
A Transcriptome and Methylome Study Comparing Tissues of Early and Late Onset Colorectal Carcinoma
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Differential Gene Expression in CRC
2.1.1. Age of Onset of CRC and Differential Gene Expression of Cancer Related Gene Sets
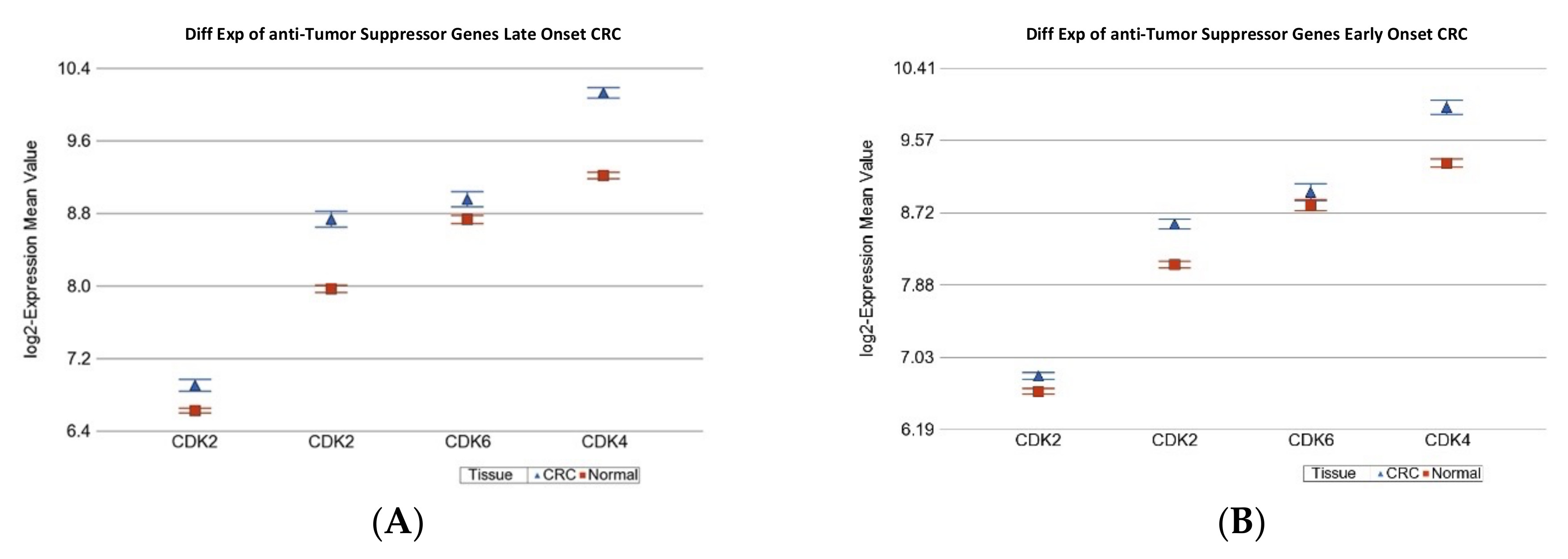
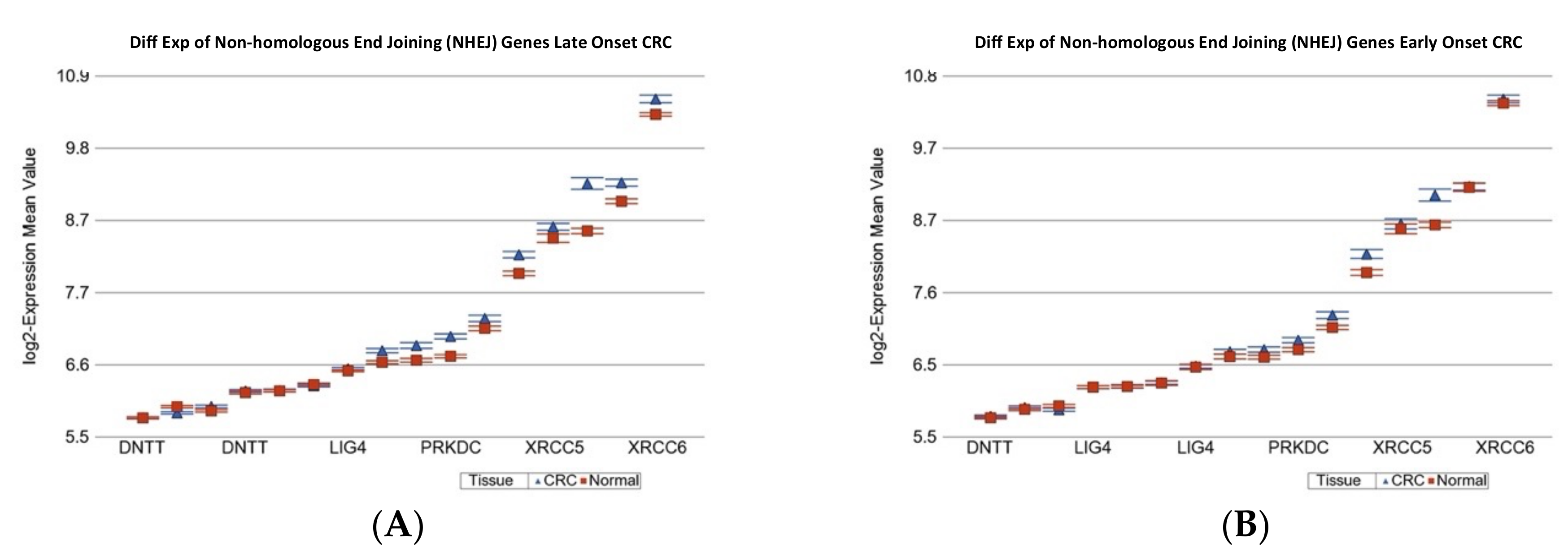
2.1.2. Age of Onset of CRC and Differential Gene Expression of DNA Damage Repair Gene Sets
2.1.3. Age of Onset of CRC and Differential Expression of Replication Stress Gene-Sets
2.1.4. Age of Onset of CRC and Differential Expression of Notch4-GATA4-IRG1 axis Gene-Sets
2.1.5. Age of Onset of CRC and Differential Expression of KEGG Pathways
2.1.6. Association of Differentially Expressed “Gene Sets” with Histology in CRC
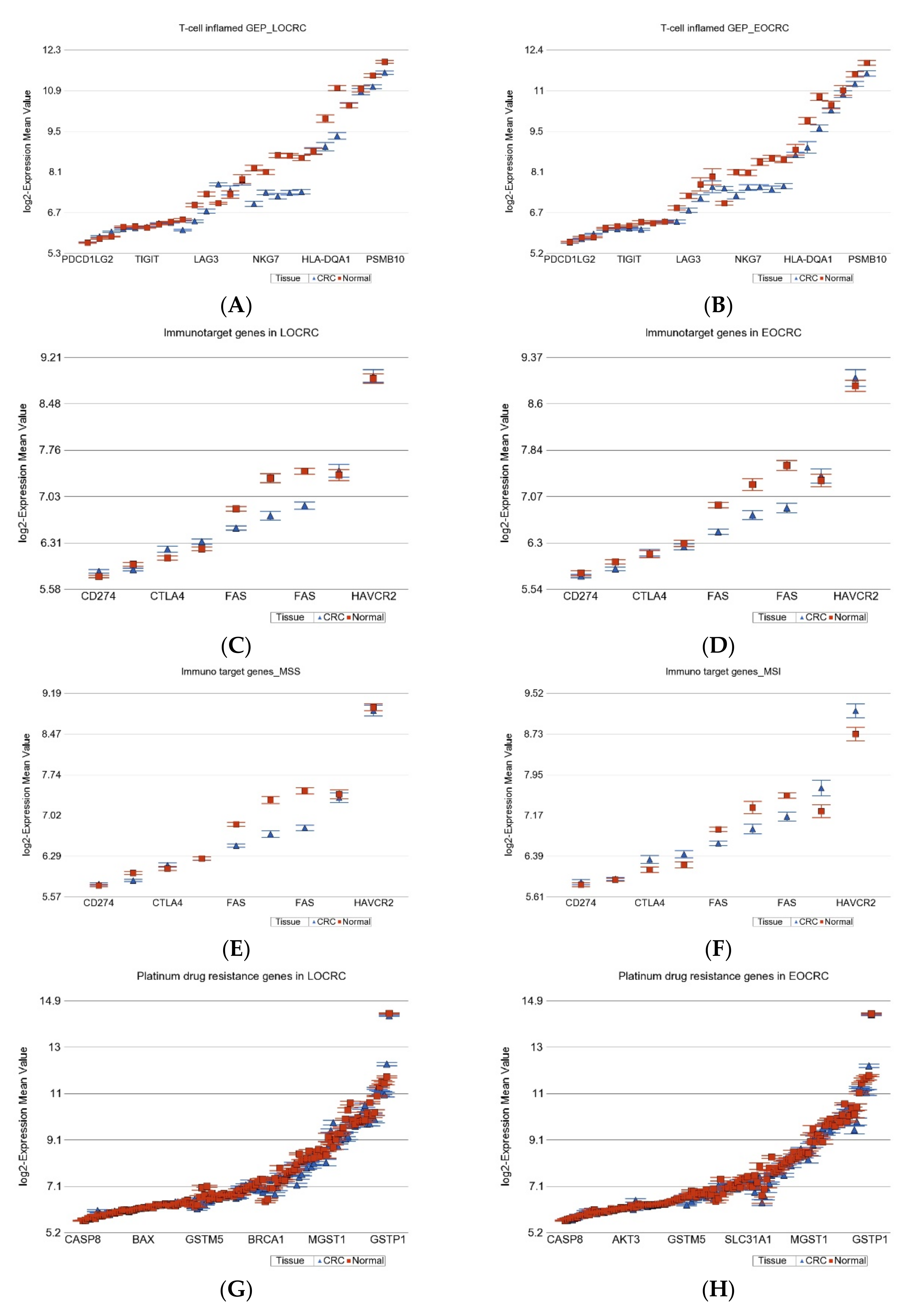
2.1.7. Gene Expression Profiling from Therapeutic Point of View
2.2. DNA Methylation
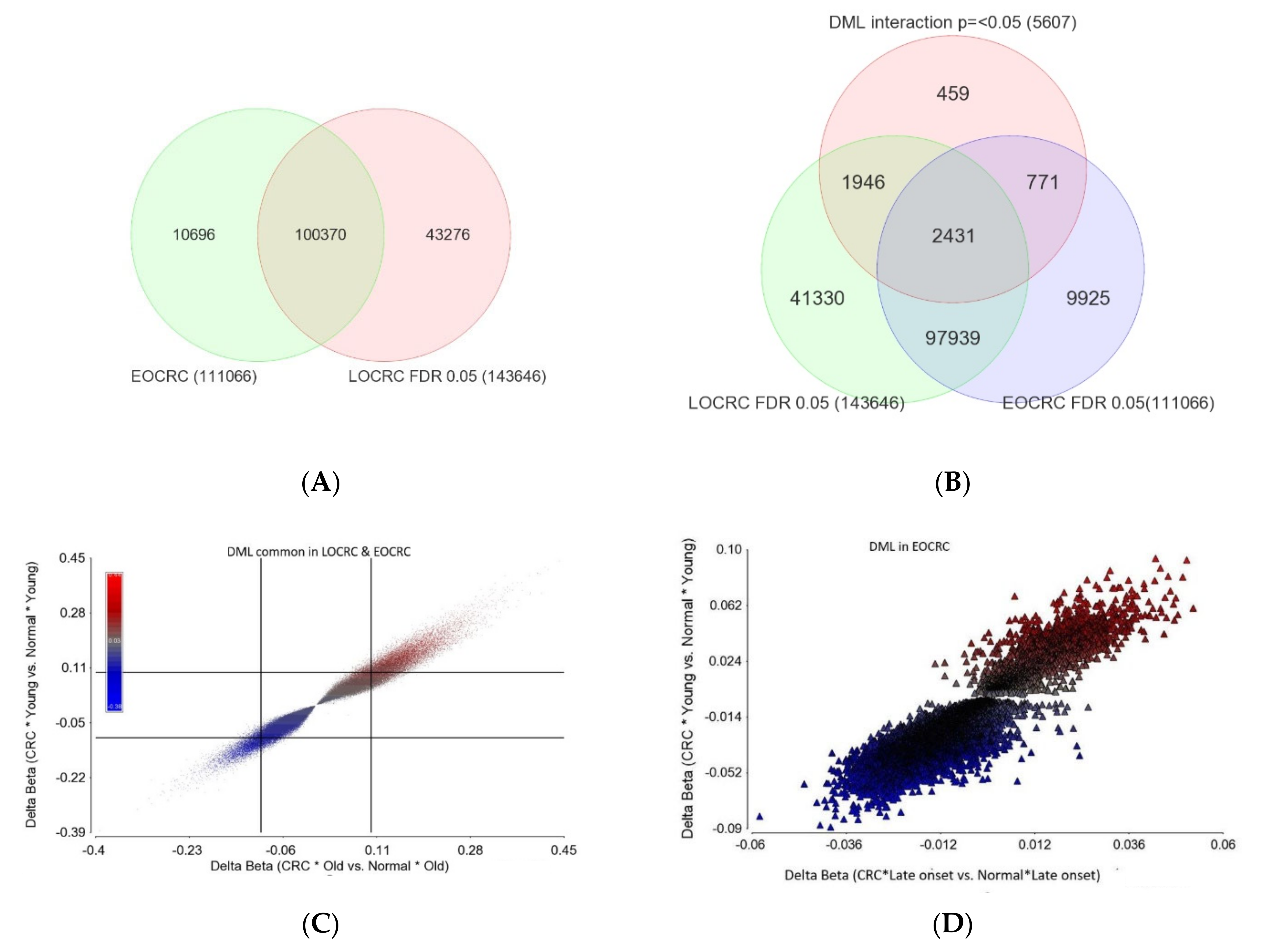
Age of Onset of CRC and Differential DNA Methylation
- I.
