
(-)-Epigallocatechin-3-Gallate Prevents IL-1β-Induced uPAR Expression and Invasiveness via the Suppression of NF-κB and AP-1 in Human Bladder Cancer Cells
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
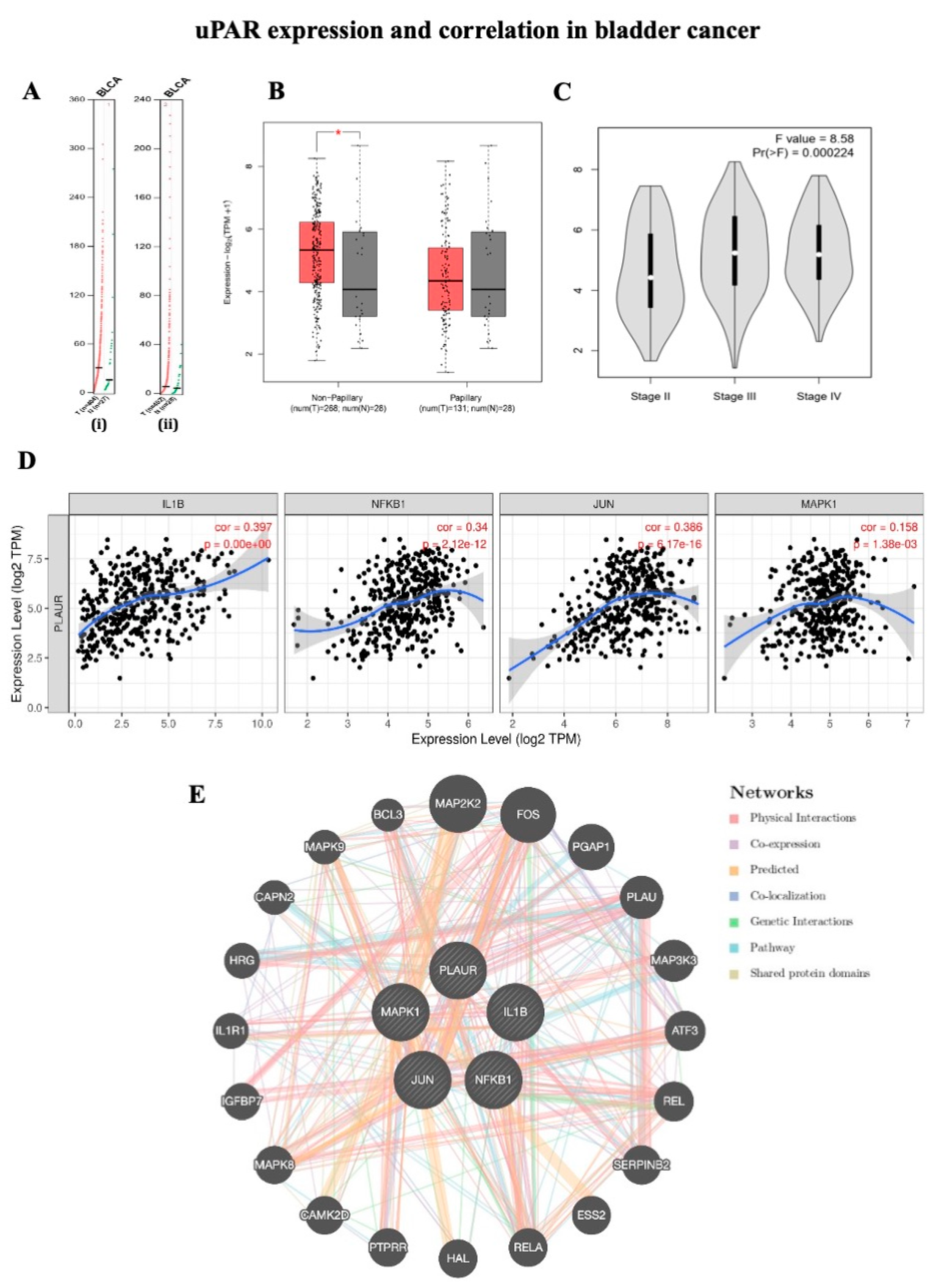
2.1. uPAR Expression and Correlation in Bladder Cancer
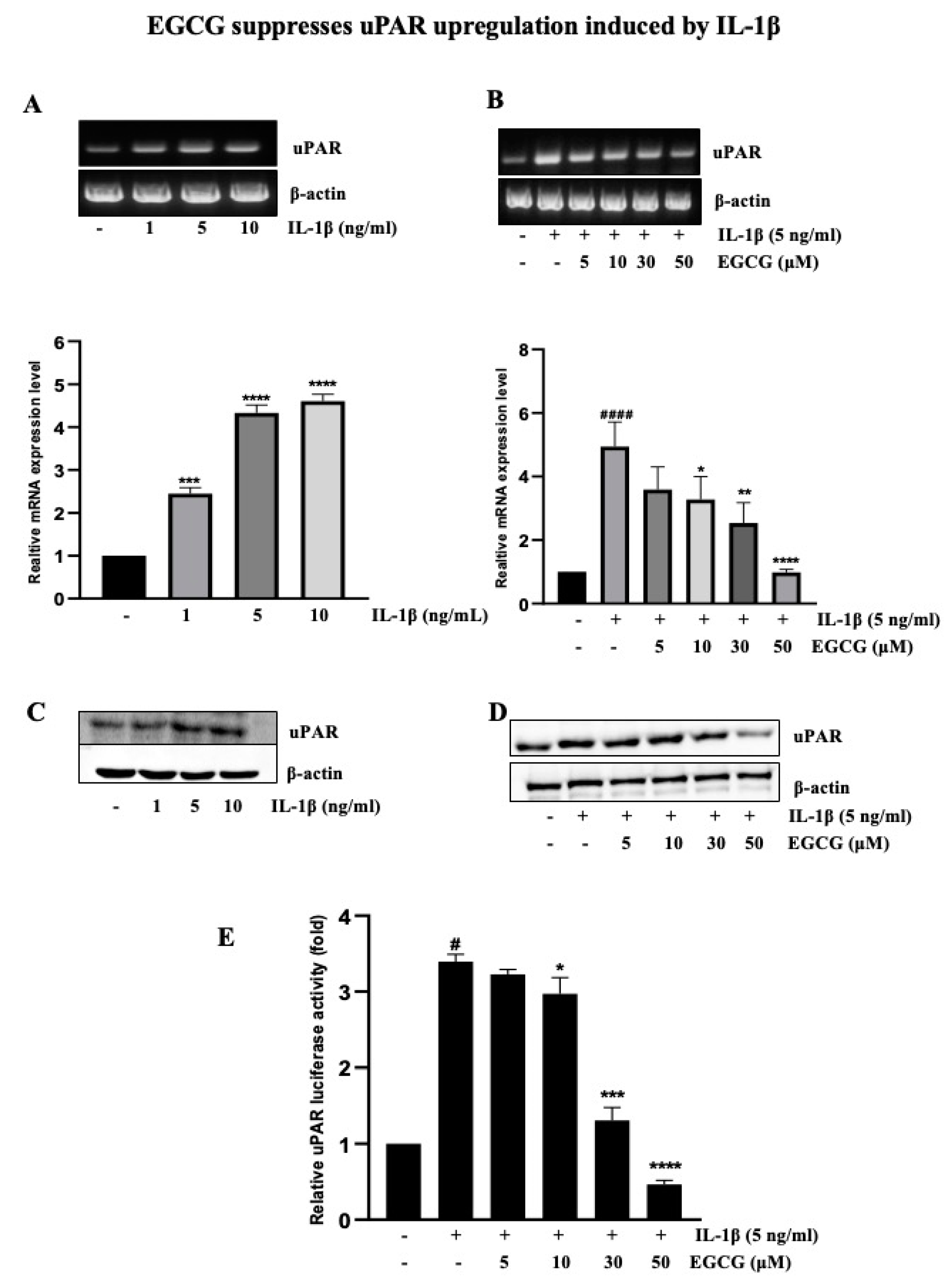
2.2. EGCG Suppresses uPAR Upregulation Induced by IL-1β
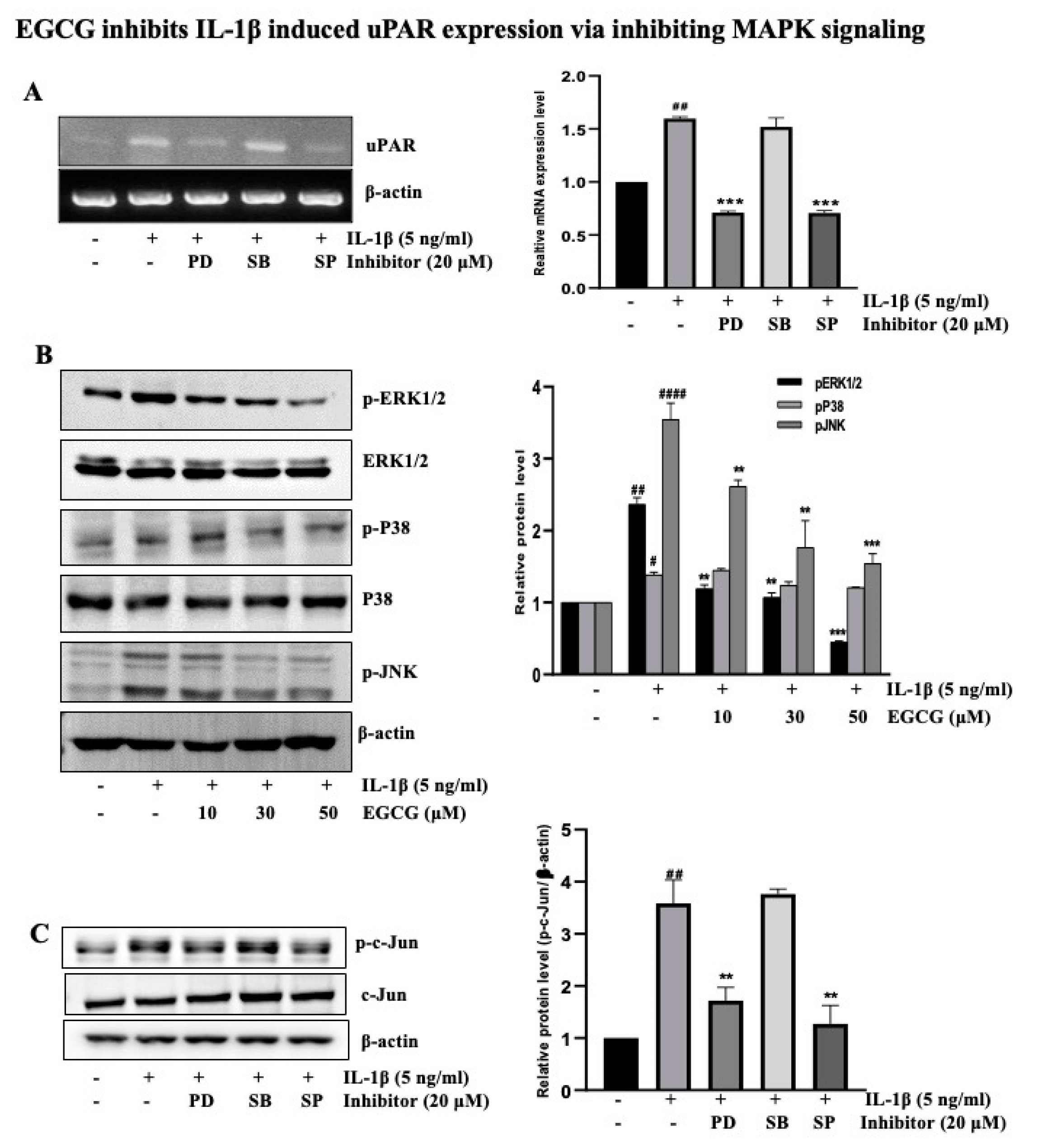
2.3. EGCG Inhibits IL-1β-Induced uPAR Expression via the Inhibition of MAPK Signaling
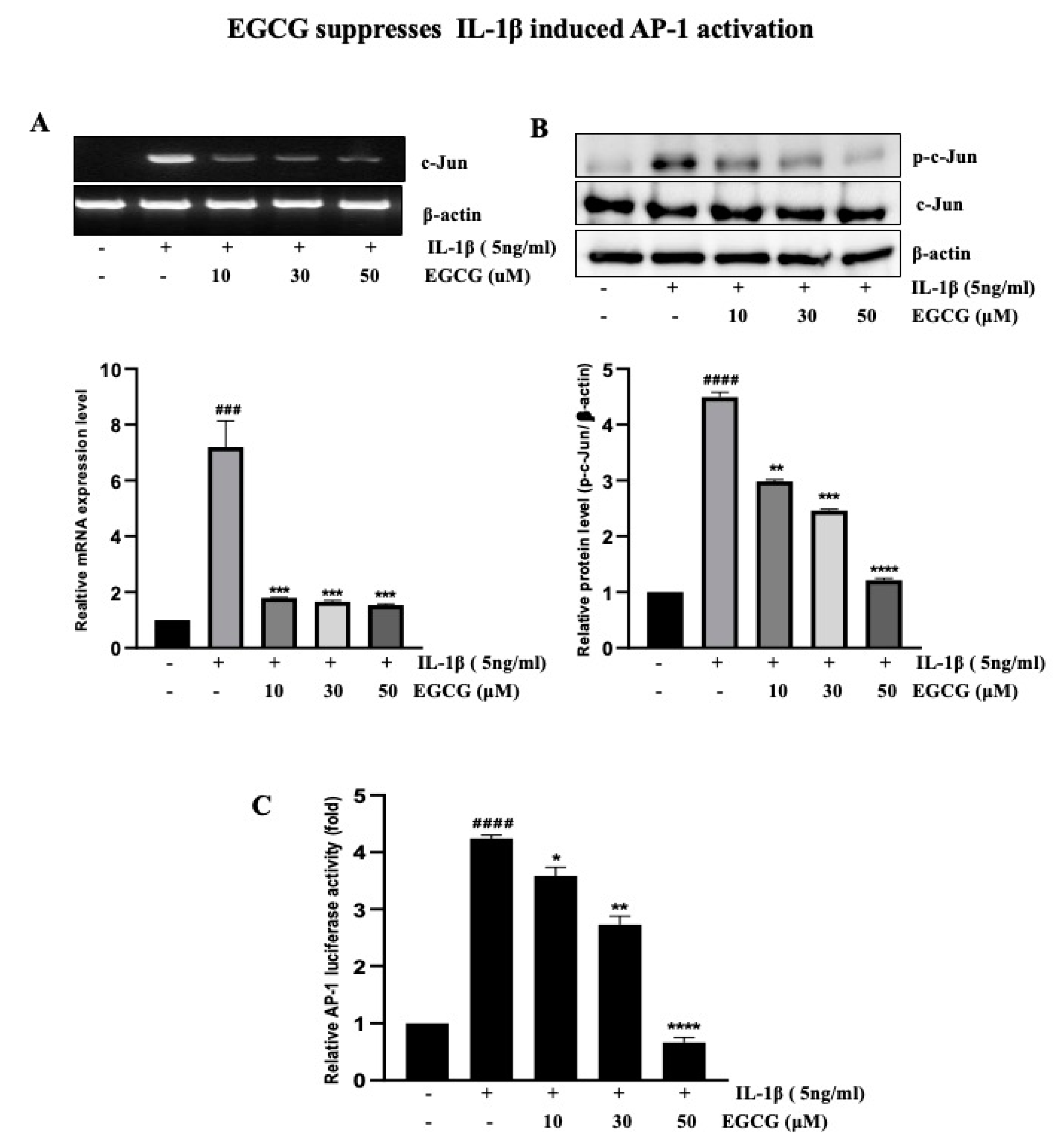
2.4. EGCG Suppresses IL-1β-Induced AP-1 Activation
2.5. EGCG Inhibits NF-κB Activity That is Involved in IL-1β-Induced uPAR Upregulation
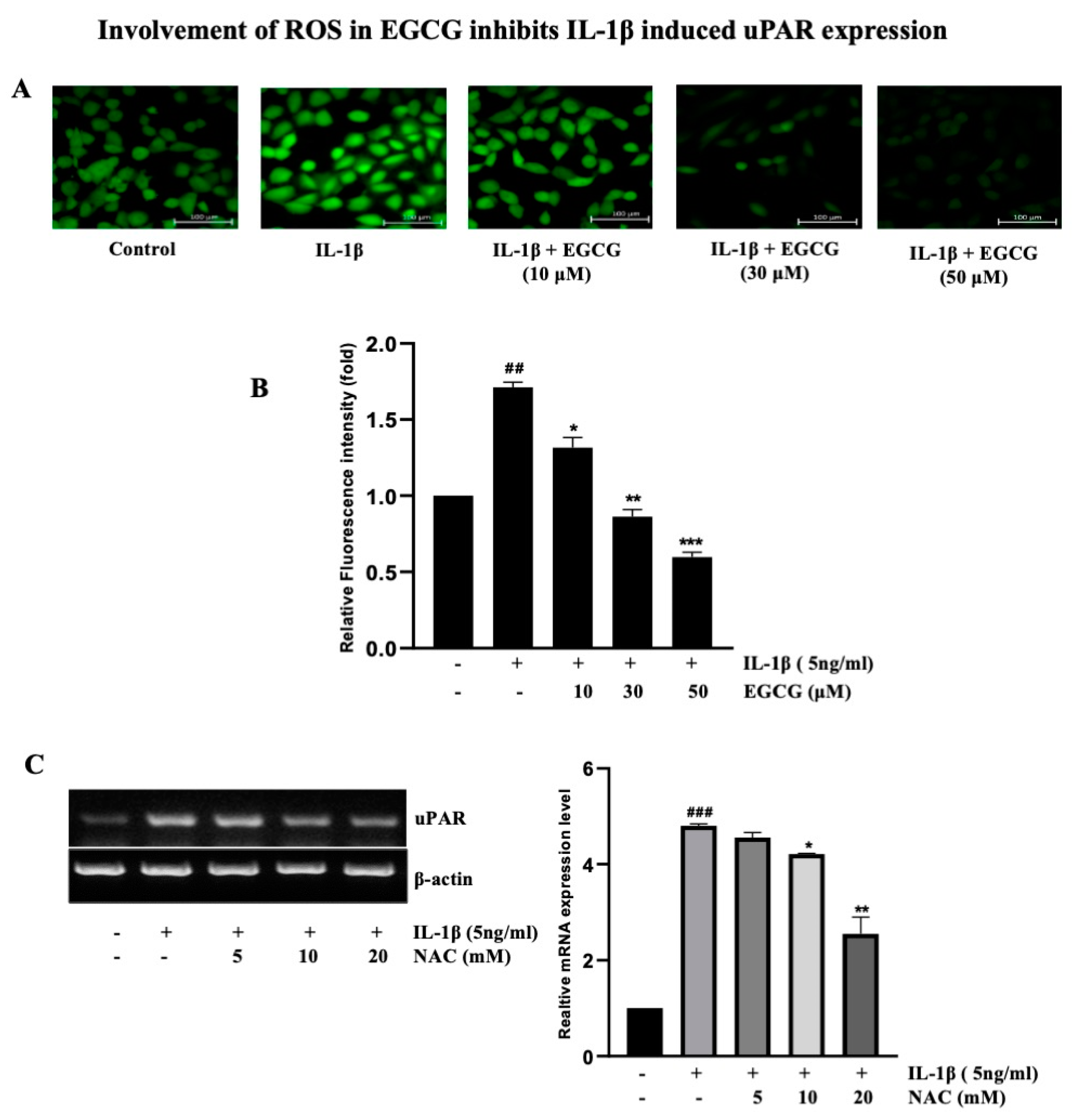
2.6. Involvement of ROS in EGCG-Induced Inhibition IL-1β-Induced uPAR Expression
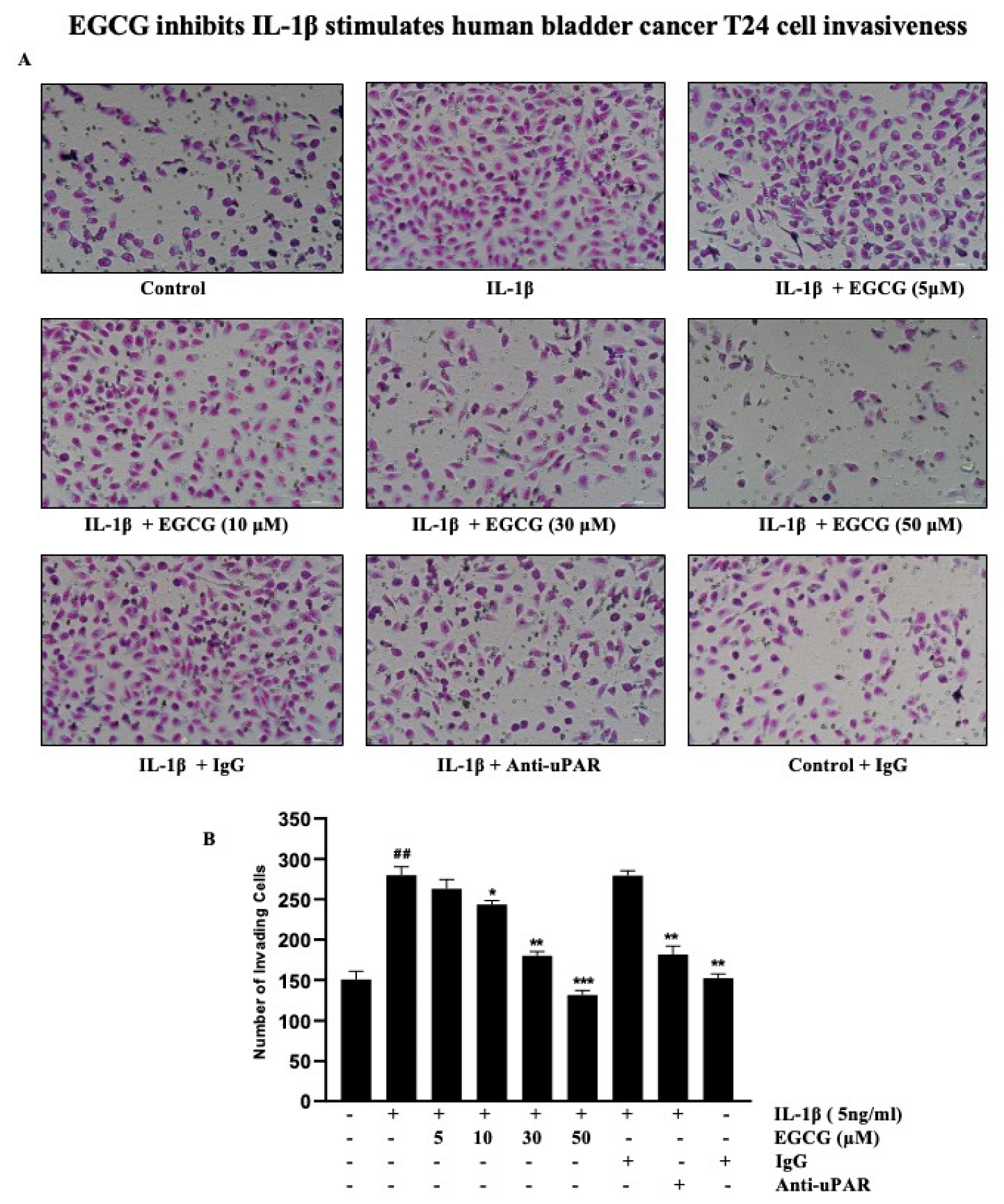
2.7. EGCG Inhibits the IL-1β Stimulation of Human Bladder Cancer T24 Cell Invasiveness
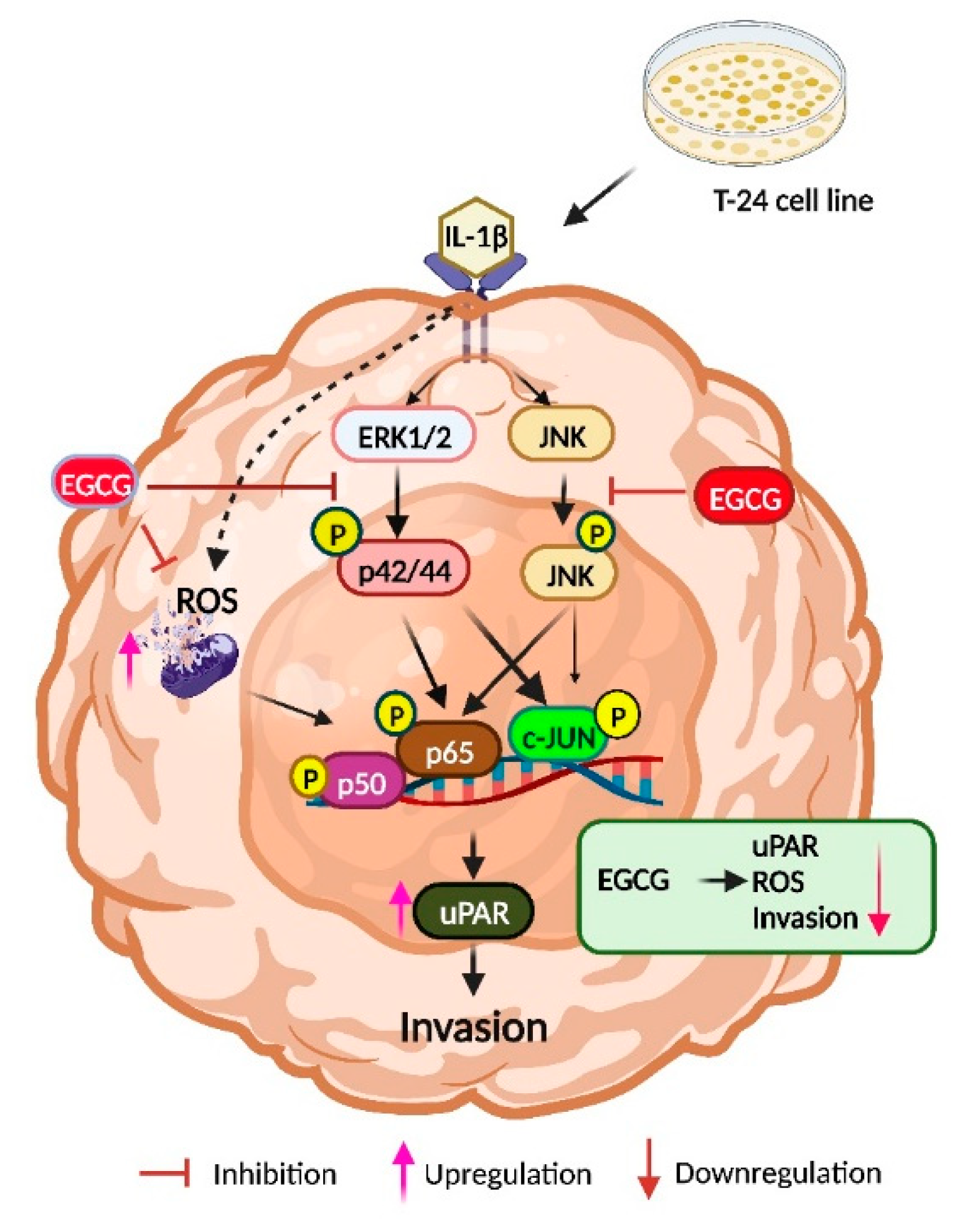
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis
4.2. Materials and Conditions of Cell Culture
4.3. Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
4.4. Western Blot Analysis
4.5. Promoter Activity Assay
4.6. Matrigel Cell Invasion Assay
4.7. Measurement of Intracellular Hydrogen Peroxide (H2O2)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoi, P.N.; Li, S.; Thuan, U.T.; Sah, D.K.; Kang, T.W.; Nguyen, T.T.; Lian, S.; Xia, Y.; Jung, Y.D. Lysophosphatidic Acid Upregulates Recepteur D’origine Nantais Expression and Cell Invasion via Egr-1, AP-1, and NF-kappaB Signaling in Bladder Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGeorge, K.C.; Holt, H.R.; Hodges, S.C. Bladder Cancer: Diagnosis and Treatment. Am. Fam. Physician 2017, 96, 507–514. [Google Scholar] [PubMed]
- Ravindranathan, D.; Master, V.A.; Bilen, M.A. Inflammatory Markers in Cancer Immunotherapy. Biology 2021, 10, 325. [Google Scholar] [CrossRef]
- Chauhan, R.; Trivedi, V. Inflammatory markers in cancer: Potential resources. Front. Biosci. 2020, 12, 1–24. [Google Scholar] [CrossRef]
