

Rejuvenation: Turning Back Time by Enhancing CISD2

 , ,
, ,
Abstract
:1. Introduction
1.1. CISD2 Is One of a Limited Number of Pro-Longevity Genes in Mammals
1.2. Functions of CISD2
1.2.1. CISD2 Is the Disease Gene of WFS2 in Humans
1.2.2. CISD2 Maintains Mitochondrial and MAM Integrity
1.2.3. CISD2 Regulates Intracellular Ca2+ Homeostasis
1.2.4. CISD2 Modulates Redox Status
2. CISD2 Mediates Lifespan and Healthspan
2.1. CISD2 Is One of a Limited Number of Pro-Longevity Genes in Mammals
2.2. CISD2 in Cardiac Aging
2.3. CISD2 in Muscle Aging
2.4. CISD2 in Liver Aging
3. CISD2 Alleviates a Range of Age-Associated Disorders
3.1. CISD2 Improves the Outcome of Alzheimer’s Disease in Mice
3.2. CISD2 Ameliorates Fatty Liver Disease
3.3. CISD2 Maintains Corneal Epithelial Homeostasis
4. Regimens or Treatments That Enhance CISD2 Gene Expression
4.1. Weight Loss Surgery Restores CISD2 Levels in Obese Subjects
4.1.1. Obesity Is One of the Major Risk Factors That Accelerate Age-Associated Diseases
4.1.2. CISD2 Is Downregulated in Subjects with Morbid Obesity, Whereas Weight Loss Surgery Restores CISD2 Expression in Obese Humans
4.2. Exercise Attenuates Aging and Enhances CISD2 Gene Expression
4.2.1. Exercise Is a Promising Lifestyle Intervention That Is Able to Slow Down Aging
4.2.2. Exercise Enhances CISD2 Expression and Attenuates Skeletal Muscle Aging
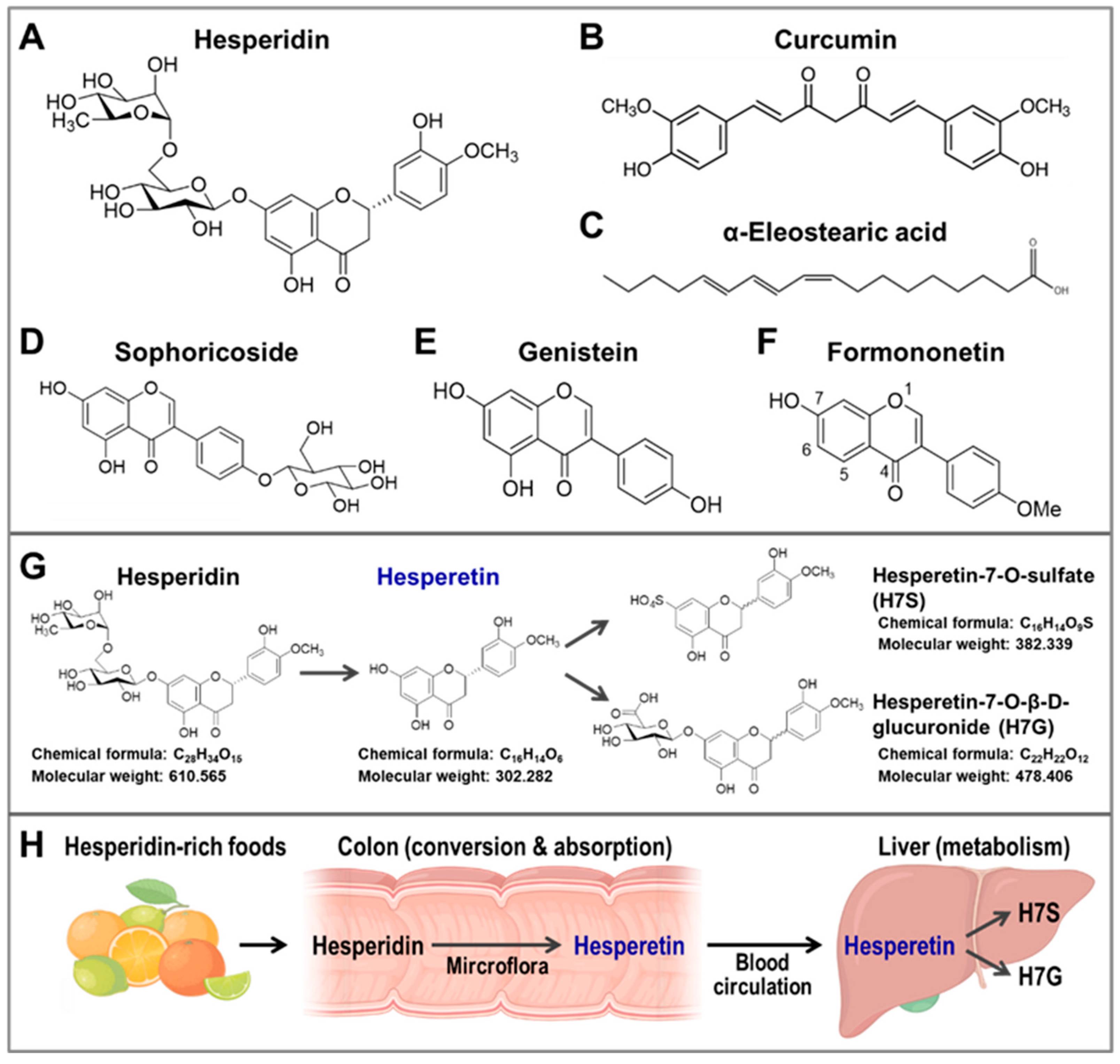
4.3. Natural Compounds That Can Upregulate CISD2 Expression
4.3.1. Curcumin Increases CISD2 Expression
4.3.2. WBM Extract, α-Eleostearic Acid, and ESM Powder

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound or Intervention | Treatment and Duration | Tissue or Cell Line | Animal Model or Human Subjects | Enhance CISD2 Expression | Reference |
|---|---|---|---|---|---|
| A. In Vitro Cell Study | |||||
| Hesperetin | 10 and 30 μM for 24 h | HEK293-CISD2 reporter cells | CISD2 reporter | [75] | |
| Hesperetin -7-O-sulfate | 30 μM for 24 h | HEK293-CISD2 reporter cells | CISD2 reporter | [75] | |
| Curcumin | 1 μM for 24 h | SH-SY5Y Rat primary astrocyte | CISD2 mRNA | [38] | |
| Wild bitter melon extract | 1 μg/mL for 24 h | Astrocyte cell line | LPS-challenged ACL | CISD2 mRNA | [73] |
| α-Eleostearic acid | 0.28 μg/mL for 24 h | Astrocyte cell line | LPS-challenged ACL | CISD2 mRNA | [73] |
| Sophoricoside | 10 and 30 μM for 24 h | HEK293-CISD2 reporter cells | CISD2 reporter | [75] | |
| Genistein | 10 and 30 μM for 24 h | HEK293-CISD2 reporter cells | CISD2 reporter | [75] | |
