
Design and Evaluation of Novel HIV-1 Protease Inhibitors Containing Phenols or Polyphenols as P2 Ligands with High Activity against DRV-Resistant HIV-1 Variants
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Chemistry
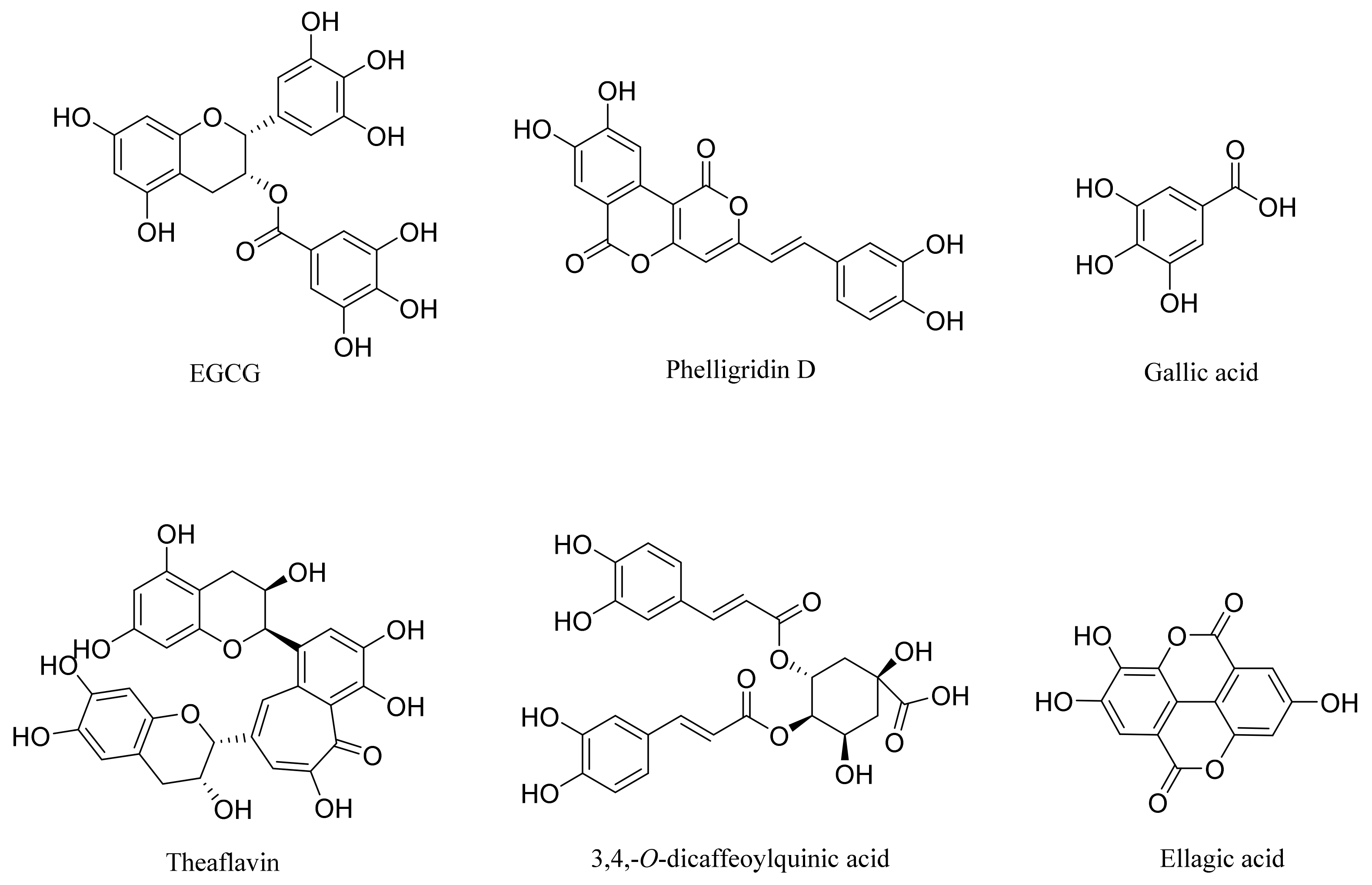
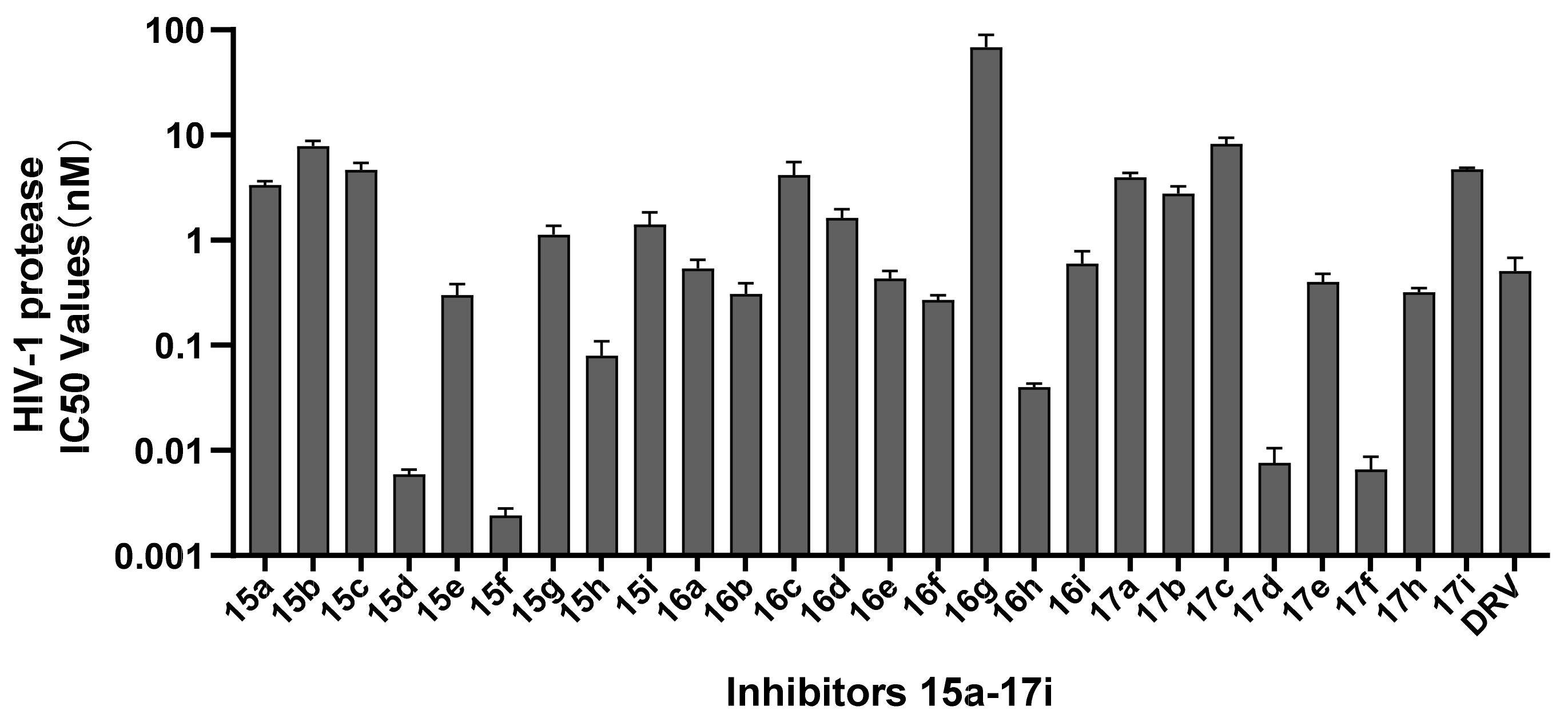
2.2. HIV-1 Enzymatic Inhibitory Activity Assay
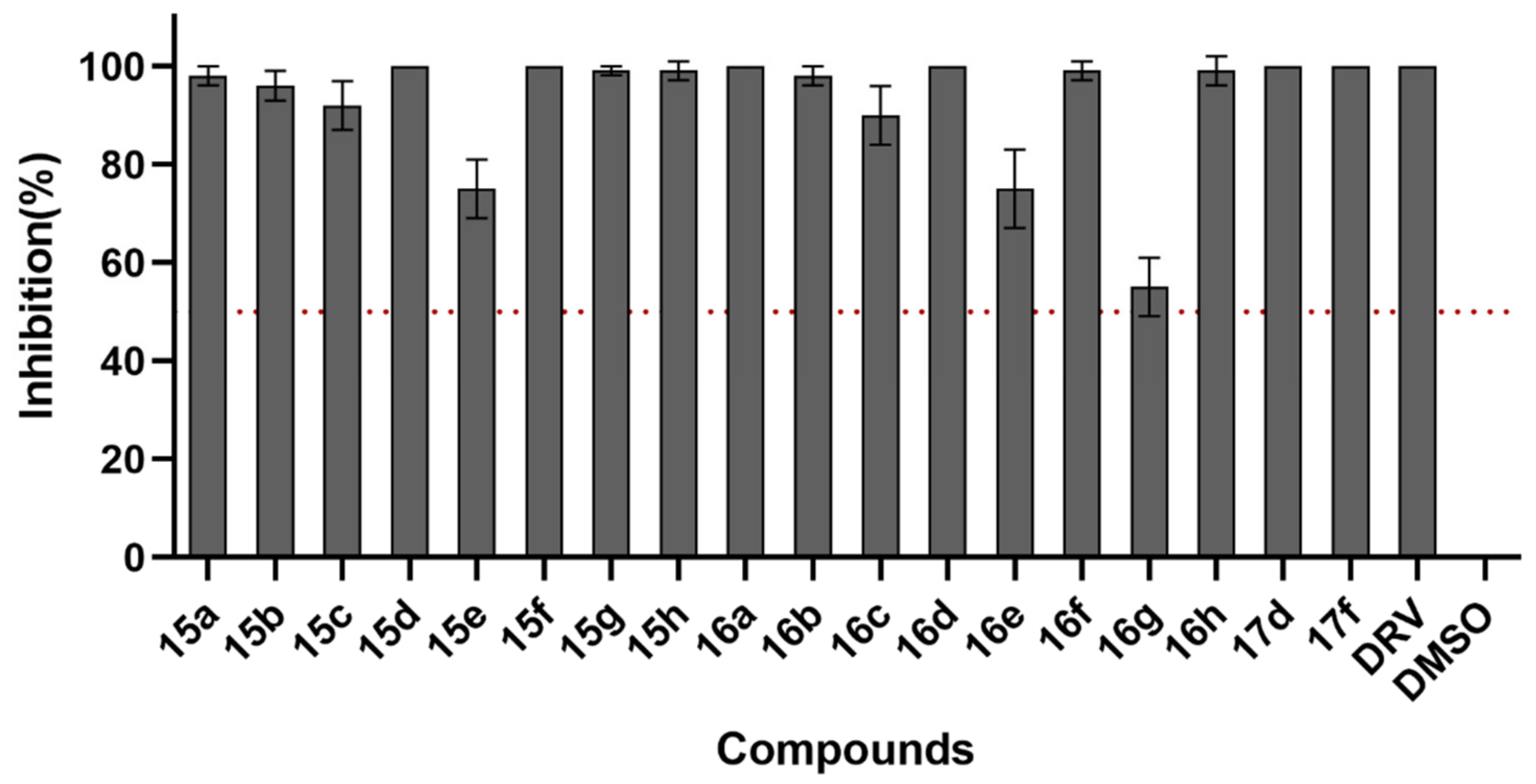
2.3. HIV-1 Infectivity Assay
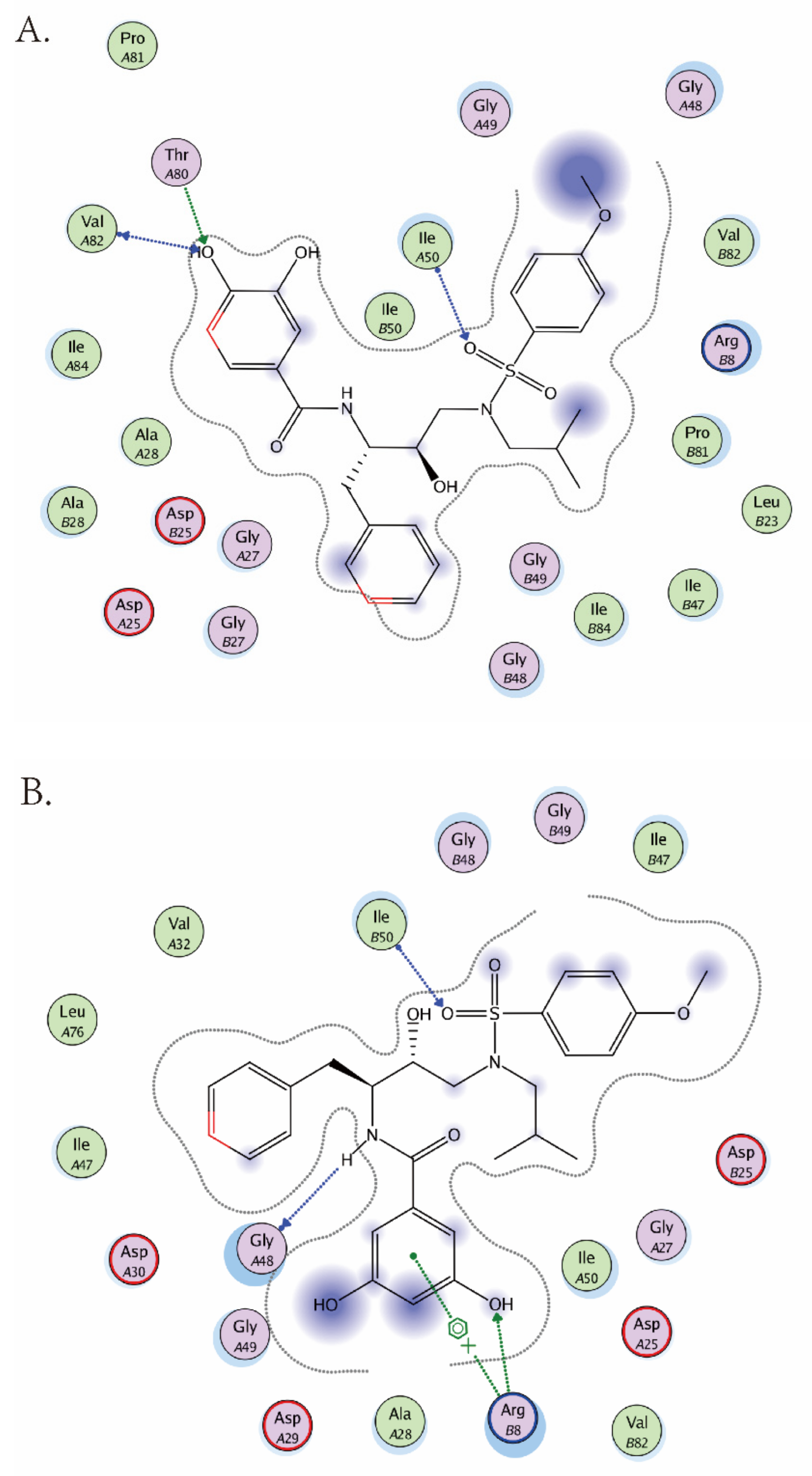
2.4. Molecular Modeling Studies
2.5. Correlation of Phenol or Polyphenol Analogs
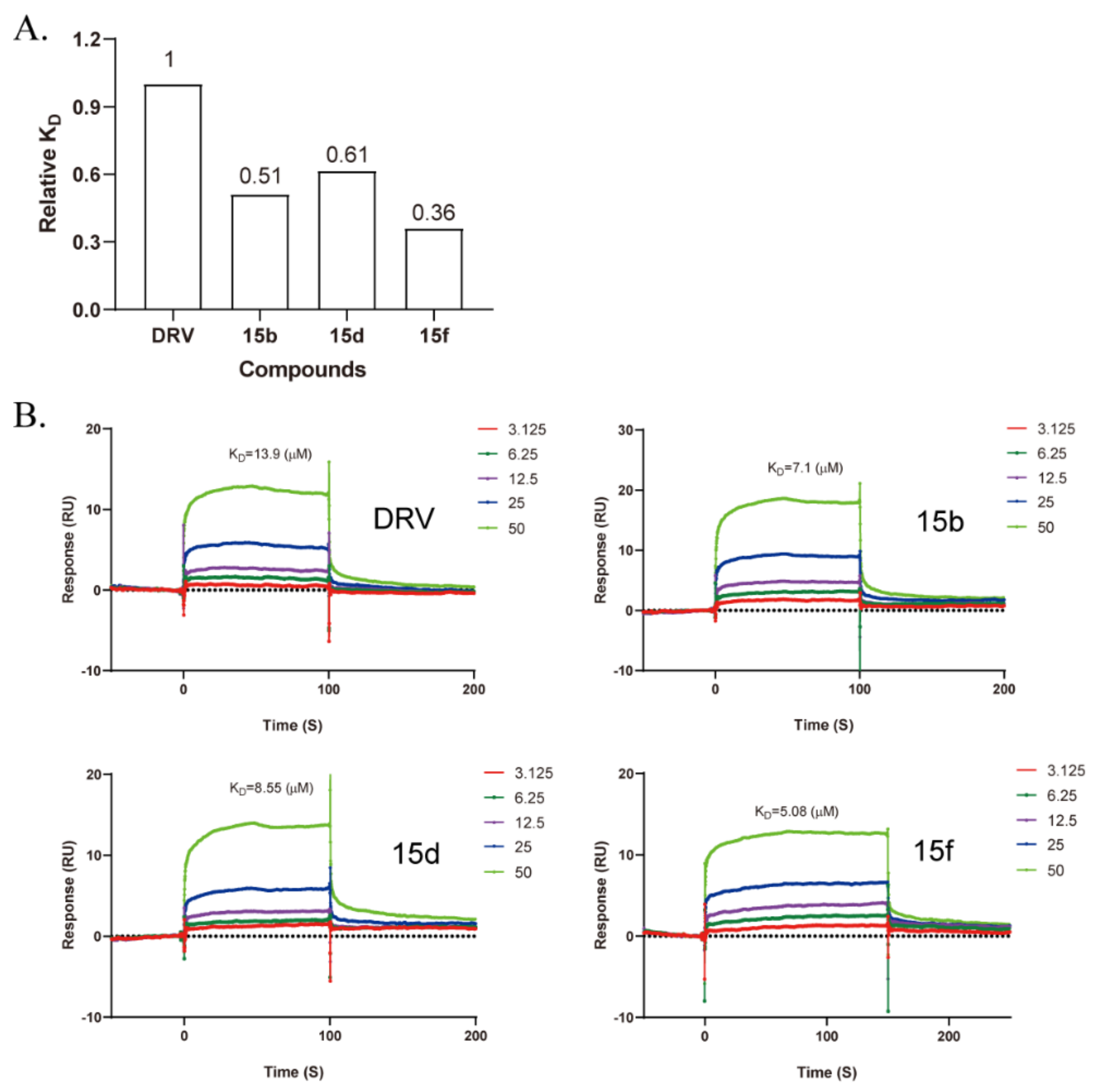
2.6. Binding Assay
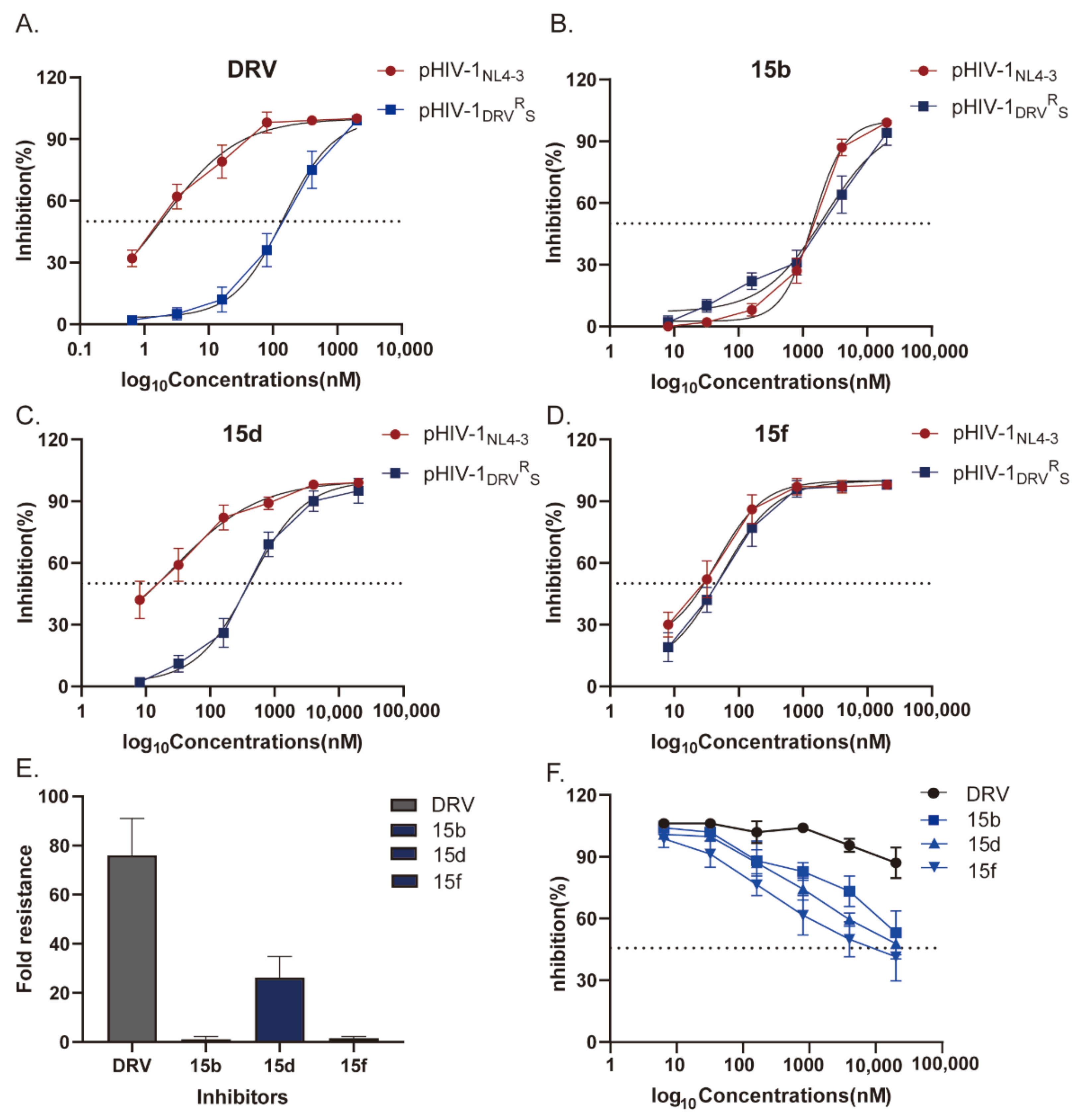
2.7. Antiviral Activity against the DRV-Resistant HIV-1 Variant
3. Discussion
4. Materials and Methods
4.1. Cells, Viruses, Plasmids, and Reagents
4.2. In Vitro Assay for HIV-1 Protease Inhibition
4.3. Cytotoxicity Assay
4.4. HIV-1 Infectivity Assay
4.5. Construction of DRV-Resistant pNL4-3-E-R- Cloning (pHIV-1DRVRS)
4.6. Molecular Modeling
4.7. Binding Assay by SPR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman-Kien, A.E. Disseminated Kaposi’s sarcoma syndrome in young homosexual men. J. Am. Acad. Dermatol. 1981, 5, 468–471. [Google Scholar] [CrossRef]
- Bevan, R.J.; Harrison, P.T.C. Threshold and non-threshold chemical carcinogens: A survey of the present regulatory landscape. Regul. Toxicol. Pharmacol. 2017, 88, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, L.; Vella, S. A brief history of antiretroviral therapy of HIV infection: Success and challenges. Ann. Ist. Super. Sanita 2011, 47, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.S.; Lima, V.D.; Barrios, R.; Yip, B.; Wood, E.; Kerr, T.; Shannon, K.; Harrigan, P.R.; Hogg, R.S.; Daly, P.; et al. Association of highly active antiretroviral therapy coverage, population viral load, and yearly new HIV diagnoses in British Columbia, Canada: A population-based study. Lancet 2010, 376, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Braitstein, P.; Brinkhof, M.W.; Dabis, F.; Schechter, M.; Boulle, A.; Miotti, P.; Wood, R.; Laurent, C.; Sprinz, E.; Seyler, C.; et al. Mortality of HIV-1-infected patients in the first year of antiretroviral therapy: Comparison between low-income and high-income countries. Lancet 2006, 367, 817–824. [Google Scholar] [PubMed]
