
Esophageal Squamous Cancer from 4NQO-Induced Mice Model: CNV Alterations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
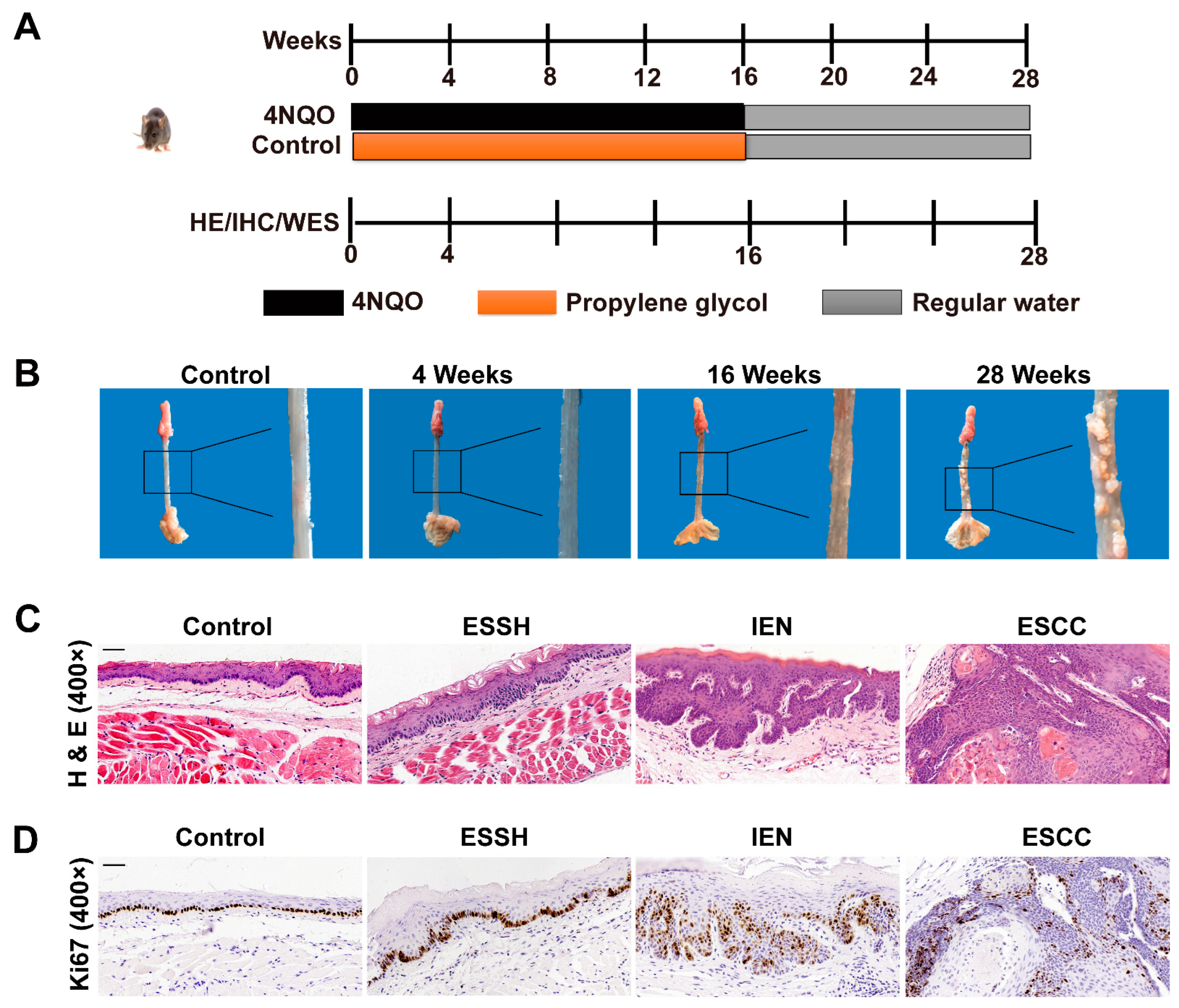
2.1. NQO Induction Esophageal Tumorigenesis in Mice