| Formononetin | 10 and 30 μM for 24 h | HEK293-CISD2 reporter cells | CISD2 reporter | [75] | |
| B. Animal Study | |||||
| Exercise | |||||
| Treadmill exercise for 8 weeks | Whole body (ventral view) | CISD2 reporter mice | CISD2 reporter | [66] | |
| Exercise with a running wheel for 4 weeks | Skeletal muscle eWAT | Male C57BL/6J mice | CISD2 protein | [67] | |
| Hesperetin | |||||
| 100 mg/kg/day (provided in food) for 5 months | Heart Skeletal muscle | Aged mice (26 months old) | CISD2 protein | [75] | |
| 100 mg/kg/day (provided in food) for 6 weeks | Whole body (ventral view) | CISD2 reporter mice | CISD2 reporter | [75] | |
| Curcumin | |||||
| 40 mg/kg/day (i.p. injection) for 2 days | Spinal cord | Aged mice (24 months old) | CISD2 protein | [38] | |
| 40 mg/kg/day (i.p. injection) for 2 days | Spinal cord | Spinal cord hemisectionin mice | CISD2 mRNA | [71] | |
| Miscellaneous | |||||
| Wild bitter melon extract | 500 mg/kg (i.p. injection) for single dose | Spinal cord | Spinal cord hemisection in mice | CISD2 mRNA and protein | [73] |
| ESM powder | Diet with 10 g/kg ESM powder for 14 days | Liver | Wistar rats | CISD2 mRNA | [74] |
| HEM powder | Diet with 10 g/kg HEM powder for 14 days | Liver | Wistar rats | CISD2 mRNA | [74] |
| C. Human Study | |||||
| RYGB surgery | 3 months post-RYGB | Skeletal muscle | Obese females (BMI > 40 kg/m2) | CISD2 protein | [59] |
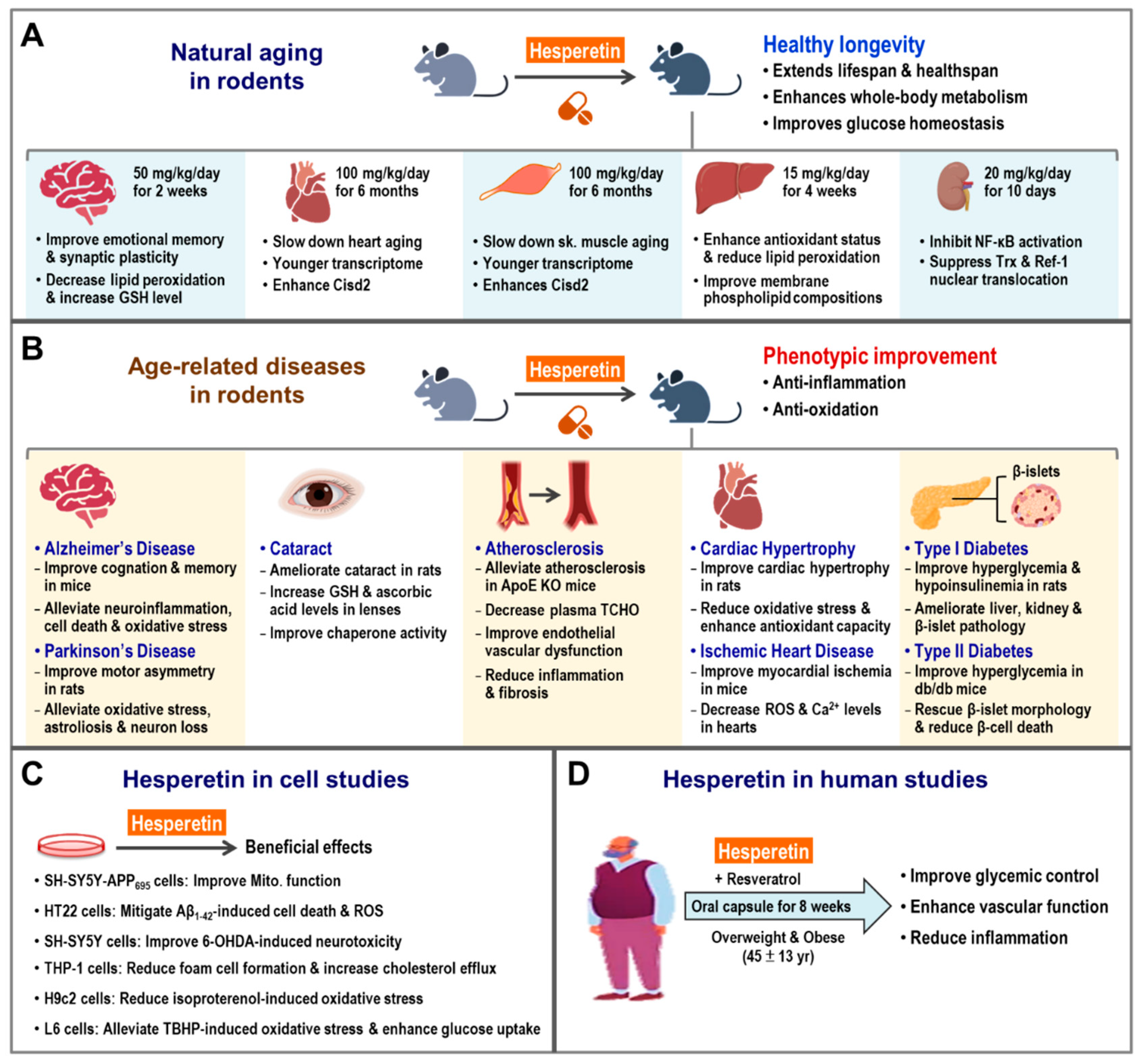
5. Hesperetin Rejuvenates Aged Organs and Promotes Longevity
5.1. Hesperetin Enhances CISD2 Expression and Promotes Longevity in Naturally Aged Mice
5.2. Hesperetin Slows Down Aging at the Organ Level
5.2.1. Hesperetin and Brain Aging
5.2.2. Hesperetin and Liver Aging
5.2.3. Hesperetin and Kidney Aging
5.3. Beneficial Effects of Hesperetin on Age-Associated Diseases
5.3.1. Hesperetin Improves Age-Associated Neurodegenerative Diseases
5.3.2. Hesperetin and AD
5.3.3. Hesperetin and PD
5.3.4. Hesperetin Protects against Age-Associated Eye Diseases
5.3.5. Hesperetin Improves Age-Associated Cardiovascular Diseases
5.3.6. Hesperetin Reduces Diabetes
5.3.7. Hesperetin Reduces Age-Associated Metabolic Dysfunction
6. The Anti-Aging Effect of Hesperetin Is Mainly Dependent on CISD2
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- United Nations DoEaSA, Population Division. World Population Prospects 2022: Summary of Results. Available online: https://www.un.org/development/desa/pd/content/World-Population-Prospects-2022 (accessed on 14 September 2022).