- Este, J.A.; Cihlar, T. Current status and challenges of antiretroviral research and therapy. Antivir. Res. 2010, 85, 25–33. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [Green Version]
- Kohl, N.; Emini, E.; Schleif, W.; Davis, L.J.; Heimbach, J.C.; Dixon, R.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686. [Google Scholar] [CrossRef] [Green Version]
- Mitsuya, Y.; Liu Tommy, F.; Rhee, S.Y.; Fessel, W.J.; Shafer Robert, W. Prevalence of Darunavir Resistance–Associated Mutations: Patterns of Occurrence and Association with Past Treatment. J. Infect. Dis. 2007, 196, 1177–1179. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rao, K.V.; Nyalapatla, P.R.; Osswald, H.L.; Martyr, C.D.; Aoki, M.; Hayashi, H.; Agniswamy, J.; Wang, Y.-F.; Bulut, H.; et al. Design and Development of Highly Potent HIV-1 Protease Inhibitors with a Crown-Like Oxotricyclic Core as the P2-Ligand To Combat Multidrug-Resistant HIV Variants. J. Med. Chem. 2017, 60, 4267–4278. [Google Scholar] [CrossRef]
- Aoki, M.; Das, D.; Hayashi, H.; Aoki-Ogata, H.; Takamatsu, Y.; Ghosh, A.K.; Mitsuya, H. Mechanism of Darunavir (DRV)’s High Genetic Barrier to HIV-1 Resistance: A Key V32I Substitution in Protease Rarely Occurs, but Once It Occurs, It Predisposes HIV-1 To Develop DRV Resistance. MBio 2018, 9, e02425-17. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Nyalapatla, P.R.; Kovela, S.; Rao, K.V.; Brindisi, M.; Osswald, H.L.; Amano, M.; Aoki, M.; Agniswamy, J.; Wang, Y.-F.; et al. Design and Synthesis of Highly Potent HIV-1 Protease Inhibitors Containing Tricyclic Fused Ring Systems as Novel P2 Ligands: Structure-Activity Studies, Biological and X-ray Structural Analysis. J. Med. Chem. 2018, 61, 4561–4577. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Williams, J.N.; Ho, R.Y.; Simpson, H.M.; Hattori, S.-I.; Hayashi, H.; Agniswamy, J.; Wang, Y.-F.; Weber, I.T.; Mitsuya, H. Design and Synthesis of Potent HIV-1 Protease Inhibitors Containing Bicyclic Oxazolidinone Scaffold as the P2 Ligands: Structure-Activity Studies and Biological and X-ray Structural Studies. J. Med. Chem. 2018, 61, 9722–9737. [Google Scholar] [CrossRef]
- Brik, A.; Wong, C.H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef]