- Rebe, C.; Ghiringhelli, F. Interleukin-1beta and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Idris, A.; Ghazali, N.B.; Koh, D. Interleukin 1beta-A Potential Salivary Biomarker for Cancer Progression? Biomark. Cancer 2015, 7, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Yuan, M.; Li, S.; Thuan, U.T.; Nguyen, T.T.; Kang, T.W.; Liao, W.; Lian, S.; Jung, Y.D. Apigenin Suppresses the IL-1beta-Induced Expression of the Urokinase-Type Plasminogen Activator Receptor by Inhibiting MAPK-Mediated AP-1 and NF-kappaB Signaling in Human Bladder Cancer T24 Cells. J. Agric. Food Chem. 2018, 66, 7663–7673. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.D.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef]
- Ogura, N.; Tobe, M.; Tamaki, H.; Nagura, H.; Abiko, Y. IL-1beta increases uPA and uPA receptor expression in human gingival fibroblasts. IUBMB Life 2001, 51, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.L.; Kassouf, W. Radical Cystectomy is the best choice for most patients with muscle-invasive bladder cancer? Opinion: Yes. Int. Braz. J. Urol. 2017, 43, 184–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.C.; Citrin, D.E.; Agarwal, P.K.; Apolo, A.B. Multimodal management of muscle-invasive bladder cancer. Curr. Probl. Cancer 2014, 38, 80–108. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.G.; Munera-Maravilla, E.; Bernardini, A.; Rubio, C.; Suarez-Cabrera, C.; Segovia, C.; Lodewijk, I.; Duenas, M.; Martinez-Fernandez, M.; Paramio, J.M. Epigenetics of Bladder Cancer: Where Biomarkers and Therapeutic Targets Meet. Front. Genet. 2019, 10, 1125. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Khoi, P.N.; Joo, Y.E.; Lee, Y.H.; Lang, S.A.; Stoeltzing, O.; Jung, Y.D. EGCG inhibits recepteur d’origine nantais expression by suppressing Egr-1 in gastric cancer cells. Int. J. Oncol. 2013, 42, 1120–1126. [Google Scholar] [CrossRef] [Green Version]
- Du, G.J.; Zhang, Z.; Wen, X.D.; Yu, C.; Calway, T.; Yuan, C.S.; Wang, C.Z. Epigallocatechin Gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 2012, 4, 1679–1691. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chen, Y.J.; Chang, W.A.; Li, W.M.; Ke, H.L.; Wu, W.J.; Kuo, P.L. Effects of Epigallocatechin Gallate (EGCG) on Urinary Bladder Urothelial Carcinoma-Next-Generation Sequencing and Bioinformatics Approaches. Medicina 2019, 55, 