elmquist, J.K.; Nestler, E.J.; et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat. Neurosci. 2008, 11, 752–753. [Google Scholar] [CrossRef] [Green Version]
- Harmatz, E.S.; Stone, L.; Lim, S.H.; Lee, G.; McGrath, A.; Gisabella, B.; Peng, X.; Kosoy, E.; Yao, J.; Liu, E.; et al. Central Ghrelin Resistance Permits the Overconsolidation of Fear Memory. Biol. Psychiatry 2017, 81, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, D.; Mao, H.; Wei, X.; Xu, M.; Zhang, S.; Jiang, Y.; Wang, C.; Xin, Q.; Chen, X.; et al. Disruption of 5-hydroxytryptamine 1A receptor and orexin receptor 1 heterodimer formation affects novel G protein-dependent signaling pathways and has antidepressant effects in vivo. Transl. Psychiatry 2022, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.Y.; Chen, W.D.; Wang, Y.D. The Apelin/APJ System in Psychosis and Neuropathy. Front. Pharmacol. 2020, 11, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, related compounds and their metabolic routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; Miller, G.E.; Carrier, E.J.; Gorzalka, B.B.; Hillard, C.J. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology 2009, 34, 1257–1262. [Google Scholar] [CrossRef] [Green Version]
- Romero-Sanchiz, P.; Nogueira-Arjona, R.; Pastor, A.; Araos, P.; Serrano, A.; Boronat, A.; Garcia-Marchena, N.; Mayoral, F.; Bordallo, A.; Alen, F.; et al. Plasma concentrations of oleoylethanolamide in a primary care sample of depressed patients are increased in those treated with selective serotonin reuptake inhibitor-type antidepressants. Neuropharmacology 2019, 149, 212–220. [Google Scholar] [CrossRef]
- Kranaster, L.; Hoyer, C.; Aksay, S.S.; Bumb, J.M.; Leweke, F.M.; Janke, C.; Thiel, M.; Lutz, B.; Bindila, L.; Sartorius, A. Electroconvulsive therapy enhances endocannabinoids in the cerebrospinal fluid of patients with major depression: A preliminary prospective study. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 781–786. [Google Scholar] [CrossRef]
- Lazary, J.; Eszlari, N.; Juhasz, G.; Bagdy, G. Genetically reduced FAAH activity may be a risk for the development of anxiety and depression in persons with repetitive childhood trauma. Eur. Neuropsychopharmacol. 2016, 26, 1020–1028. [Google Scholar] [CrossRef]
- Domschke, K.; Dannlowski, U.; Ohrmann, P.; Lawford, B.; Bauer, J.; Kugel, H.; Heindel, W.; Young, R.; Morris, P.; Arolt, V.; et al. Cannabinoid receptor 1 (CNR1) gene: Impact on antidepressant treatment response and emotion processing in major depression. Eur. Neuropsychopharmacol. 2008, 18, 751–759. [Google Scholar] [CrossRef]
- Aso, E.; Ozaita, A.; Valdizán, E.M.; Ledent, C.; Pazos, A.; Maldonado, R.; Valverde, O. BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J. Neurochem. 2008, 105, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Realini, N.; Vigano, D.; Guidali, C.; Zamberletti, E.; Rubino, T.; Parolaro, D. Chronic URB597 treatment at adulthood reverted most depressive-like symptoms induced by adolescent exposure to THC in female rats. Neuropharmacology 2011, 60, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X. FAAH inhibition produces antidepressant-like efforts of mice to acute stress via synaptic long-term depression. Behav. Brain Res. 2017, 324, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, W.; Zhong, P.; Liu, S.J.; Long, J.Z.; Zhao, L.; Gao, H.Q.; Cravatt, B.F.; Liu, Q.S. Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus 2015, 25, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, B.; Shilpa, B.M.; Shah, R.; Goyal, A.; Xie, S.; Bakalian, M.J.; Suckow, R.F.; Cooper, T.B.; Mann, J.J.; Arango, V.; et al. Dual pharmacological inhibitor of endocannabinoid degrading enzymes reduces depressive-like behavior in female rats. J. Psychiatr. Res. 2020, 120, 103–112. [Google Scholar] [CrossRef]
- Rodríguez-Gaztelumendi, A.; Rojo, M.L.; Pazos, A.; Díaz, A. Altered CB receptor-signaling in prefrontal cortex from an animal model of depression is reversed by chronic fluoxetine. J. Neurochem. 2009, 108, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Umathe, S.N.; Manna, S.S.; Jain, N.S. Involvement of endocannabinoids in antidepressant and anti-compulsive effect of fluoxetine in mice. Behav. Brain Res. 2011, 223, 125–134. [Google Scholar] [CrossRef]