- Lefebvre, E.; Schiffer, C.A. Resilience to resistance of HIV-1 protease inhibitors: Profile of darunavir. AIDS Rev. 2008, 10, 131–142. [Google Scholar]
- Berti, F.; Frecer, V.; Miertus, S. Inhibitors of HIV-protease from computational design. A history of theory and synthesis still to be fully appreciated. Curr. Pharm. Des. 2014, 20, 3398–3411. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc. Chem. Res. 2008, 41, 78–86. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Sridhar, P.R.; Leshchenko, S.; Hussain, A.K.; Li, J.; Kovalevsky, A.Y.; Walters, D.E.; Wedekind, J.E.; Grum-Tokars, V.; Das, D.; et al. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J. Med. Chem. 2006, 49, 5252–5261. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Ramu Sridhar, P.; Kumaragurubaran, N.; Koh, Y.; Weber, I.T.; Mitsuya, H. Bis-Tetrahydrofuran: A Privileged Ligand for Darunavir and a New Generation of HIV Protease Inhibitors That Combat Drug Resistance. ChemMedChem 2006, 1, 939–950. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Anderson, D.D.; Weber, I.T.; Mitsuya, H. Enhancing protein backbone binding--a fruitful concept for combating drug-resistant HIV. Angew. Chem. 2012, 51, 1778–1802. [Google Scholar] [CrossRef]
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Yu, X.; Osswald, H.L.; Agniswamy, J.; Wang, Y.-F.; Amano, M.; Weber, I.T.; Mitsuya, H. Structure-Based Design of Potent HIV-1 Protease Inhibitors with Modified P1-Biphenyl Ligands: Synthesis, Biological Evaluation, and Enzyme–Inhibitor X-ray Structural Studies. J. Med. Chem. 2015, 58, 5334–5343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agniswamy, J.; Shen, C.-H.; Wang, Y.-F.; Ghosh, A.K.; Rao, K.V.; Xu, C.-X.; Sayer, J.M.; Louis, J.M.; Weber, I.T. Extreme Multidrug Resistant HIV-1 Protease with 20 Mutations Is Resistant to Novel Protease Inhibitors with P1′-Pyrrolidinone or P2-Tris-tetrahydrofuran. J. Med. Chem. 2013, 56, 4017–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meher, B.R.; Wang, Y. Interaction of I50V mutant and I50L/A71V double mutant HIV-protease with inhibitor TMC114 (darunavir): Molecular dynamics simulation and binding free energy studies. J. Phys. Chem. B. 2012, 116, 1884–1900. [Google Scholar] [CrossRef] [PubMed]