- Biesemann, N.; Ried, J.S.; Ding-Pfennigdorff, D.; Dietrich, A.; Rudolph, C.; Hahn, S.; Hennerici, W.; Asbrand, C.; Leeuw, T.; Strubing, C. High throughput screening of mitochondrial bioenergetics in human differentiated myotubes identifies novel enhancers of muscle performance in aged mice. Sci. Rep. 2018, 8, 9408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perls, T.T.; Wilmoth, J.; Levenson, R.; Drinkwater, M.; Cohen, M.; Bogan, H.; Joyce, E.; Brewster, S.; Kunkel, L.; Puca, A. Life-long sustained mortality advantage of siblings of centenarians. Proc. Natl. Acad. Sci. USA 2002, 99, 8442–8447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westendorp, R.G.; van Heemst, D.; Rozing, M.P.; Frolich, M.; Mooijaart, S.P.; Blauw, G.J.; Beekman, M.; Heijmans, B.T.; de Craen, A.J.; Slagboom, P.E.; et al. Nonagenarian siblings and their offspring display lower risk of mortality and morbidity than sporadic nonagenarians: The Leiden longevity study. J. Am. Geriatr. Soc. 2009, 57, 1634–1637. [Google Scholar] [CrossRef]
- Terry, D.F.; Wilcox, M.A.; McCormick, M.A.; Perls, T.T. Cardiovascular disease delay in centenarian offspring. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Terry, D.F.; Wilcox, M.A.; McCormick, M.A.; Pennington, J.Y.; Schoenhofen, E.A.; Andersen, S.L.; Perls, T.T. Lower all-cause, cardiovascular, and cancer mortality in centenarians’ offspring. J. Am. Geriatr. Soc. 2004, 52, 2074–2076. [Google Scholar] [CrossRef]
- Nadeau, J.H.; Topol, E.J. The genetics of health. Nat. Genet. 2006, 38, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.R.; Nayee, S.; Topol, E.J. Protective alleles and modifier variants in human health and disease. Nat. Rev. Genet. 2015, 16, 689–701. [Google Scholar] [CrossRef]
- Beekman, M.; Nederstigt, C.; Suchiman, H.E.; Kremer, D.; van der Breggen, R.; Lakenberg, N.; Alemayehu, W.G.; de Craen, A.J.; Westendorp, R.G.; Boomsma, D.I.; et al. Genome-wide association study (GWAS)-identified disease risk alleles do not compromise human longevity. Proc. Natl. Acad. Sci. USA 2010, 107, 18046–18049. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. New drugs cost US$2.6 billion to develop. Nat. Rev. Drug Discov. 2014, 13, 877. [Google Scholar] [CrossRef]
- Human Aging Genomic Resources. Available online: http://genomics.senescence.info/genes/search.php?search=&show=5&sort=1&organism=Mus+musculus&long_influence=pro&lifespan_effect=increase_decrease&search=&page=1 (accessed on 14 September 2022).