768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Liu, Z.; Li, J.; Zhang, Q.; Zhong, P.; Teng, T.; Chen, M.; Xie, Z.; Ji, A.; Li, Y. Epigallocatechin-3-gallate inhibits the growth and increases the apoptosis of human thyroid carcinoma cells through suppression of EGFR/RAS/RAF/MEK/ERK signaling pathway. Cancer Cell Int. 2019, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Moreau, M.; Mourah, S.; Dosquet, C. beta-Catenin and NF-kappaB cooperate to regulate the uPA/uPAR system in cancer cells. Int. J. Cancer 2011, 128, 1280–1292. [Google Scholar] [CrossRef]
- Chang, H.J.; Kim, M.H.; Baek, M.K.; Park, J.S.; Chung, I.J.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. Triptolide Inhibits Tumor Promoter-induced uPAR Expression via Blocking NF-κB Signaling in Human Gastric AGS Cells. Anticancer Res. 2007, 27, 3411. [Google Scholar] [PubMed]
- Oka, S.-i.; Kamata, H.; Kamata, K.; Yagisawa, H.; Hirata, H. N-Acetylcysteine suppresses TNF-induced NF-κB activation through inhibition of IκB kinases. FEBS Lett. 2000, 472, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Kretzmann, N.A.; Chiela, E.; Matte, U.; Marroni, N.; Marroni, C.A. N-acetylcysteine improves antitumoural response of Interferon alpha by NF-kB downregulation in liver cancer cells. Comp. Hepatol. 2012, 11, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Bhori, M.; Kasu, Y.A.; Bhat, G.; Marar, T. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity—Exploring the armoury of obscurity. Saudi Pharm. J. 2018, 26, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Kaefer, C.M.; Milner, J.A. Herbs and Spices in Cancer Prevention and Treatment. In Herbal Medicine: Biomolecular and Clinical Aspects; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRS Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Wang, H.; Khor, T.O.; Shu, L.; Su, Z.Y.; Fuentes, F.; Lee, J.H.; Kong, A.N. Plants vs. cancer: A review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med. Chem. 2012, 12, 1281–1305. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhang, Z.; Han, Y.; Wang, J.; Wang, Y.; Chen, X.; Shao, Y.; Cheng, Y.; Zhou, W.; Lu, X.; et al. A review on anti-cancer effect of green tea catechins. J. Funct. Foods 2020, 74, 104172. [Google Scholar] [CrossRef]