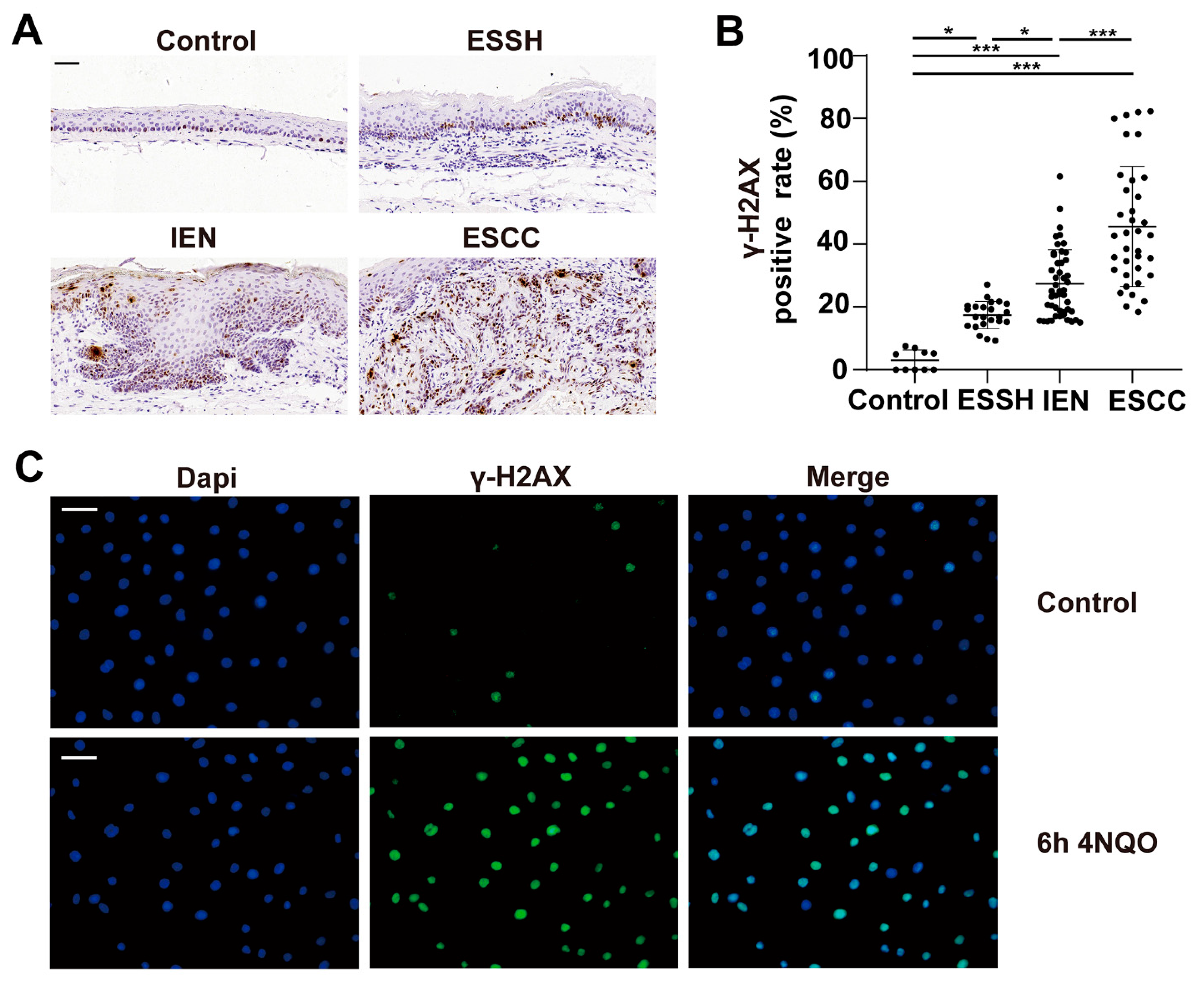
2.2. DNA Damage Induced by 4NQO in Both Mice and NE2 Cell Line