- DML common in both EOCRC and LOCRC and the magnitude of delta beta was not different (n = 97,939) as the interaction p was >=0.05. This represents the largest group of DML in CRC. The scatterplot in Figure 7C shows the magnitude of delta beta of these DML in LOCRC and EOCRC and the cut-off lines on both axes show a large number of loci with delta beta > 0.1 or <−0.1, indicating hyper- or hypo-methylation exceeding 10%.
- II.
- DML common in both EOCRC and LOCRC, but the magnitude of delta beta was significantly different between EOCRC and LOCRC patients (n = 2431).
- III.
- DML found only in EOCRC, but the magnitude of the delta beta was not different between the EOCRC and LOCRC (n = 9925). The scatterplot in Figure 7D shows that the magnitudes of delta beta of these DML were low and not different among LOCRC and EOCRC.
- IV.
- DML specific to EOCRC (n = 771): these DML are differentially methylated only in EOCRC, and the magnitude of the delta beta is significantly more pronounced from that seen in LOCRC. The details of these DML are presented in Supplementary Table S6. The scatterplot in Figure 7E shows the magnitudes of delta beta of these DML; only a few exceeded 0.1 or 10% differential methylation. Regardless of the low magnitudes of delta beta of these 771 EOCRC specific DML, because of specificity, these markers were able to separate CRC from normal among the EOCRC patients (see the PCA plot in Figure 7F). The only hypermethylated DML with delta beta >= 0.1 was the SOX8 gene. Among the three hypomethylated loci with delta beta =< 0.1, one was the TACC1 gene (see Figure 7E), which is known to be associated with other cancers. The methylation status of the TACC1 gene in LOCRC and EOCRC is shown in Figure 7G,H, respectively, showing that TACC1 was hypomethylated only in EOCRC, but not in LOCRC.
- V.
- DML specific to old age onset CRC (1946): these DML are differentially methylated only in LOCRC, and the magnitude of the delta beta is significantly more pronounced from that seen in EOCRC. The details of these DML are presented in Supplementary Table S7. For many of these DML, the magnitudes of delta beta exceeded 0.1 or 10% differential methylation.