- Parai, M.K.; Huggins, D.J.; Cao, H.; Nalam, M.N.L.; Ali, A.; Schiffer, C.A.; Tidor, B.; Rana, T.M. Design, synthesis, and biological and structural evaluations of novel HIV-1 protease inhibitors to combat drug resistance. J. Med. Chem. 2012, 55, 6328–6341. [Google Scholar] [CrossRef] [Green Version]
- Mirani, A.; Kundaikar, H.; Velhal, S.; Patel, V.; Bandivdekar, A.; Degani, M.; Patravale, V. Evaluation of Phytopolyphenols for their gp120-CD4 Binding Inhibitory Properties by In Silico Molecular Modelling & In Vitro Cell Line Studies. Curr. HIV Res. 2019, 17, 102–113. [Google Scholar] [CrossRef]
- Calland, N.; Sahuc, M.-E.; Belouzard, S.; Pène, V.; Bonnafous, P.; Mesalam, A.A.; Deloison, G.; Descamps, V.; Sahpaz, S.; Wychowski, C.; et al. Polyphenols Inhibit Hepatitis C Virus Entry by a New Mechanism of Action. J. Virol. 2015, 89, 10053–10063. [Google Scholar] [CrossRef] [Green Version]
- Fassina, G.; Buffa, A.; Benelli, R.; Varnier, O.E.; Noonan, D.M.; Albini, A. Polyphenolic antioxidant (-)-epigallocatechin-3-gallate from green tea as a candidate anti-HIV agent. AIDS 2002, 16, 939–941. [Google Scholar] [CrossRef]
- Hwang, B.S.; Lee, I.-K.; Choi, H.J.; Yun, B.-S. Anti-influenza activities of polyphenols from the medicinal mushroom Phellinus baumii. Bioorg. Med. Chem. Lett. 2015, 25, 3256–3260. [Google Scholar] [CrossRef]
- Kratz, J.M.; Andrighetti-Fröhner, C.R.; Kolling, D.J.; Leal, P.C.; Cirne-Santos, C.C.; Yunes, R.A.; Nunes, R.J.; Trybala, E.; Bergström, T.; Frugulhetti, I.C.P.P.; et al. Anti-HSV-1 and anti-HIV-1 activity of gallic acid and pentyl gallate. Mem. Inst. Oswaldo Cruz. 2008, 103, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Ohba, M.; Oka, T.; Ando, T.; Arahata, S.; Ikegaya, A.; Takagi, H.; Ogo, N.; Zhu, C.; Owada, K.; Kawamori, F.; et al. Antiviral effect of theaflavins against caliciviruses. J. Antibiot. 2016, 70, 443–447. [Google Scholar] [CrossRef]
- de Oliveira, A.; Prince, D.; Lo, C.Y.; Lee, L.H.; Chu, T.C. Antiviral activity of theaflavin digallate against herpes simplex virus type 1. Antivir. Res. 2015, 118, 56–67. [Google Scholar] [CrossRef]
- Zu, M.; Yang, F.; Zhou, W.; Liu, A.; Du, G.; Zheng, L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antivir. Res. 2012, 94, 217–224. [Google Scholar] [CrossRef]