- Blessing, E.M.; Steenkamp, M.M.; Manzanares, J.; Marmar, C.R. Cannabidiol as a Potential Treatment for Anxiety Disorders. Neurotherapeutics 2015, 12, 825–836. [Google Scholar] [CrossRef]
- Porter, B.E.; Jacobson, C. Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav. 2013, 29, 574–577. [Google Scholar] [CrossRef] [Green Version]
- Réus, G.Z.; Stringari, R.B.; Ribeiro, K.F.; Luft, T.; Abelaira, H.M.; Fries, G.R.; Aguiar, B.W.; Kapczinski, F.; Hallak, J.E.; Zuardi, A.W.; et al. Administration of cannabidiol and imipramine induces antidepressant-like effects in the forced swimming test and increases brain-derived neurotrophic factor levels in the rat amygdala. Acta Neuropsychiatr. 2011, 23, 241–248. [Google Scholar] [CrossRef]
- Kudova, E. Rapid effects of neurosteroids on neuronal plasticity and their physiological and pathological implications. Neurosci. Lett. 2021, 750, 135771. [Google Scholar] [CrossRef] [PubMed]
- Botelho, A.F.M.; Pierezan, F.; Soto-Blanco, B.; Melo, M.M. A review of cardiac glycosides: Structure, toxicokinetics, clinical signs, diagnosis and antineoplastic potential. Toxicon 2019, 158, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Nesher, M.; Shpolansky, U.; Rosen, H.; Lichtstein, D. The digitalis-like steroid hormones: New mechanisms of action and biological significance. Life Sci. 2007, 80, 2093–2107. [Google Scholar] [CrossRef]
- Goldstein, I.; Lax, E.; Gispan-Herman, I.; Ovadia, H.; Rosen, H.; Yadid, G.; Lichtstein, D. Neutralization of endogenous digitalis-like compounds alters catecholamines metabolism in the brain and elicits anti-depressive behavior. Eur. Neuropsychopharmacol. 2012, 22, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.L.; Lingrel, J.B.; Moseley, A.E.; Vorhees, C.V.; Williams, M.T. Targeted mutations in the Na,K-ATPase alpha 2 isoform confer ouabain resistance and result in abnormal behavior in mice. Synapse 2011, 65, 520–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, I.; Levy, T.; Galili, D.; Ovadia, H.; Yirmiya, R.; Rosen, H.; Lichtstein, D. Involvement of Na(+), K(+)-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol. Psychiatry 2006, 60, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Lerer, E.; Laiba, E.; Mallet, J.; Mujaheed, M.; Laurent, C.; Rosen, H.; Ebstein, R.P.; Lichtstein, D. Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol. Psychiatry 2009, 65, 985–991. [Google Scholar] [CrossRef]
- Amaral, L.S.; Martins Ferreira, J.; Predes, D.; Abreu, J.G.; Noel, F.; Quintas, L.E.M. Telocinobufagin and Marinobufagin Produce Different Effects in LLC-PK1 Cells: A Case of Functional Selectivity of Bufadienolides. Int. J. Mol. Sci. 2018, 19, 2769. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.J. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Qin, Y.J.; Xu, C.; Xia, T.; Du, Z.W.; Zheng, L.P.; Li, A.A.; Meng, F.; Zhang, Y.; Zhang, J.; et al. Design of fast-onset antidepressant by dissociating SERT from nNOS in the DRN. Science 2022, 378, 390–398. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, G.; Zhang, M.; Wang, Y.; Yu, M.; Zhou, Y. Potential of Heterogeneous Compounds as Antidepressants: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 13776. https://doi.org/10.3390/ijms232213776
Hu G, Zhang M, Wang Y, Yu M, Zhou Y. Potential of Heterogeneous Compounds as Antidepressants: A Narrative Review. International Journal of Molecular Sciences. 2022; 23(22):13776. https://doi.org/10.3390/ijms232213776
Chicago/Turabian StyleHu, Gonghui, Meng Zhang, Yuyang Wang, Ming Yu, and Yu Zhou. 2022. "Potential of Heterogeneous Compounds as Antidepressants: A Narrative Review" International Journal of Molecular Sciences 23, no. 22: 13776. https://doi.org/10.3390/ijms232213776
APA StyleHu, G., Zhang, M., Wang, Y., Yu, M., & Zhou, Y. (2022). Potential of Heterogeneous Compounds as Antidepressants: A Narrative Review. International Journal of Molecular Sciences, 23(22), 13776. https://doi.org/10.3390/ijms232213776