- Pedro de Magalhaes, J.; Thompson, L.; de Lima, I.; Gaskill, D.; Li, X.; Thornton, D.; Yang, C.; Palmer, D. A reassessment of genes modulating aging in mice using demographic measurements of the rate of aging. Genetics 2018, 208, 1617–1630. [Google Scholar] [CrossRef]
- Finch, C.E.; Pike, M.C. Maximum life span predictions from the Gompertz mortality model. J. Gerontol. A Biol. Sci. Med. Sci. 1996, 51, B183–B194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Chen, Y.F.; Wang, C.H.; Kao, C.H.; Zhuang, H.W.; Chen, C.C.; Chen, L.K.; Kirby, R.; Wei, Y.H.; Tsai, S.F.; et al. A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum. Mol. Genet. 2012, 21, 3956–3968. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Chou, T.Y.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upregulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Wu, C.Y.; Kirby, R.; Kao, C.H.; Tsai, T.F. A role for the CISD2 gene in lifespan control and human disease. Ann. N. Y. Acad. Sci. 2010, 1201, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Shen, Z.Q.; Wu, C.Y.; Teng, Y.C.; Liao, C.C.; Kao, C.H.; Chen, L.K.; Lin, C.H.; Tsai, T.F. Comparative proteomic profiling reveals a role for Cisd2 in skeletal muscle aging. Aging Cell 2018, 17, e12705. [Google Scholar] [CrossRef]
- Yeh, C.H.; Shen, Z.Q.; Hsiung, S.Y.; Wu, P.C.; Teng, Y.C.; Chou, Y.J.; Fang, S.W.; Chen, C.F.; Yan, Y.T.; Kao, L.S.; et al. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019, 17, e3000508. [Google Scholar] [CrossRef] [Green Version]
- Karmi, O.; Marjault, H.B.; Bai, F.; Roy, S.; Sohn, Y.S.; Darash Yahana, M.; Morcos, F.; Ioannidis, K.; Nahmias, Y.; Jennings, P.A.; et al. A VDAC1-mediated NEET protein chain transfers [2Fe-2S] clusters between the mitochondria and the cytosol and impacts mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2022, 119, e2121491119. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Huang, Y.L.; Teng, Y.C.; Wang, T.W.; Kao, C.H.; Yeh, C.H.; Tsai, T.F. CISD2 maintains cellular homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118954. [Google Scholar] [CrossRef]
- Hu, X.; Jogasuria, A.; Wang, J.; Kim, C.; Han, Y.; Shen, H.; Wu, J.; You, M. MitoNEET Deficiency Alleviates experimental alcoholic steatohepatitis in mice by stimulating endocrine adiponectin-Fgf15 Axis. J. Biol. Chem. 2016, 291, 22482–22495. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, W.J.; Benkovic, S.A.; Lin, L.; Yonutas, H.M.; Crish, S.D.; Sullivan, P.G.; Darvesh, A.S.; Brown, C.M.; Richardson, J.R. MitoNEET (CISD1) knockout mice show signs of striatal mitochondrial dysfunction and a Parkinson’s disease phenotype. ACS Chem. Neurosci. 2017, 8, 2759–2765. [Google Scholar] [CrossRef]
- Furihata, T.; Takada, S.; Kakutani, N.; Maekawa, S.; Tsuda, M.; Matsumoto, J.; Mizushima, W.; Fukushima, A.; Yokota, T.; Enzan, N.; et al. Cardiac-specific loss of mitoNEET expression is linked with age-related heart failure. Commun. Biol. 2021, 4, 138. [Google Scholar] [CrossRef]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Ajlouni, K.; Jarrah, N.; El-Khateeb, M.; El-Zaheri, M.; El Shanti, H.; Lidral, A. Wolfram syndrome: Identification of a phenotypic and genotypic variant from Jordan. Am. J. Med. Genet. 2002, 115, 61–65. [Google Scholar] [CrossRef]
- Chen, Y.F.; Kao, C.H.; Chen, Y.T.; Wang, C.H.; Wu, C.Y.; Tsai, C.Y.; Liu, F.C.; Yang, C.W.; Wei, Y.H.; Hsu, M.T.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Kao, C.H.; Kirby, R.; Tsai, T.F. Cisd2 mediates mitochondrial integrity and life span in mammals. Autophagy 2009, 5, 1043–1045. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.H.; Chou, Y.J.; Kao, C.H.; Tsai, T.F. Mitochondria and calcium homeostasis: Cisd2 as a big player in cardiac ageing. Int. J. Mol. Sci. 2020, 21, 9238. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S.; et al. CISD2 haploinsufficiency disrupts calcium homeostasis, causes nonalcoholic fatty liver disease, and promotes hepatocellular carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Chen, Y.F.; Wu, C.Y.; Wu, P.C.; Huang, Y.L.; Kao, C.H.; Lin, C.H.; Kao, L.S.; Tsai, T.F.; Wei, Y.H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [Green Version]
- Loncke, J.; Vervliet, T.; Parys, J.B.; Kaasik, A.; Bultynck, G. Uniting the divergent Wolfram syndrome-linked proteins WFS1 and CISD2 as modulators of Ca(2+) signaling. Sci. Signal 2021, 14, eabc6165. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C.; Nguyen, M.; Shore, G.C. BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy 2012, 8, 856–857. [Google Scholar] [CrossRef]
- Wang, Y.; Landry, A.P.; Ding, H. The mitochondrial outer membrane protein mitoNEET is a redox enzyme catalyzing electron transfer from FMNH2 to oxygen or ubiquinone. J. Biol. Chem. 2017, 292, 10061–10067. [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Teng, Y.C.; Lin, C.H.; Tsai, T.F. Cisd2 protects the liver from oxidative stress and ameliorates Western diet-induced nonalcoholic fatty liver disease. Antioxidants 2021, 10, 559. [Google Scholar] [CrossRef]
- Lin, C.C.; Chiang, T.H.; Sun, Y.Y.; Lin, M.S. Protective effects of CISD2 and influence of curcumin on CISD2 expression in aged animals and inflammatory Cell Model. Nutrients 2019, 11, 700. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.H.; Huang, Y.L.; Shen, Z.Q.; Lin, C.H.; Tsai, T.F. Cisd2 preserves the youthful pattern of the liver proteome during natural aging of mice. Biomedicines 2021, 9, 1229. [Google Scholar] [CrossRef]