3. Discussion
4. Materials and Methods
4.1. DNA and RNA Extraction and Quality Control
4.2. Relative Telomere Length (RTL) Measurement
4.3. Genome-Wide Gene Expression Assay
4.4. Genome-Wide Methylation Assay
4.5. Microsatellite Instability (MSI) Detection
4.6. KRAS and BRAF Mutation Detection:
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vuik, F.E.; Nieuwenburg, S.A.; Bardou, M.; Lansdorp-Vogelaar, I.; Dinis-Ribeiro, M.; Bento, M.J.; Zadnik, D.; Pellisé, M.; Esteban, L.; Kaminski, M.F.; et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut 2019, 68, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Burnett-Hartman, A.N.; Powers, J.D.; Chubak, J.; Corley, D.A.; Ghai, N.R.; McMullen, C.K.; Pawloski, P.A.; Sterrett, A.T.; Feigelson, H.S. Treatment patterns and survival differ between early-onset and late-onset colorectal cancer patients: The patient outcomes to advance learning network. Cancer Causes Control 2019, 30, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, A.B.; Roche, L.M.; Johnson, L.M.; Pawlish, K.S.; Paddock, L.E.; Stroup, A.M. Trends in colorectal cancer incidence among younger adults-Disparities by age, sex, race, ethnicity, and subsite. Cancer Med. 2018, 7, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.R.; Kuk, J.K.; Kim, T.; Shin, E.J. Comparison of oncological outcomes of right-sided colon cancer versus left-sided colon cancer after curative resection: Which side is better outcome? Medicine 2017, 96, e8241. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.; García, J.L.; Corchete, L.; Tapial, S.; Olmedillas-López, S.; Vivas, A.; García-Olmo, D.; Urioste, M.; Goel, A.; González-Sarmiento, R. A clinico-pathological and molecular analysis reveals differences between solitary (early and late-onset) and synchronous rectal cancer. Sci. Rep. 2021, 11, 2202. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.; Pai, R.K.; Rybicki, L.A.; Dimaio, M.A.; Limaye, M.; Jayachandran, P.; Koong, A.; Kunz, P.A.; Fisher, G.A.; Ford, J.M.; et al. Clinicopathologic and molecular features of sporadic early-onset colorectal adenocarcinoma: An adenocarcinoma with frequent signet ring cell differentiation, rectal and sigmoid involvement, and adverse morphologic features. Mod. Pathol. 2012, 25, 1128–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.T.; Huang, K.C.; Cheng, A.L.; Jeng, Y.M.; Wu, M.S.; Wang, S.M. Clinicopathological and molecular biological features of colorectal cancer in patients less than 40 years of age. Br. J. Surg. 2003, 90, 205–214. [Google Scholar] [CrossRef] [PubMed]
- O′Connell, J.B.; Maggard, M.A.; Liu, J.H.; Etzioni, D.A.; Livingston, E.H.; Ko, C.Y. Do young colon cancer patients have worse outcomes? World J. Surg. 2004, 28, 558–562. [Google Scholar] [CrossRef]
- Li, H.; Boakye, D.; Chen, X.; Hoffmeister, M.; Brenner, H. Association of Body Mass Index With Risk of Early-Onset Colorectal Cancer: Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2021, 116, 2173–2183. [Google Scholar] [CrossRef]
- Liu, P.-H.; Wu, K.; Ng, K.; Zauber, A.G.; Nguyen, L.; Song, M.; He, X.; Fuchs, C.S.; Ogino, S.; Willett, W.C.; et al. Association of obesity with risk of early-onset colorectal cancer among women. JAMA Oncol. 2019, 5, 37–44. [Google Scholar] [CrossRef]
- Jasmine, F.; Haq, Z.; Kamal, M.; Raza, M.; da Silva, G.; Gorospe, K.; Paul, R.; Strzempek, P.; Ahsan, H.; Kibriya, M.G. Interaction between Microsatellite Instability (MSI) and Tumor DNA methylation in the pathogenesis of colorectal carcinoma. Cancers 2021, 13, 4956. [Google Scholar] [CrossRef]
- Saltzstein, S.L.; Behling, C.A. Age and time as factors in the left-to-right shift of the subsite of colorectal adenocarcinoma: A study of 213,383 cases from the California Cancer Registry. J. Clin. Gastroenterol. 2007, 41, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Bs, A.K.; George, T.; Hughes, S.J.; Delitto, D.; Allegra, C.J.; Hall, W.A.; Chang, G.J.; Tan, S.A.; Shaw, C.M.; Iqbal, A. Rectal cancer patients younger than 50 years lack a survival benefit from NCCN guideline-directed treatment for stage II and III disease. Cancer 2018, 124, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Saraste, D.; Järås, J.; Martling, A. Population-based analysis of outcomes with early-age colorectal cancer. Br. J. Surg. 2020, 107, 301–309. [Google Scholar] [CrossRef] [PubMed]
- REACCT Collaborative; Zaborowski, A.M.; Abdile, A.; Adamina, M.; Aigner, F.; D’Allens, L.; Allmer, C.; Álvarez, A.; Anula, R.; Andric, M.; et al. Characteristics of Early-Onset vs Late-Onset Colorectal Cancer: A review. JAMA Surg. 2021, 156, 865–874. [Google Scholar] [CrossRef]