- Guo, Y.; Zhi, F.; Chen, P.; Zhao, K.; Xiang, H.; Mao, Q.; Wang, X.; Zhang, X. Green tea and the risk of prostate cancer: A systematic review and meta-analysis. Medicine 2017, 96, e6426. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.; Oh, J.H.; Chang, H.J.; Sohn, D.K.; Shin, A.; Kim, J. Protective Effect of Green Tea Consumption on Colorectal Cancer Varies by Lifestyle Factors. Nutrients 2019, 11, 2612. [Google Scholar] [CrossRef]
- Chen, B.-H.; Hsieh, C.-H.; Tsai, S.-Y.; Wang, C.-Y.; Wang, C.-C. Anticancer effects of epigallocatechin-3-gallate nanoemulsion on lung cancer cells through the activation of AMP-activated protein kinase signaling pathway. Sci. Rep. 2020, 10, 5163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Veeramachaneni, N. Targeting interleukin-1beta and inflammation in lung cancer. Biomark. Res. 2022, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Tsuda, M.; Yoshida, K.; Tanino, M.; Kimura, T.; Nishihara, H.; Abe, T.; Shinohara, N.; Nonomura, K.; Tanaka, S. Aldo-keto reductase 1C1 induced by interleukin-1beta mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci. Rep. 2016, 6, 34625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, T.; Zhao, Y.; Jiang, X.; Yuan, H.; Wang, H.; Cui, X.; Xu, J.; Zhao, J.; Wang, J. uPAR: An Essential Factor for Tumor Development. J. Cancer 2021, 12, 7026–7040. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, Z.; An, J.; Mao, X.; Lin, F.; Sun, P. Effect of a synthetic inhibitor of urokinase plasminogen activator on the migration and invasion of human cervical cancer cells in vitro. Mol. Med. Rep. 2018, 17, 4273–4280. [Google Scholar] [CrossRef] [Green Version]
- Lian, S.; Xia, Y.; Ung, T.T.; Khoi, P.N.; Yoon, H.J.; Lee, S.G.; Kim, K.K.; Jung, Y.D. Prostaglandin E2 stimulates urokinase-type plasminogen activator receptor via EP2 receptor-dependent signaling pathways in human AGS gastric cancer cells. Mol. Carcinog. 2017, 56, 664–680. [Google Scholar] [CrossRef]
- Khoi, P.N.; Xia, Y.; Lian, S.; Kim, H.D.; Kim, D.H.; Joo, Y.E.; Chay, K.O.; Kim, K.K.; Jung, Y.D. Cadmium induces urokinase-type plasminogen activator receptor expression and the cell invasiveness of human gastric cancer cells via the ERK-1/2, NF-kappaB, and AP-1 signaling pathways. Int. J. Oncol. 2014, 45, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Khoi, P.N.; Park, J.S.; Kim, N.H.; Jung, Y.D. Nicotine stimulates urokinase-type plasminogen activator receptor expression and cell invasiveness through mitogen-activated protein kinase and reactive oxygen species signaling in ECV304 endothelial cells. Toxicol. Appl. Pharmacol. 2012, 259, 248–256. [Google Scholar] [CrossRef]