- Chowdhury, P.; Sahuc, M.-E.; Rouillé, Y.; Rivière, C.; Bonneau, N.; Vandeputte, A.; Brodin, P.; Goswami, M.; Bandyopadhyay, T.; Dubuisson, J.; et al. Theaflavins, polyphenols of black tea, inhibit entry of hepatitis C virus in cell culture. PLoS ONE 2018, 13, e0198226. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, L.; Tan, S.; Jin, H.; Qiu, J.; Mao, Q.; Li, R.; Xia, C.; Jiang, Z.-H.; Jiang, S.; et al. A natural theaflavins preparation inhibits HIV-1 infection by targeting the entry step: Potential applications for preventing HIV-1 infection. Fitoterapia 2012, 83, 348–355. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Hao, B.-J.; Cao, H.-C.; Xu, W.; Li, Y.-J.; Li, L.-J. Anti-hepatitis B virus effect and possible mechanism of action of 3,4-o-dicaffeoylquinic Acid in vitro and in vivo. Evid. Based Complement. Alternat. Med. 2012, 2012, 356806. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.; Kang, E.; Lee, Y. A flavonoid from medicinal plants blocks hepatitis B virus-e antigen secretion in HBV-infected hepatocytes. Antivir. Res. 2005, 67, 163–168. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Leshchenko, S.; Noetzel, M. Stereoselective Photochemical 1,3-Dioxolane Addition to 5-Alkoxymethyl-2(5H)-furanone: Synthesis of Bis-tetrahydrofuranyl Ligand for HIV Protease Inhibitor UIC-94017 (TMC-114). J. Org. Chem. 2004, 69, 7822–7829. [Google Scholar] [CrossRef]
- Zhu, M.; Ma, L.; Zhou, H.; Dong, B.; Wang, Y.; Wang, Z.; Zhou, J.; Zhang, G.; Wang, J.; Liang, C.; et al. Preliminary SAR and biological evaluation of potent HIV-1 protease inhibitors with pyrimidine bases as novel P2 ligands to enhance activity against DRV-resistant HIV-1 variants. Eur. J. Med. Chem. 2020, 185, 111866. [Google Scholar] [CrossRef]
- Zhu, M.; Ma, L.; Wen, J.; Dong, B.; Wang, Y.; Wang, Z.; Zhou, J.; Zhang, G.; Wang, J.; Guo, Y.; et al. Rational design and Structure-Activity relationship of coumarin derivatives effective on HIV-1 protease and partially on HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2020, 186, 111900. [Google Scholar] [CrossRef]
- Nakano, M.; Sato, Y. Rearrangement of (substituted benzyl)trimethylammonium ylides in a nonbasic medium: The improved Sommelet-Hauser rearrangement. J. Org. Chem. 1987, 52, 1844–1847. [Google Scholar] [CrossRef]