- Zaborowski, A.M.; Murphy, B.; Creavin, B.; Rogers, A.C.; Kennelly, R.; Hanly, A.; Martin, S.T.; O’Connell, P.R.; Sheahan, K.; Winter, D.C. Clinicopathological features and oncological outcomes of patients with young-onset rectal cancer. Br. J. Surg. 2020, 107, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losi, L.; Di Gregorio, C.; Pedroni, M.; Ponti, G.; Roncucci, L.; Scarselli, A.; Genuardi, M.; Baglioni, S.; Marino, M.; Rossi, G.; et al. Molecular genetic alterations and clinical features in early-onset colorectal carcinomas and their role for the recognition of hereditary cancer syndromes. Am. J. Gastroenterol. 2005, 100, 2280–2287. [Google Scholar] [CrossRef]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Macheret, M.; Bhowmick, R.; Sobkowiak, K.; Padayachy, L.; Mailler, J.; Hickson, I.D.; Halazonetis, T.D. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. 2020, 30, 997–1008. [Google Scholar] [CrossRef]
- Qiao, L.; Wong, B.C. Role of Notch signaling in colorectal cancer. Carcinogenesis 2009, 30, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Scheurlen, K.M.; Chariker, J.H.; Kanaan, Z.; Littlefield, A.B.; George, J.B.; Seraphine, C.; Rochet, A.; Rouchka, E.C.; Galandiuk, S. The NOTCH4-GATA4-IRG1 axis as a novel target in early-onset colorectal cancer. Cytokine Growth Factor Rev. 2022, 67, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Ogawa, A.; Kirkner, G.J.; Loda, M.; Fuchs, C.S. Fatty acid synthase overexpression in colorectal cancer is associated with microsatellite instability, independent of CpG island methylator phenotype. Hum. Pathol. 2007, 38, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Abubaker, J.; Al-Sanea, N.; Abduljabbar, A.; Ashari, L.H.; Alhomoud, S.; Al-Dayel, F.; Bavi, P.; et al. High prevalence of fatty acid synthase expression in colorectal cancers in Middle Eastern patients and its potential role as a therapeutic target. Am. J. Gastroenterol. 2009, 104, 1790–1801. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.; Young, L.E.A.; Scott, T.L.; Kelson, C.O.; He, D.; Liu, J.; Wu, Y.; Wang, C.; Weiss, H.L.; Fan, T.; et al. Tissue-specific downregulation of fatty acid synthase suppresses intestinal adenoma formation via coordinated reprograming of transcriptome and metabolism in the mouse model of Apc-driven colorectal cancer. Int. J. Mol. Sci. 2022, 23, 6510. [Google Scholar] [CrossRef]
- Lu, T.; Sun, L.; Wang, Z.; Zhang, Y.; He, Z.; Xu, C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. Onco Targets Ther. 2019, 12, 3339–3347. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; McClanahan, T.K. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Merchant, S.A.; Jensen, C.D.; Murphy, C.C.; Udaltsova, N.; Corley, D.A. Rising early-onset colorectal cancer incidence is not an artifact of increased screening colonoscopy use in a large, diverse healthcare system. Gastroenterology 2022, 162, 325–327. [Google Scholar] [CrossRef]
- Nikolic, N.; Spasic, J.; Stanic, N.; Nikolic, V.; Radosavljevic, D. Young-onset colorectal cancer in Serbia: Tertiary cancer center experience. J. Adolesc. Young Adult Oncol. 2022. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaiano, A.; Berretta, M.; Von Arx, C.; Capozzi, M.; Caraglia, M. Editorial: The Treatment of RAS or BRAF Mutated Metastatic Colorectal Cancer: Challenges and Perspectives. Front. Oncol. 2022, 12, 852445. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.I.; Lewin, D.N.; Wang, H.L.; Lauwers, G.Y.; Srivastava, A.; Shyr, Y.; Shakhtour, B.; Revetta, F.; Washington, M.K. Lymphovascular invasion in colorectal cancer: An interobserver variability study. Am. J. Surg. Pathol. 2008, 32, 1816–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebig, C.; Ayala, G.; Wilks, J.; Verstovsek, G.; Liu, H.; Agarwal, N.; Berger, D.H.; Albo, D. Perineural invasion is an independent predictor of outcome in colorectal cancer. J. Clin. Oncol. 2009, 27, 5131–5137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, A.L.; Penney, M.D.; Allison, M.C. The prevalence of iron deficiency among patients presenting with colorectal cancer. Color. Dis. 2005, 7, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.G.M.T.; Wallace, R.; McKee, R.F.; Anderson, J.H.; Going, J.J.; Edwards, J.; Horgan, P.G. The relationship between tumour site, clinicopathological characteristics and cancer-specific survival in patients undergoing surgery for colorectal cancer. Color. Dis. 2012, 14, 1493–1499. [Google Scholar] [CrossRef]