- Pagan, L.U.; Gomes, M.J.; Gatto, M.; Mota, G.A.F.; Okoshi, K.; Okoshi, M.P. The role of oxidative stress in the aging heart. Antioxidants 2022, 11, 336. [Google Scholar] [CrossRef]
- Yeh, C.H.; Chou, Y.J.; Chu, T.K.; Tsai, T.F. Rejuvenating the aging heart by enhancing the expression of the Cisd2 prolongevity Gene. Int. J. Mol. Sci. 2021, 22, 11487. [Google Scholar] [CrossRef]
- Marzetti, E. Musculoskeletal aging and sarcopenia in the elderly. Int. J. Mol. Sci. 2022, 23, 2808. [Google Scholar] [CrossRef]
- Protasi, F.; Pietrangelo, L.; Boncompagni, S. Improper remodeling of organelles deputed to Ca(2+) handling and aerobic ATP production underlies muscle dysfunction in ageing. Int. J. Mol. Sci. 2021, 22, 6195. [Google Scholar] [CrossRef]
- Zampino, M.; Tanaka, T.; Ubaida-Mohien, C.; Fantoni, G.; Candia, J.; Semba, R.D.; Ferrucci, L. A plasma proteomic signature of skeletal muscle mitochondrial function. Int. J. Mol. Sci. 2020, 21, 9540. [Google Scholar] [CrossRef]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar]
- Sinha-Hikim, I.; Sinha-Hikim, A.P.; Parveen, M.; Shen, R.; Goswami, R.; Tran, P.; Crum, A.; Norris, K.C. Long-term supplementation with a cystine-based antioxidant delays loss of muscle mass in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 749–759. [Google Scholar] [CrossRef]
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of aging in the liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161. [Google Scholar] [CrossRef]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Lin, C.H.; Tsai, T.F. Cisd2 slows down liver aging and attenuates age-related metabolic dysfunction in male mice. Aging Cell 2021, 20, e13523. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Chitturi, S.; Farrell, G.C. Mechanisms and implications of age-related changes in the liver: Nonalcoholic Fatty liver disease in the elderly. Curr. Gerontol. Geriatr. Res. 2011, 2011, 831536. [Google Scholar] [CrossRef]
- Mohamad, B.; Shah, V.; Onyshchenko, M.; Elshamy, M.; Aucejo, F.; Lopez, R.; Hanouneh, I.A.; Alhaddad, R.; Alkhouri, N. Characterization of hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease (NAFLD) patients without cirrhosis. Hepatol. Int. 2016, 10, 632–639. [Google Scholar] [CrossRef]
- Loomba, R.; Ratziu, V.; Harrison, S.A.; Group, N.C.T.D.I.W. Expert panel review to compare FDA and EMA guidance on drug development and endpoints in nonalcoholic steatohepatitis. Gastroenterology 2022, 162, 680–688. [Google Scholar] [CrossRef]
- Gipson, I.K. Age-related changes and diseases of the ocular surface and cornea. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF48-53. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.C.; Lee, S.Y.; Kao, C.H.; Chen, L.H.; Shen, Z.Q.; Lai, C.H.; Tzeng, T.Y.; Pang, J.S.; Chiu, W.T.; Tsai, T.F. Cisd2 plays an essential role in corneal epithelial regeneration. EBioMedicine 2021, 73, 103654. [Google Scholar] [CrossRef]
- Tam, B.T.; Morais, J.A.; Santosa, S. Obesity and ageing: Two sides of the same coin. Obes. Rev. 2020, 21, e12991. [Google Scholar] [CrossRef]
- Campbell, L.E.; Langlais, P.R.; Day, S.E.; Coletta, R.L.; Benjamin, T.R.; De Filippis, E.A.; Madura, J.A., 2nd; Mandarino, L.J.; Roust, L.R.; Coletta, D.K. Identification of novel changes in human skeletal muscle proteome after Roux-en-Y gastric bypass surgery. Diabetes 2016, 65, 2724–2731. [Google Scholar] [CrossRef] [Green Version]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73. [Google Scholar] [CrossRef]
- Carapeto, P.V.; Aguayo-Mazzucato, C. Effects of exercise on cellular and tissue aging. Aging 2021, 13, 14522–14543. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Li, C.; Song, Y.; Wang, Y.; Bo, H.; Zhang, Y. Impact of exercise and aging on mitochondrial homeostasis in skeletal muscle: Roles of ROS and epigenetics. Cells 2022, 11, 2086. [Google Scholar] [CrossRef]
- Roberts, S.B.; Silver, R.E.; Das, S.K.; Fielding, R.A.; Gilhooly, C.H.; Jacques, P.F.; Kelly, J.M.; Mason, J.B.; McKeown, N.M.; Reardon, M.A.; et al. Healthy aging-nutrition matters: Start early and screen often. Adv. Nutr. 2021, 12, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Gao, Z.J.; Yu, X.; Wang, P. Dietary regulation in health and disease. Signal Transduct. Target Ther. 2022, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Lages, A.; Lopes, V.; Horta, J.; Espregueira-Mendes, J.; Andrade, R.; Rebelo-Marques, A. Therapeutics that can potentially replicate or augment the anti-aging effects of physical exercise. Int. J. Mol. Sci. 2022, 23, 9957. [Google Scholar] [CrossRef]
- Teng, Y.C.; Wang, J.Y.; Chi, Y.H.; Tsai, T.F. Exercise and the Cisd2 prolongevity gene: Two promising strategies to delay the aging of skeletal muscle. Int. J. Mol. Sci. 2020, 21, 9059. [Google Scholar] [CrossRef]
- Yokokawa, T.; Kido, K.; Suga, T.; Sase, K.; Isaka, T.; Hayashi, T.; Fujita, S. Exercise training increases CISD family protein expression in murine skeletal muscle and white adipose tissue. Biochem. Biophys. Res. Commun. 2018, 506, 571–577. [Google Scholar] [CrossRef]
- Benameur, T.; Soleti, R.; Panaro, M.A.; La Torre, M.E.; Monda, V.; Messina, G.; Porro, C. Curcumin as prospective anti-aging natural compound: Focus on brain. Molecules 2021, 26, 4794. [Google Scholar] [CrossRef]
- Mahjoob, M.; Stochaj, U. Curcumin nanoformulations to combat aging-related diseases. Ageing Res. Rev. 2021, 69, 101364. [Google Scholar] [CrossRef]