- Matayoshi, E.; Wang, G.; Krafft, G.; Erickson, J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science 1990, 247, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Swanson, L.M.; Cho, H.; Leshchenko, S.; Hussain, K.A.; Kay, S.; Walters, D.E.; Koh, Y.; Mitsuya, H. Structure-Based Design: Synthesis and Biological Evaluation of a Series of Novel Cycloamide-Derived HIV-1 Protease Inhibitors. J. Med. Chem. 2005, 48, 3576–3585. [Google Scholar] [CrossRef]
- Garcia, J.-M.; Gao, A.; He, P.-L.; Choi, J.; Tang, W.; Bruzzone, R.; Schwartz, O.; Naya, H.; Nan, F.-J.; Li, J.; et al. High-throughput screening using pseudotyped lentiviral particles: A strategy for the identification of HIV-1 inhibitors in a cell-based assay. Antivir. Res. 2009, 81, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, A.F.; Abdel-Moty, S.G.; Hussein, M.A.; Abdel-Alim, A.A. Design, synthesis and molecular docking of some new 1,2,4-triazolobenzimidazol-3-yl acetohydrazide derivatives with anti-inflammatory-analgesic activities. Arch. Pharm. Res. 2013, 36, 1465–1479. [Google Scholar] [CrossRef]
- Ganguly, A.K.; Alluri, S.S.; Wang, C.-H.; Antropow, A.; White, A.; Caroccia, D.; Biswas, D.; Kang, E.; Zhang, L.-K.; Carroll, S.S.; et al. Structural optimization of cyclic sulfonamide based novel HIV-1 protease inhibitors to picomolar affinities guided by X-ray crystallographic analysis. Tetrahedron 2014, 70, 2894–2904. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rao, K.V.; Nyalapatla, P.R.; Kovela, S.; Brindisi, M.; Osswald, H.L.; Reddy, B.S.; Agniswamy, J.; Wang, Y.-F.; Aoki, M.; et al. Design of Highly Potent, Dual-Acting and Central-Nervous-System-Penetrating HIV-1 Protease Inhibitors with Excellent Potency against Multidrug-Resistant HIV-1 Variants. ChemMedChem 2018, 13, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Rusere, L.N.; Lockbaum, G.J.; Lee, S.-K.; Henes, M.; Kosovrasti, K.; Spielvogel, E.; Nalivaika, E.A.; Swanstrom, R.; Yilmaz, N.K.; Schiffer, C.A.; et al. HIV-1 Protease Inhibitors Incorporating Stereochemically Defined P2’ Ligands To Optimize Hydrogen Bonding in the Substrate Envelope. J. Med. Chem. 2019, 62, 8062–8079. [Google Scholar] [CrossRef]