- Khan, S.A.; Morris, M.; Idrees, K.; Gimbel, M.I.; Rosenberg, S.; Zeng, Z.; Li, F.; Gan, G.; Shia, J.; LaQuaglia, M.P.; et al. Colorectal cancer in the very young: A comparative study of tumor markers, pathology and survival in early onset and adult onset patients. J. Pediatr. Surg. 2016, 51, 1812–1817. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, N.; Yau, C.; Sakthianandeswaren, A.; Mouradov, D.; Gibbs, P.; Suraweera, N.; Cazier, J.-B.; Polanco-Echeverry, G.; Ghosh, A.; Thaha, M.; et al. Analysis of colorectal cancers in British Bangladeshi identifies early onset, frequent mucinous histotype and a high prevalence of RBFOX1 deletion. Mol. Cancer 2013, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Nikberg, M.; Chabok, A.; Letocha, H.; Kindler, C.; Glimelius, B.; Smedh, K. Lymphovascular and perineural invasion in stage II rectal cancer: A report from the Swedish colorectal cancer registry. Acta Oncol. 2016, 55, 1418–1424. [Google Scholar] [CrossRef] [Green Version]
- Tunca, B.; Tezcan, G.; Cecener, G.; Egeli, U.; Zorluoglu, A.; Yilmazlar, T.; Ak, S.; Yerci, O.; Ozturk, E.; Umut, G.; et al. Overexpression of CK20, MAP3K8 and EIF5A correlates with poor prognosis in early-onset colorectal cancer patients. J. Cancer Res. Clin. Oncol. 2013, 139, 691–702. [Google Scholar] [CrossRef]
- Hong, Y.; Ho, K.S.; Eu, K.W.; Cheah, P.Y. A susceptibility gene set for early onset colorectal cancer that integrates diverse signaling pathways: Implication for tumorigenesis. Clin. Cancer Res. 2007, 13, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandova, J.; Xu, W.; Nfonsam, V. Sporadic early-onset colon cancer expresses unique molecular features. J. Surg. Res. 2016, 204, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Ågesen, T.H.; Thiis-Evensen, E.; Merok, M.A.; Teixeira, M.R.; Vatn, M.H.; Nesbakken, A.; Skotheim, R.I.; Lothe, R.A.; the INFAC-Study Group. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol. Cancer 2010, 9, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agesen, T.H.; Berg, M.P.V.D.; Clancy, T.; Thiisevensen, E.; Cekaite, L.; Lind, G.E.; Nesland, J.M.; Bakka, A.; Mala, T.; Hauss, H.J.; et al. CLC and IFNAR1 are differentially expressed and a global immunity score is distinct between early- and late-onset colorectal cancer. Genes Immun. 2011, 12, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, X.; Su, Z.; Yang, B.; Zeng, Z.; Lei, S.; Qiao, H. Identification of key genes involved in the development and progression of early-onset colorectal cancer by co-expression network analysis. Oncol. Lett. 2020, 19, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Cava, A.; Alviggi, C.; Matarese, G. Unraveling the multiple roles of leptin in inflammation and autoimmunity. J. Mol. Med. 2004, 82, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, M.; Maruoka, S.; Katayose, S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur. J. Endocrinol. 2002, 147, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.; Clendenning, M.; Wong, E.; Rosty, C.; Mahmood, K.; Georgeson, P.; Winship, I.; Preston, S.; Win, A.; Dugué, P.-A.; et al. DNA methylation signatures and the contribution of age-associated methylomic drift to carcinogenesis in early-onset colorectal cancer. Cancers 2021, 13, 2589. [Google Scholar] [CrossRef]
- Hamilton, S.R. Tumours of the Colon and Rectum; World Health Organization Classification of Tumours-Pathology and Genetics of Tumours of the Digestive System; World Health Organization: Lyon, France, 2000. [Google Scholar]
- Jasmine, F.; Shinkle, J.; Sabarinathan, M.; Ahsan, H.; Pierce, B.L.; Kibriya, M.G. A novel pooled-sample multiplex luminex assay for high-throughput measurement of relative telomere length. Am. J. Hum. Biol. 2018, 30, e23118. [Google Scholar] [CrossRef]
- Kibriya, M.G.; Jasmine, F.; Roy, S.; Ahsan, H.; Pierce, B. Measurement of telomere length: A new assay using QuantiGene chemistry on a Luminex platform. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2667–2672. [Google Scholar] [CrossRef]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, 1333. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Raza, M.; Kamal, M.; Haq, Z.; Paul, R.; Mareczko, A.; Pierce, B.L.; Ahsan, H.; Jasmine, F. Relative telomere length change in colorectal carcinoma and its association with tumor characteristics, gene expression and microsatellite instability. Cancers 2022, 14, 2250. [Google Scholar] [CrossRef] [PubMed]