- Zia, A.; Farkhondeh, T.; Pourbagher-Shahri, A.M.; Samarghandian, S. The role of curcumin in aging and senescence: Molecular mechanisms. Biomed. Pharmacother. 2021, 134, 111119. [Google Scholar] [CrossRef]
- Lin, C.C.; Chiang, T.H.; Chen, W.J.; Sun, Y.Y.; Lee, Y.H.; Lin, M.S. CISD2 serves a novel role as a suppressor of nitric oxide signalling and curcumin increases CISD2 expression in spinal cord injuries. Injury 2015, 46, 2341–2350. [Google Scholar] [CrossRef]
- Kung, W.M.; Lin, M.S. Beneficial impacts of alpha-eleostearic Acid from wild bitter melon and curcumin on promotion of CDGSH iron-sulfur domain 2: Therapeutic roles in CNS injuries and diseases. Int. J. Mol. Sci. 2021, 22, 3289. [Google Scholar] [CrossRef]
- Kung, W.M.; Lin, C.C.; Kuo, C.Y.; Juin, Y.C.; Wu, P.C.; Lin, M.S. Wild bitter melon exerts anti-inflammatory effects by upregulating injury-attenuated CISD2 expression following spinal cord injury. Behav. Neurol. 2020, 2020, 1080521. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.J.; Saito, K.J.; Aw, W.P.; Takahashi, S.; Hanate, M.; Hasebe, Y.; Kato, H. Transcriptional profiling in rats and an ex vivo analysis implicate novel beneficial function of egg shell membrane in liver fibrosis. J. Funct. Foods 2013, 5, 1611–1619. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.H.; Shen, Z.Q.; Wang, T.W.; Kao, C.H.; Teng, Y.C.; Yeh, T.K.; Lu, C.K.; Tsai, T.F. Hesperetin promotes longevity and delays aging via activation of Cisd2 in naturally aged mice. J. Biomed. Sci. 2022, 29, 53. [Google Scholar] [CrossRef] [PubMed]
- Pyrzynska, K. Hesperidin: A review on extraction methods, stability and biological activities. Nutrients 2022, 14, 2387. [Google Scholar] [CrossRef] [PubMed]
- Roohbakhsh, A.; Parhiz, H.; Soltani, F.; Rezaee, R.; Iranshahi, M. Neuropharmacological properties and pharmacokinetics of the citrus flavonoids hesperidin and hesperetin—A mini-review. Life Sci. 2014, 113, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Cruz-Martins, N.; Butnariu, M.; Sarac, I.; Bagiu, I.C.; Ezzat, S.M.; Wang, J.; Koay, A.; Sheridan, H.; Adetunji, C.O.; et al. Hesperetin’s health potential: Moving from preclinical to clinical evidence and bioavailability issues, to upcoming strategies to overcome current limitations. Crit. Rev. Food Sci. Nutr. 2022, 62, 4449–4464. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Walkowiak, J.; Pietrzak, R.; Bazan-Wozniak, A.; Cielecka-Piontek, J. Bioavailability of hesperidin and its aglycone hesperetin-compounds found in citrus fruits as a parameter conditioning the pro-health potential (neuroprotective and antidiabetic activity)-mini-review. Nutrients 2022, 14, 2647. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Xu, S.; Ren, J.; Tang, L.; Gong, J.; Lin, Y.; Fang, H.; Su, D. Hesperetin, a promising treatment option for diabetes and related complications: A literature review. J. Agric. Food Chem. 2022, 70, 8582–8592. [Google Scholar] [CrossRef]
- Ortiz, A.C.; Fideles, S.O.M.; Reis, C.H.B.; Bellini, M.Z.; Pereira, E.; Pilon, J.P.G.; de Marchi, M.A.; Detregiachi, C.R.P.; Flato, U.A.P.; Trazzi, B.F.M.; et al. Therapeutic effects of citrus flavonoids neohesperidin, hesperidin and its aglycone, hesperetin on bone health. Biomolecules 2022, 12, 626. [Google Scholar] [CrossRef]
- Aron, L.; Zullo, J.; Yankner, B.A. The adaptive aging brain. Curr. Opin. Neurobiol. 2022, 72, 91–100. [Google Scholar] [CrossRef]
- Luo, Y.; Fan, H.; Tan, X.; Li, Z. Hesperetin rescues emotional memory and synaptic plasticity deficit in aged rats. Behav. Neurosci. 2021, 135, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Li, Q.; Li, J. Understanding the unique microenvironment in the aging liver. Front. Med. 2022, 9, 842024. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Adeniji, N.T.; Fan, W.; Kunimoto, K.; Torok, N.J. Non-alcoholic fatty liver disease and liver fibrosis during aging. Aging Dis. 2022, 13, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Miler, M.; Zivanovic, J.; Ajdzanovic, V.; Orescanin-Dusic, Z.; Milenkovic, D.; Konic-Ristic, A.; Blagojevic, D.; Milosevic, V.; Sosic-Jurjevic, B. Citrus flavanones naringenin and hesperetin improve antioxidant status and membrane lipid compositions in the liver of old-aged Wistar rats. Exp. Gerontol. 2016, 84, 49–60. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Denic, A.; Glassock, R.J.; Rule, A.D. The kidney in normal aging: A comparison with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2022, 17, 137–139. [Google Scholar] [CrossRef]
- Franzin, R.; Stasi, A.; Ranieri, E.; Netti, G.S.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Castellano, G. Targeting premature renal aging: From molecular mechanisms of cellular senescence to senolytic trials. Front. Pharmacol. 2021, 12, 630419. [Google Scholar] [CrossRef]
- Kim, J.Y.; Jung, K.J.; Choi, J.S.; Chung, H.Y. Modulation of the age-related nuclear factor-kappaB (NF-kappaB) pathway by hesperetin. Aging Cell 2006, 5, 401–411. [Google Scholar] [CrossRef]
- Chen, J.; Xu, J.; Huang, P.; Luo, Y.; Shi, Y.; Ma, P. The potential applications of traditional Chinese medicine in Parkinson’s disease: A new opportunity. Biomed. Pharmacother. 2022, 149, 112866. [Google Scholar] [CrossRef]