- Baek, M.K.; Park, J.S.; Park, J.H.; Kim, M.H.; Kim, H.D.; Bae, W.K.; Chung, I.J.; Shin, B.A.; Jung, Y.D. Lithocholic acid upregulates uPAR and cell invasiveness via MAPK and AP-1 signaling in colon cancer cells. Cancer Lett. 2010, 290, 123–128. [Google Scholar] [CrossRef]
- Kim, M.H.; Park, J.S.; Chang, H.J.; Baek, M.K.; Kim, H.R.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. Lysophosphatidic acid promotes cell invasion by up-regulating the urokinase-type plasminogen activator receptor in human gastric cancer cells. J. Cell. Biochem. 2008, 104, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Choi, E.Y.; Koh, S.; Kim, M.K.; Jang, B.I.; Kim, S.W.; Kim, J.-R. IL-1β-stimulated urokinase plasminogen activator expression through NF-κB in gastric cancer after HGF treatment. Oncol. Rep. 2014, 31, 2123–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, I.A.; Malik, A.; Adhami, V.M.; Asim, M.; Hafeez, B.B.; Sarfaraz, S.; Mukhtar, H. Green tea polyphenol EGCG sensitizes human prostate carcinoma LNCaP cells to TRAIL-mediated apoptosis and synergistically inhibits biomarkers associated with angiogenesis and metastasis. Oncogene 2008, 27, 2055–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Mukhtar, H. Multitargeted therapy of cancer by green tea polyphenols. Cancer Lett. 2008, 269, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namiki, K.; Wongsirisin, P.; Yokoyama, S.; Sato, M.; Rawangkan, A.; Sakai, R.; Iida, K.; Suganuma, M. (-)-Epigallocatechin gallate inhibits stemness and tumourigenicity stimulated by AXL receptor tyrosine kinase in human lung cancer cells. Sci. Rep. 2020, 10, 2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zan, L.; Chen, Q.; Zhang, L.; Li, X. Epigallocatechin gallate (EGCG) suppresses growth and tumorigenicity in breast cancer cells by downregulation of miR-25. Bioengineered 2019, 10, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Khoi, P.N.; Park, J.S.; Kim, J.H.; Xia, Y.; Kim, N.H.; Kim, K.K.; Jung, Y.D. (-)-Epigallocatechin-3-gallate blocks nicotine-induced matrix metalloproteinase-9 expression and invasiveness via suppression of NF-kappaB and AP-1 in endothelial cells. Int. J. Oncol. 