- Janavicius, R.; Matiukaite, D.; Jakubauskas, A.; Griskevicius, L. Microsatellite instability detection by high-resolution melting analysis. Clin. Chem. 2010, 56, 1750–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibriya, M.G.; Raza, M.; Jasmine, F.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Dodsworth, C.; Rakibuz-Zaman, M.; Kamal, M.; Ahsan, H. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med. Genom. 2011, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschoolmeester, V.; Baay, M.; Wuyts, W.; Van Marck, E.; Van Damme, N.; Vermeulen, P.; Lukaszuk, K.; Lardon, F.; Vermorken, J.B. Detection of microsatellite instability in colorectal cancer using an alternative multiplex assay of quasi-monomorphic mononucleotide markers. J. Mol. Diagn. 2008, 10, 154–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findeisen, P.; Kloor, M.; Merx, S.; Sutter, C.; Woerner, S.M.; Dostmann, N.; Benner, A.; Dondog, B.; Pawlita, M.; Dippold, W.; et al. T25 repeat in the 3’ untranslated region of the CASP2 gene: A sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res. 2005, 65, 8072–8078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhart, C. The assumptions underlying the analysis of variance. Biometrics 1947, 3, 1–21. [Google Scholar] [CrossRef]
- Downey, T. Analysis of a multifactor microarray study using Partek genomics solution. Methods Enzymol. 2006, 411, 256–270. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Category | Late-Onset (>40 Years) | Early-Onset (<40 Years) | Chi-Square Test |
|---|---|---|---|---|
| p-Value | ||||
| Sex | Male | 57 (58.2%) | 39 (58.2%) | 0.995 |
| Female | 41 (41.8%) | 28 (41.8%) | ||
| Location | Right Colon | 22 (22.4%) | 11 (16.4%) | 0.327 |
| Left Colon | 20 (20.4%) | 10 (14.9%) | ||
| Rectum | 56 (57.1%) | 46 (68.7%) | ||
| Stage | Stage-1 | 30 (30.6%) | 7 (10.4%) | 0.004 * |
| Stage-2 | 26 (26.5%) | 16 (23.9%) | ||
| Stage-3 | 42 (42.9%) | 44 (65.7%) | ||
| Grade | Low | 51 (52%) | 27 (40.3%) | 0.138 |
| High | 47 (48%) | 40 (59.7%) | ||
| Lymph Node | Present | 56 (57.1%) | 23 (34.3%) | 0.004 * |
| Absent | 42 (42.9%) | 44 (65.7%) | ||
| TIL | 0 | 58 (59.2%) | 41 (61.2%) | 0.796 |
| 1 | 40 (40.8%) | 26 (38.8%) | ||
| Signet Ring | Absent | 70 (71.4%) | 43 (64.2%) | 0.325 |
| Present | 28 (28.6%) | 24 (35.8%) | ||
| LV Invasion | Absent | 71 (72.4%) | 36 (53.7%) | 0.013 * |
| Present | 27 (27.6%) | 31 (46.3%) | ||
| PN Invasion | Absent | 90 (91.8%) | 53 (79.1%) | 0.018 * |
| Present | 8 (8.2%) | 14 (20.9%) | ||
| CEA (ng/mL) | Mean | 50.221 | 30.133 | 0.180 # |
| (SD) | 100.584 | 65.732 | ||
| Microsatellite | MSI | 26 (26.5%) | 15 (22.4%) | 0.545 |
| MSS | 72 (73.5%) | 52 (77.6%) | ||
| KRAS (rs112445441) | Wild | 68 (70.8%) | 47 (70.1%) | 0.925 |
| Mutant | 28 (29.2%) | 20 (29.9% | ||
| BRAFV600E | Wild | 89 (92.7%) | 62 (93.9%) | 0.76 |
| Mutant | 7 (7.3%) | 4 (6.1%) | ||
| EGFR | Wild | 96 (100%) | 67 (100%) | NA |
| Mutant | 0 (0%) | 0 (0%) | ||
| Telomere | Absent | 28 (30.8%) | 23 (37.7%) | 0.375 |
| Shortening | Present | 63 (69.2%) | 38 (62.3%) |
| Characteristic | Stage | Status | Late-Onset (>40 Years) | Early-Onset (<40 Years) | Chi-Square Test p-Value |
|---|---|---|---|---|---|
| LV Invasion | Stage-1 | LVI Absent | 26 (86.7%) | 6 (85.7%) | 0.947 |
| LVI Present | 4 (13.3%) | 1 (14.3%) | |||
| Stage-2 | LVI Absent | 23 (88.5%) | 9 (56.3%) | 0.017 * | |
| LVI Present | 3 (11.5%) | 7 (43.8%) | |||
| Stage-3 | LVI Absent | 22 (52.4%) | 21 (47.7%) | 0.666 | |
| LVI Present | 20 (47.6%) | 23 (52.3%) | |||
| PN Invasion | Stage-1 | PNI Absent | 29 (96.7%) | 7 (100%) | 0.624 |
| PNI Present | 1 (3.3%) | 0 (0%) | |||
| Stage-2 | PNI Absent | 24 (92.3%) | 15 (93.8%) | 0.860 | |
| PNI Present | 2 (7.7%) | 1 (6.3%) | |||
| Stage-3 | PNI Absent | 37 (88.1%) | 31 (70.5%) | 0.044 * | |
| PNI Present | 5 (11.9%) | 13 (29.5%) |
| Gene Set | Interaction p | Late-Onset (>40 Years) | Early-Onset (<40 Years) | ||||
|---|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | p | Fold Change | (95% CI) | p | ||
| Growth Factor Receptors | 6.64 × 10−3 | −1.12 | (−1.16 to −1.08) | 1.21 × 10−8 | −1.02 | (−1.08 to 1.03) | 0.34 |
| Anti-TSG | 7.59 × 10−3 | 1.46 | (1.38 to 1.55) | 5.23 × 10−35 | 1.29 | (1.20 to 1.39) | 3.8 × 10−11 |
| DNA Repair | 0.06 | 1.07 | (1.05 to 1.08) | 2.28 × 10−19 | 1.04 | (1.02 to 1.06) | 5.4 × 10−6 |
| Pro-Apoptosis | 0.10 | 1.03 | (1.01 to 1.05) | 7.46 × 10−3 | −1.00 | (−1.03 to 1.03) | 0.99 |
| Tumor Suppressor Gene | 0.10 | −1.18 | (−1.23 to −1.13) | 3.41 × 10−12 | −1.11 | (−1.17 to −1.04) | 8.1 × 10−4 |
| Hexokinase | 0.32 | −1.20 | (−1.26 to −1.14) | 5.64 × 10−13 | −1.15 | (−1.23 to −1.08) | 1.6 × 10−5 |
| Warburg Effect | 0.36 | 1.32 | (1.21 to 1.43) | 1.22 × 10−10 | 1.24 | (1.11 to 1.38) | 1.2 × 10−4 |
| Anti-Apoptosis | 0.58 | −1.10 | (−1.15 to −1.05) | 1.27 × 10−4 | −1.07 | (−1.14 to −1.01) | 0.02 |
| Caspases Initiator | 0.69 | 1.03 | (1.01 to 1.05) | 1.27 × 10−3 | 1.02 | (−1.00 to 1.05) | 0.05 |
| p53 suppressor | 0.76 | 1.00 | (−1.01 to 1.02) | 0.66 | −1.00 | (−1.02 to 1.02) | 9.6 × 10−1 |
| Caspases Executor | 0.77 | −1.18 | (−1.23 to −1.14) | 4.29 × 10−18 | −1.19 | (−1.25 to −1.14) | 2.2 × 10−12 |
| Growth Factors | 0.78 | −1.18 | (−1.26 to −1.10) | 3.51 × 10−6 | −1.20 | (−1.31 to −1.09) | 9.1 × 10−5 |
| Gene Set | Interaction p | Late-Onset (>40 Years) | Early-Onset (<40 Years) | ||||