- Evans, J.A.; Mendonca, P.; Soliman, K.F.A. Neuroprotective effects and therapeutic potential of the citrus flavonoid hesperetin in neurodegenerative diseases. Nutrients 2022, 14, 2228. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Hahm, J.R.; Kim, M.O. Antioxidant and anti-inflammatory effects of citrus flavonoid hesperetin: Special focus on neurological disorders. Antioxidants 2020, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin confers neuroprotection by regulating Nrf2/TLR4/NF-kappaB signaling in an Abeta mouse model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef] [PubMed]
- Babylon, L.; Grewal, R.; Stahr, P.L.; Eckert, R.W.; Keck, C.M.; Eckert, G.P. Hesperetin nanocrystals improve mitochondrial function in a cell model of early Alzheimer disease. Antioxidants 2021, 10, 1003. [Google Scholar] [CrossRef] [PubMed]
- Kiasalari, Z.; Khalili, M.; Baluchnejadmojarad, T.; Roghani, M. Protective effect of oral hesperetin against unilateral striatal 6-hydroxydopamine damage in the rat. Neurochem. Res. 2016, 41, 1065–1072. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Wang, L.; Gu, Z.W.; Huang, Z.G.; Fug, H.; Liu, Q.S. Hesperetin protects SH-SY5Y cells against 6-hydroxydopamine-induced neurotoxicity via activation of NRF2/ARE signaling pathways. Trop. J. Pharm. Res. 2020, 19, 1197–1201. [Google Scholar] [CrossRef]
- Heruye, S.H.; Maffofou Nkenyi, L.N.; Singh, N.U.; Yalzadeh, D.; Ngele, K.K.; Njie-Mbye, Y.F.; Ohia, S.E.; Opere, C.A. Current trends in the pharmacotherapy of cataracts. Pharmaceuticals 2020, 13, 15. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, Y.; Oka, M.; Tamura, H.; Takehana, M. Effect of hesperetin on chaperone activity in selenite-induced cataract. Open Med. 2016, 11, 183–189. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Oka, M.; Bando, M.; Takehana, M. Hesperetin prevents selenite-induced cataract in rats. Mol. Vis. 2015, 21, 804–810. [Google Scholar]
- Nakazawa, Y.; Pauze, M.; Fukuyama, K.; Nagai, N.; Funakoshi-Tago, M.; Sugai, T.; Tamura, H. Effect of hesperetin derivatives on the development of seleniteinduced cataracts in rats. Mol. Med. Rep. 2018, 18, 1043–1050. [Google Scholar]
- Najjar, R.S.; Feresin, R.G. Protective role of polyphenols in heart failure: Molecular targets and cellular mechanisms underlying their therapeutic potential. Int. J. Mol. Sci. 2021, 22, 1668. [Google Scholar] [CrossRef]
- Sugasawa, N.; Katagi, A.; Kurobe, H.; Nakayama, T.; Nishio, C.; Takumi, H.; Higashiguchi, F.; Aihara, K.I.; Shimabukuro, M.; Sata, M.; et al. Inhibition of atherosclerotic plaque development by oral administration of alpha-glucosyl hesperidin and water-dispersible hesperetin in apolipoprotein E knockout mice. J. Am. Coll. Nutr. 2019, 38, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zou, D.; Chen, X.; Wu, H.; Xu, D. Hesperetin inhibits foam cell formation and promotes cholesterol efflux in THP-1-derived macrophages by activating LXRalpha signal in an AMPK-dependent manner. J. Physiol. Biochem. 2021, 77, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, P.; Mohan, T.; Ravi, D.B.; Kishore Kumar, S.N.; Srinivasan, A.; Chakrapani, L.N.; Singh, A.; Varadharaj, S.; Kalaiselvi, P. Targeting the Nrf2/ARE signalling pathway to mitigate isoproterenol-induced cardiac hypertrophy: Plausible role of hesperetin in redox homeostasis. Oxid Med. Cell Longev 2020, 2020, 9568278. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, J.; Liu, M.; Zhang, M.; Xue, Y.; Zhang, Y.; Han, X.; Jing, X.; Chu, L. Hesperetin modulates the Sirt1/Nrf2 signaling pathway in counteracting myocardial ischemia through suppression of oxidative stress, inflammation, and apoptosis. Biomed. Pharmacother. 2021, 139, 111552. [Google Scholar] [CrossRef] [PubMed]
- Korac, B.; Kalezic, A.; Pekovic-Vaughan, V.; Korac, A.; Jankovic, A. Redox changes in obesity, metabolic syndrome, and diabetes. Redox. Biol. 2021, 42, 101887. [Google Scholar] [CrossRef]
- Jayaraman, R.; Subramani, S.; Sheik Abdullah, S.H.; Udaiyar, M. Antihyperglycemic effect of hesperetin, a citrus flavonoid, extenuates hyperglycemia and exploring the potential role in antioxidant and antihyperlipidemic in streptozotocin-induced diabetic rats. Biomed. Pharmacother. 2018, 97, 98–106. [Google Scholar] [CrossRef]
- Wang, S.W.; Sheng, H.; Bai, Y.F.; Weng, Y.Y.; Fan, X.Y.; Zheng, F.; Fu, J.Q.; Zhang, F. Inhibition of histone acetyltransferase by naringenin and hesperetin suppresses Txnip expression and protects pancreatic beta cells in diabetic mice. Phytomedicine 2021, 88, 153454. [Google Scholar] [CrossRef]
- Dhanya, R.; Jayamurthy, P. In vitro evaluation of antidiabetic potential of hesperidin and its aglycone hesperetin under oxidative stress in skeletal muscle cell line. Cell Biochem. Funct. 2020, 38, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Reversal of insulin resistance in overweight and obese subjects by trans-resveratrol and hesperetin combination-link to dysglycemia, blood pressure, dyslipidemia, and low-grade inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved glycemic control and vascular function in overweight and obese subjects by glyoxalase 1 inducer formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Gosmain, Y.; Dif, N.; Berbe, V.; Loizon, E.; Rieusset, J.; Vidal, H.; Lefai, E. Regulation of SREBP-1 expression and transcriptional action on HKII and FAS genes during fasting and refeeding in rat tissues. J. Lipid Res. 2005, 46, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.M.; Lee, S.H.; Jung, Y.; Lee, Y.; Yoon, J.H.; Choi, J.Y.; Hwang, C.Y.; Son, Y.H.; Park, S.S.; Hwang, G.S.; et al. FABP3-mediated membrane lipid saturation alters fluidity and induces ER stress in skeletal muscle with aging. Nat. Commun. 2020, 11, 5661. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Hesselink, M.K. The role of uncoupling protein 3 in fatty acid metabolism: Protection against lipotoxicity? Proc. Nutr. Soc. 2004, 63, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, D.; Hashidate, T.; Shimizu, T.; Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014, 55, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, M. Chaperonin-containing TCP-1 subunit 2-mediated aggrephagy: A potential target for treating neurodegeneration. Clin. Transl. Med. 2022, 12, e1027. [Google Scholar] [CrossRef]