2013, 43, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-kappaB and AP-1 connection: Mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef] [Green Version]
- Rex, J.; Lutz, A.; Faletti, L.E.; Albrecht, U.; Thomas, M.; Bode, J.G.; Borner, C.; Sawodny, O.; Merfort, I. IL-1beta and TNFalpha Differentially Influence NF-kappaB Activity and FasL-Induced Apoptosis in Primary Murine Hepatocytes During LPS-Induced Inflammation. Front. Physiol. 2019, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- Kundu, J.K.; Surh, Y.J. Epigallocatechin gallate inhibits phorbol ester-induced activation of NF-kappa B and CREB in mouse skin: Role of p38 MAPK. Ann. N. Y. Acad. Sci. 2007, 1095, 504–512. [Google Scholar] [CrossRef]
- Hong, M.H.; Kim, M.H.; Chang, H.J.; Kim, N.H.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. (-)-Epigallocatechin-3-gallate inhibits monocyte chemotactic protein-1 expression in endothelial cells via blocking NF-kappaB signaling. Life Sci. 2007, 80, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Afaq, F.; Adhami, V.M.; Ahmad, N.; Mukhtar, H. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea Constituent (-)-epigallocatechin-3-gallate. Oncogene 2003, 22, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, D.S.; Catravas, J.D.; Odoms, K.; Denenberg, A.; Malhotra, V.; Wong, H.R. Epigallocatechin-3-gallate, a green tea-derived polyphenol, inhibits IL-1 beta-dependent proinflammatory signal transduction in cultured respiratory epithelial cells. J. Nutr. 2004, 134, 1039–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda-Yamamoto, M.; Suzuki, N.; Sawai, Y.; Miyase, T.; Sano, M.; Hashimoto-Ohta, A.; Isemura, M. Association of suppression of extracellular signal-regulated kinase phosphorylation by epigallocatechin gallate with the reduction of matrix metalloproteinase activities in human fibrosarcoma HT1080 cells. J. Agric. Food Chem. 2003, 51, 1858–1863. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, M.H.; Jeong, M.; Hwang, Y.S.; Lim, S.H.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. EGCG blocks tumor promoter-induced MMP-9 expression via suppression of MAPK and AP-1 activation in human gastric AGS cells. Anticancer Res. 2004, 24, 747–753. [Google Scholar]