|---|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | p | Fold Change | (95% CI) | p | ||
| Mismatch Repair (MMR) | 1.16 × 10−4 | 1.12 | (1.10 to 1.14) | 2.02 × 10−33 | 1.06 | (1.03 to 1.08) | 7.01 × 10−6 |
| Fanconi Anemia (FA) | 4.86 × 10−4 | 1.15 | (1.13 to 1.17) | 1.50 × 10−46 | 1.09 | (1.06 to 1.12) | 1.42 × 10−11 |
| Non-homologous end joining (NHEJ) | 6.13 × 10−4 | 1.12 | (1.10 to 1.14) | 5.60 × 10−34 | 1.06 | (1.04 to 1.09) | 3.28 × 10−7 |
| Microhomology mediated end joining (MMEJ) | 9.09 × 10−4 | 1.26 | (1.22 to 1.30) | 1.29 × 10−49 | 1.16 | (1.12 to 1.21) | 4.24 × 10−14 |
| Translesion Synthesis (TLS) | 0.03 | 1.01 | (−1.01 to 1.03) | 0.30 | −1.02 | (−1.05 to 1.00) | 0.05 |
| Homologous Recombination (HR) | 0.15 | 1.09 | (1.07 to 1.10) | 5.92 × 10−39 | 1.07 | (1.05 to 1.09) | 1.82 × 10−16 |
| Nucleotide Excision Repair (NER) | 0.19 | 1.08 | (1.07 to 1.09) | 1.76 × 10−41 | 1.07 | (1.05 to 1.09) | 2.56 × 10−18 |
| Checkpoint Signaling | 0.21 | 1.08 | (1.06 to 1.09) | 6.56 × 10−18 | 1.06 | (1.03 to 1.08) | 4.55 × 10−7 |
| Base Excision Repair (BER) | 0.40 | 1.06 | (1.04 to 1.07) | 4.12 × 10−15 | 1.05 | (1.03 to 1.07) | 7.80 × 10−7 |
| Direct Reversal Repair (DRR) | 0.49 | 1.20 | (1.12 to 1.28) | 2.96 × 10−7 | 1.15 | (1.05 to 1.26) | 2.01 × 10−3 |
| Gene Set | Interaction p | Late-Onset (>40 Years) | Early-Onset (<40 Years) | ||||
|---|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | p | Fold Change | (95% CI) | p | ||
| DNA Replication Repair | 2.54 × 10−8 | 1.14 | (1.13 to 1.15) | 9.40 × 10−184 | 1.09 | (1.08 to 1.10) | 2.60 × 10−53 |
| Metabolism | 5.96 × 10−6 | 1.12 | (1.10 to 1.14) | 1.34 × 10−24 | 1.03 | (1.00 to 1.06) | 0.03 |
| Cell Movement | 6.86 × 10−4 | −1.06 | (−1.07 to −1.05) | 3.59 × 10−26 | −1.03 | (−1.04 to −1.01) | 1.25 × 10−4 |
| RNA Processing | 0.11 | 1.05 | (1.03 to 1.06) | 6.29 × 10−13 | 1.06 | (1.05 to 1.08) | 5.88 × 10−14 |
| GF Signaling | 0.13 | −1.04 | (−1.06 to −1.02) | 8.99 × 10−5 | −1.01 | (−1.04 to 1.01) | 0.28 |
| Development Regulation | 0.16 | 1.05 | (1.03 to 1.08) | 4.04 × 10−7 | 1.03 | (1.00 to 1.06) | 0.04 |
| Cell Survival | 0.27 | 1.02 | (1.01 to 1.03) | 3.17 × 10−4 | 1.01 | (−1.00 to 1.02) | 0.18 |
| Immune Regulation | 0.63 | −1.02 | (−1.03 to −1.01) | 6.66 × 10−3 | −1.02 | (−1.04 to −1.01) | 7.48 × 10−3 |
| Protein Translation | 0.65 | −1.04 | (−1.06 to −1.03) | 4.68 × 10−9 | −1.05 | (−1.07 to −1.03) | 4.50 × 10−7 |
| Stress Responses | 0.73 | 1.04 | (1.02 to 1.06) | 4.78 × 10−5 | 1.04 | (1.01 to 1.06) | 7.67 × 10−3 |
| Cell Cycle | 0.8 | 1.04 | (1.03 to 1.06) | 1.08 × 10−8 | 1.04 | (1.02 to 1.06) | 5.14 × 10−5 |
| Angiogenesis | 0.91 | −1.03 | (−1.07 to 1.01) | 0.18 | −1.03 | (−1.08 to 1.03) | 0.37 |
| Chromatin TF Transcription | 0.93 | 1.01 | (−1.00 to 1.02) | 0.21 | 1.01 | (−1.01 to 1.02) | 0.40 |
| Stratification | Base Excision Repair Genes | Mismatch Repair Genes | Non-Homologous End-Joining | |||
|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | Fold Change | (95% CI) | Fold Change | (95% CI) | |
| LV Invasion | ||||||
| Absent | 1.13 | (1.11 to 1.14) | 1.18 | (1.16 to 1.19) | 1.11 | (1.09 to 1.13) |
| Present | 1.07 | (1.05 to 1.08) | 1.12 | (1.11 to 1.14) | 1.07 | (1.05 to 1.09) |
| p | 1.39 × 10−8 | 1.02 × 10−5 | 1.60 × 10−3 | |||
| PN Invasion | ||||||
| Absent | 1.11 | (1.09 to 1.12) | 1.16 | (1.15 to 1.17) | 1.09 | (1.08 to 1.11) |
| Present | 1.07 | (1.05 to 1.09) | 1.12 | (1.09 to 1.15) | 1.07 | (1.04 to 1.10) |
| p | 2.93 × 10−3 | 6.58 × 10−3 | 0.23 | |||
| Stage | ||||||
| Stage-1 | 1.13 | (1.1 to 1.15) | 1.18 | (1.16 to 1.21) | 1.11 | (1.08 to 1.15) |
| Stage-2 | 1.1 | (1.08 to 1.12) | 1.15 | (1.13 to 1.17) | 1.09 | (1.06 to 1.11) |
| Stage-3 | 1.09 | (1.07 to 1.10) | 1.14 | (1.13 to 1.16) | 1.08 | (1.06 to 1.10) |
| p | 0.01 | 0.04 | 0.18 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibriya, M.G.; Raza, M.; Quinn, A.; Kamal, M.; Ahsan, H.; Jasmine, F. A Transcriptome and Methylome Study Comparing Tissues of Early and Late Onset Colorectal Carcinoma. Int. J. Mol. Sci. 2022, 23, 14261. https://doi.org/10.3390/ijms232214261
Kibriya MG, Raza M, Quinn A, Kamal M, Ahsan H, Jasmine F. A Transcriptome and Methylome Study Comparing Tissues of Early and Late Onset Colorectal Carcinoma. International Journal of Molecular Sciences. 2022; 23(22):14261. https://doi.org/10.3390/ijms232214261
Chicago/Turabian StyleKibriya, Muhammad G, Maruf Raza, Anthony Quinn, Mohammed Kamal, Habibul Ahsan, and Farzana Jasmine. 2022. "A Transcriptome and Methylome Study Comparing Tissues of Early and Late Onset Colorectal Carcinoma" International Journal of Molecular Sciences 23, no. 22: 14261. https://doi.org/10.3390/ijms232214261