- Miller, J.F.; Andrews, C.W.; Brieger, M.; Furfine, E.S.; Hale, M.R.; Hanlon, M.H.; Hazen, R.J.; Kaldor, I.; McLean, E.W.; Reynolds, D.; et al. Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: The discovery of GW0385. Bioorg. Med. Chem. Lett. 2006, 16, 1788–1794. [Google Scholar] [CrossRef]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J. Virol. 1996, 70, 2581–2585. [Google Scholar] [CrossRef] [Green Version]
- Dergousova, N.I.; Amerik, A.; Volynskaya, A.M.; Rumsh, L.D. HIV-I protease. Cloning, expression, and purification. Appl. Biochem. Biotechnol. 1996, 61, 97–107. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
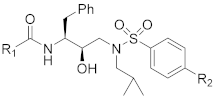
| Comp. | R1 | R2 | IC50 (nM) a | CC50 (μM) b |
|---|---|---|---|---|
| 15a |  | OCH3 | 3.34 ± 0.28 | >100 |
| 15b |  | OCH3 | 7.82 ± 0.93 | 33.86 |
| 15c |  | OCH3 | 4.68 ± 0.74 | >100 |
| 15d | 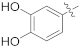 | OCH3 | 0.0059 ± 0.0007 | 38.22 |
| 15e | 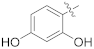 | OCH3 | 0.30 ± 0.08 | 78.76 |
| 15f | 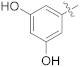 | OCH3 | 0.0024 ± 0.0004 | >100 |
| 15g |  | OCH3 | 1.13 ± 0.23 | >100 |
| 15h | 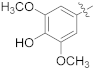 | OCH3 | 0.08 ± 0.03 | 84.66 |
| 15i | 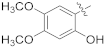 | OCH3 | 1.41 ± 0.43 | >100 |
| 16a | 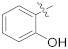 | NH2 | 0.54 ± 0.11 | >100 |
| 16b | 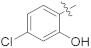 | NH2 | 0.31 ± 0.08 | >100 |
| 16c |  | NH2 | 4.14 ± 1.38 | >100 |
| 16d | 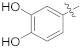 | NH2 | 1.63 ± 0.33 | >100 |
| 16e | 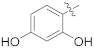 | NH2 | 0.43 ± 0.08 | >100 |
| 16f | 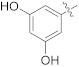 | NH2 | 0.27 ± 0.03 | >100 |
| 16g |  | NH2 | 68.16 ± 21.35 | 57.64 |
| 16h | 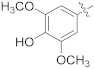 | NH2 | 0.04 ± 0.003 | >100 |
| 16i | 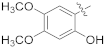 | NH2 | 0.60 ± 0.18 | >100 |
| 17a |  | SCH3 | 3.97 ± 0.41 | >100 |
| 17b | 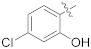 | SCH3 | 2.79 ± 0.44 | >100 |
| 17c | 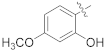 | SCH3 | 8.21 ± 1.20 | >100 |
| 17d | 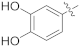 | SCH3 | 0.0076 ± 0.0029 | >100 |
| 17e | 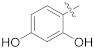 | SCH3 | 0.40 ± 0.08 | >100 |
| 17f | 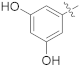 | SCH3 | 0.0066 ± 0.0021 | >100 |
| 17h | 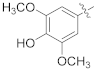 | SCH3 | 0.32 ± 0.03 | >100 |