- Chen, N.Y.; Ma, W.Y.; Yang, C.S.; Dong, Z. Inhibition of arsenite-induced apoptosis and AP-1 activity by epigallocatechin-3-gallate and theaflavins. J. Environ. Pathol. Toxicol. Oncol. 2000, 19, 287–295. [Google Scholar]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.X.; Wei, F.; Li, N.; Li, J.L.; Chen, L.J.; Liu, Y.Y.; Luo, F.; Xiong, H.R.; Hou, W.; Yang, Z.Q. Amelioration of influenza virus-induced reactive oxygen species formation by epigallocatechin gallate derived from green tea. Acta Pharmacol. Sin. 2012, 33, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Kucera, O.; Mezera, V.; Moravcova, A.; Endlicher, R.; Lotkova, H.; Drahota, Z.; Cervinkova, Z. In vitro toxicity of epigallocatechin gallate in rat liver mitochondria and hepatocytes. Oxidative Med. Cell. Longev. 2015, 2015, 476180. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Fang, Y.; Wei, S.M. Effect and mechanism of epigallocatechin-3-gallate (EGCG). against the hydrogen peroxide-induced oxidative damage in human dermal fibroblasts. J. Cosmet. Sci. 2013, 64, 35–44. [Google Scholar]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Cho, H.S.; Jung, M.; Hong, M.H.; Lee, S.K.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. Extracellular signal-regulated kinase and AP-1 pathways are involved in reactive oxygen species-induced urokinase plasminogen activator receptor expression in human gastric cancer cells. Int. J. Oncol. 2005, 26, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Carmi, Y.; Dvorkin, T.; White, R.M.; Gayvoronsky, L.; et al. Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 2006, 42, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. Role of IL-1-mediated inflammation in tumor angiogenesis. Adv. Exp. Med. Biol. 2007, 601, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1beta activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.W.; Kuzuya, M.; Kanda, S.; Maeda, K.; Sasaki, T.; Wang, Q.L.; Tamaya-Mori, N.; Shibata, T.; Iguchi, A. Epigallocatechin-3-gallate binding to MMP-2 inhibits gelatinolytic activity without influencing the attachment to extracellular matrix proteins but enhances MMP-2 binding to TIMP-2. Arch. Biochem. Biophys. 2003, 415, 126–132. [Google Scholar] [CrossRef]