- de Araujo, M.E.; Stasyk, T.; Taub, N.; Ebner, H.L.; Furst, B.; Filipek, P.; Weys, S.R.; Hess, M.W.; Lindner, H.; Kremser, L.; et al. Stability of the endosomal scaffold protein LAMTOR3 depends on heterodimer assembly and proteasomal degradation. J. Biol. Chem. 2013, 288, 18228–18242. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, G.; Hu, J.S.; Zhang, G.Q.; Chen, H.Z.; Yuan, Y.; Li, Y.L.; Lv, X.J.; Tian, F.Y.; Pan, S.H.; et al. RB1CC1-enhanced autophagy facilitates PSCs activation and pancreatic fibrogenesis in chronic pancreatitis. Cell Death Dis. 2018, 9, 952. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Li, Y.; Wang, G.; Zhou, Z.; Song, G.; Feng, Q.; Zhao, Y.; Mi, W.; Ma, Z.; Dong, C. Molecular basis for recognition of Gly/N-degrons by CRL2(ZYG11B) and CRL2(ZER1). Mol. Cell 2021, 81, 3262–3274.e3. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Z.; Li, L.; Qin, Y.R.; Liu, H.; Jiang, C.; Zeng, T.T.; Li, M.Q.; Xie, D.; Li, Y.; et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-kappaB signaling. Nat. Commun. 2018, 9, 1423. [Google Scholar] [CrossRef] [Green Version]
- Rennie, M.J.; Bowtell, J.L.; Bruce, M.; Khogali, S.E. Interaction between glutamine availability and metabolism of glycogen, tricarboxylic acid cycle intermediates and glutathione. J. Nutr. 2001, 131 (Suppl. 9), 2488S–2490S; discussion 2496S–2497S. [Google Scholar] [CrossRef] [Green Version]
- Yien, Y.Y.; Robledo, R.F.; Schultz, I.J.; Takahashi-Makise, N.; Gwynn, B.; Bauer, D.E.; Dass, A.; Yi, G.; Li, L.; Hildick-Smith, G.J.; et al. TMEM14C is required for erythroid mitochondrial heme metabolism. J. Clin. Investig. 2014, 124, 4294–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.Q.; Huang, Y.L.; Tsai, T.F. Cisd2 haploinsufficiency: A driving force for hepatocellular carcinoma. Mol. Cell Oncol. 2018, 5, e1441627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.L.; Shen, Z.Q.; Tsai, T.F. Enhancing CISD2 expression to retard liver aging. Aging 2022, 14, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Demontis, F.; Piccirillo, R.; Goldberg, A.L.; Perrimon, N. The influence of skeletal muscle on systemic aging and lifespan. Aging Cell 2013, 12, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Wang, L.; You, W.; Shan, T. Myokines mediate the cross talk between skeletal muscle and other organs. J. Cell Physiol. 2021, 236, 2393–2412. [Google Scholar] [CrossRef]



| CISD2 Dependent | CISD2 Independent | |
|---|---|---|
| A. Lipid Metabolism | ||
| Lipogenesis | Srebp1 | |
| Fatty acid oxidation | Slc22a5, Fabp3, Cpt1, and Ucp3 | |
| Malonyl CoA metabolism | Mlycd | |
| Pyruvate metabolism | Pdk4 | |
| Phospholipid synthesis | Agpat1 | Agpat3 and Pla2g12a |
| Others | Abcd3, Scd2, Pdss2, Cot7, Insig1, Synj2, Nceh1, and Pigq | |
| B. Proteostasis | ||
| Ubiquitin proteasome system | Zyg11b, Tspan15, Hspbp1 | |
| Autophagy-lysosome system | Rb1cc1, Cct2, Rab4a, Scarb2, and Lamtor3 | |
| Others | Serp1, Paip1, Gspt1, Rpl34, Eif3g, Lrpprc, Mrps9, Cct4, Sugt1, Cox17, Cd24a, Rap1gds1, Kank1, Spag9, Pdss2, Nceh1, Fiz1, Adck1, Cript, and Ccdc47 | |
| C. Nitrogen, Protein & Amino Acid Metabolism | ||
| Methionine | Mat2b | |
| Glutamate | Slc25a22 | |
| Proline | Pycrl | |
| Protein transport | Timm17b | |
| Others | Irf2, Smndc1, Zhx2, Cct4, Srebf1, Csde1, Mrps9, Tbx15, Ddx1, Pura, Acot7, Rap1gds1, Rb1cc1, Fiz1, Gspt1, Gm20390, Cct2, Hnrpll, Hist2h2aa1, and Lrpprc | Cops2, Hfe2, Taf9b, Slu7, and Tmem14c |
| D. Miscellaneous (Aging and Mitochondrial Function) | ||
| Aging | Serp1 and Coq7 | |
| Mitochondrial function | Cox17, Slc22a5, Immt, Cd24a, Synj2, Coq7, Lrpprc, Cox4i1, and Ndufb9 | |
| Immune response and inflammation | Ddx1, Pdk4, Fgfbp1, Slc22a5, Sugt1, and Cd24a | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, C.-H.; Shen, Z.-Q.; Lin, C.-C.; Lu, C.-K.; Tsai, T.-F. Rejuvenation: Turning Back Time by Enhancing CISD2. Int. J. Mol. Sci. 2022, 23, 14014. https://doi.org/10.3390/ijms232214014
Yeh C-H, Shen Z-Q, Lin C-C, Lu C-K, Tsai T-F. Rejuvenation: Turning Back Time by Enhancing CISD2. International Journal of Molecular Sciences. 2022; 23(22):14014. https://doi.org/10.3390/ijms232214014
Chicago/Turabian StyleYeh, Chi-Hsiao, Zhao-Qing Shen, Ching-Cheng Lin, Chung-Kuang Lu, and Ting-Fen Tsai. 2022. "Rejuvenation: Turning Back Time by Enhancing CISD2" International Journal of Molecular Sciences 23, no. 22: 14014. https://doi.org/10.3390/ijms232214014
APA StyleYeh, C.-H., Shen, Z.-Q., Lin, C.-C., Lu, C.-K., & Tsai, T.-F. (2022). Rejuvenation: Turning Back Time by Enhancing CISD2. International Journal of Molecular Sciences, 23(22), 14014. https://doi.org/10.3390/ijms232214014