| 17i | 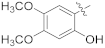 | SCH3 | 4.71 ± 0.18 | >100 |
| DRV | - | - | 0.51 ± 0.17 | >100 |
| Compounds | Inhibition (%) (10 μM) a | Compounds | Inhibition (%) (10 μM) a |
|---|---|---|---|
| 15a | 98 ± 2 | 16c | 90 ± 6 |
| 15b | 96 ± 3 | 16d | 100 |
| 15c | 92 ± 5 | 16e | 75 ± 8 |
| 15d | 100 | 16f | 99 ± 2 |
| 15e | 75 ± 6 | 16g | 55 ± 6 |
| 15f | 100 | 16h | 99 ± 3 |
| 15g | 99 ± 1 | 17d | 100 |
| 15h | 99 ± 2 | 17f | 100 |
| 16a | 100 | DRV | 100 |
| 16b | 98 ± 2 | DMSO | 0 |
| Compounds | cLogP a | Mean EC50 (nM, ± SD) b | Fold Resistance c | |
|---|---|---|---|---|
| HIV-1NL4−3 | HIVDRVRs | |||
| 15b | 6.695 | 1402 ± 23.00 | 1843 ± 39.60 | 1.31 |
| 15d | 4.303 | 15.36 ± 2.17 | 402.4 ± 47.32 | 26.19 |
| 15f | 4.372 | 28.89 ± 9.21 | 45.53 ± 12.11 | 1.57 |
| DRV | 2.887 | 1.80 ± 0.73 | 136.80 ± 1.12 | 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, L.; Wen, J.; Dong, B.; Zhou, J.; Hu, S.; Wang, J.; Wang, Y.; Zhu, M.; Cen, S. Design and Evaluation of Novel HIV-1 Protease Inhibitors Containing Phenols or Polyphenols as P2 Ligands with High Activity against DRV-Resistant HIV-1 Variants. Int. J. Mol. Sci. 2022, 23, 14178. https://doi.org/10.3390/ijms232214178
Ma L, Wen J, Dong B, Zhou J, Hu S, Wang J, Wang Y, Zhu M, Cen S. Design and Evaluation of Novel HIV-1 Protease Inhibitors Containing Phenols or Polyphenols as P2 Ligands with High Activity against DRV-Resistant HIV-1 Variants. International Journal of Molecular Sciences. 2022; 23(22):14178. https://doi.org/10.3390/ijms232214178
Chicago/Turabian StyleMa, Ling, Jiajia Wen, Biao Dong, Jinming Zhou, Shangjiu Hu, Juxian Wang, Yucheng Wang, Mei Zhu, and Shan Cen. 2022. "Design and Evaluation of Novel HIV-1 Protease Inhibitors Containing Phenols or Polyphenols as P2 Ligands with High Activity against DRV-Resistant HIV-1 Variants" International Journal of Molecular Sciences 23, no. 22: 14178. https://doi.org/10.3390/ijms232214178
APA StyleMa, L., Wen, J., Dong, B., Zhou, J., Hu, S., Wang, J., Wang, Y., Zhu, M., & Cen, S. (2022). Design and Evaluation of Novel HIV-1 Protease Inhibitors Containing Phenols or Polyphenols as P2 Ligands with High Activity against DRV-Resistant HIV-1 Variants. International Journal of Molecular Sciences, 23(22), 14178. https://doi.org/10.3390/ijms232214178