- Demeule, M.; Brossard, M.; Page, M.; Gingras, D.; Beliveau, R. Matrix metalloproteinase inhibition by green tea catechins. Biochim. Biophys. Acta 2000, 1478, 51–60. [Google Scholar] [CrossRef]
- Kim, C.H.; Moon, S.K. Epigallocatechin-3-gallate causes the p21/WAF1-mediated G(1)-phase arrest of cell cycle and inhibits matrix metalloproteinase-9 expression in TNF-alpha-induced vascular smooth muscle cells. Arch. Biochem. Biophys. 2005, 435, 264–272. [Google Scholar] [CrossRef]
- Braicu, C.; Gherman, C.D.; Irimie, A.; Berindan-Neagoe, I. Epigallocatechin-3-Gallate (EGCG) inhibits cell proliferation and migratory behaviour of triple negative breast cancer cells. J. Nanosci. Nanotechnol. 2013, 13, 632–637. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ung, T.T.; Li, S.; Lian, S.; Xia, Y.; Park, S.Y.; Do Jung, Y. Metformin inhibits lithocholic acid-induced interleukin 8 upregulation in colorectal cancer cells by suppressing ROS production and NF-kB activity. Sci. Rep. 2019, 9, 2003. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sah, D.K.; Khoi, P.N.; Li, S.; Arjunan, A.; Jeong, J.-U.; Jung, Y.D. (-)-Epigallocatechin-3-Gallate Prevents IL-1β-Induced uPAR Expression and Invasiveness via the Suppression of NF-κB and AP-1 in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2022, 23, 14008. https://doi.org/10.3390/ijms232214008
Sah DK, Khoi PN, Li S, Arjunan A, Jeong J-U, Jung YD. (-)-Epigallocatechin-3-Gallate Prevents IL-1β-Induced uPAR Expression and Invasiveness via the Suppression of NF-κB and AP-1 in Human Bladder Cancer Cells. International Journal of Molecular Sciences. 2022; 23(22):14008. https://doi.org/10.3390/ijms232214008
Chicago/Turabian StyleSah, Dhiraj Kumar, Pham Ngoc Khoi, Shinan Li, Archana Arjunan, Jae-Uk Jeong, and Young Do Jung. 2022. "(-)-Epigallocatechin-3-Gallate Prevents IL-1β-Induced uPAR Expression and Invasiveness via the Suppression of NF-κB and AP-1 in Human Bladder Cancer Cells" International Journal of Molecular Sciences 23, no. 22: 14008. https://doi.org/10.3390/ijms232214008