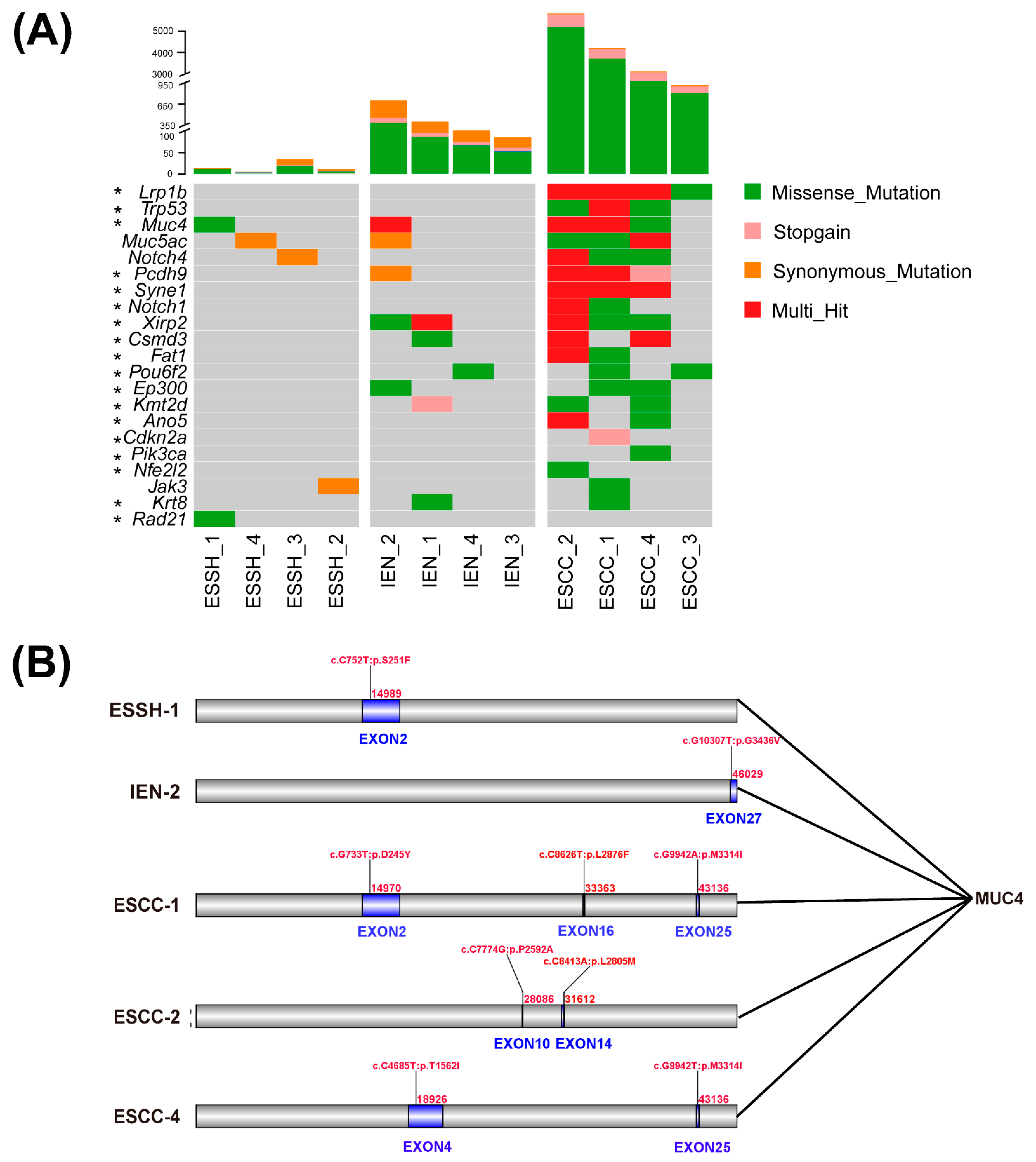
2.3. Mutated Genes in 4NQO-Induced ESCC Development
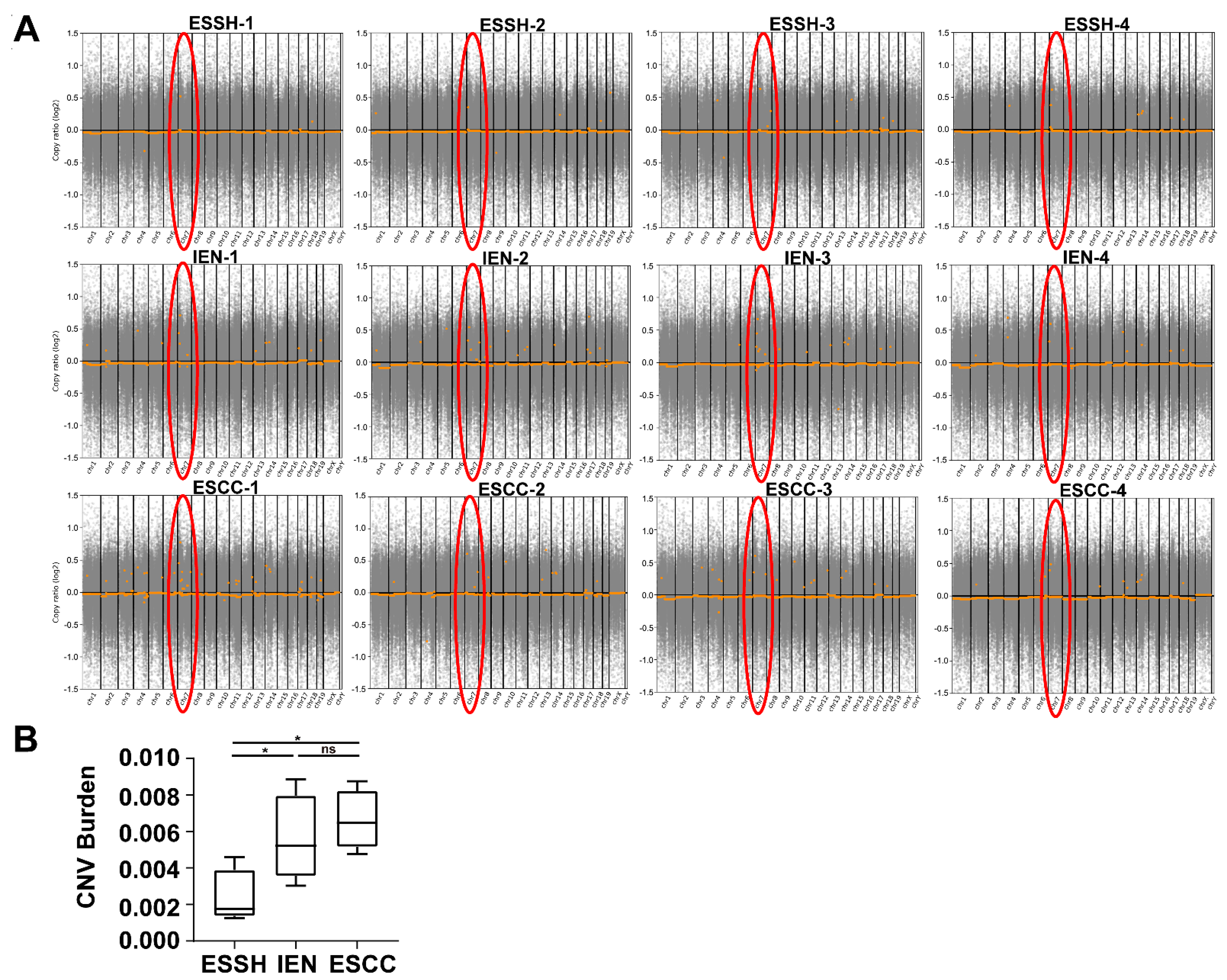
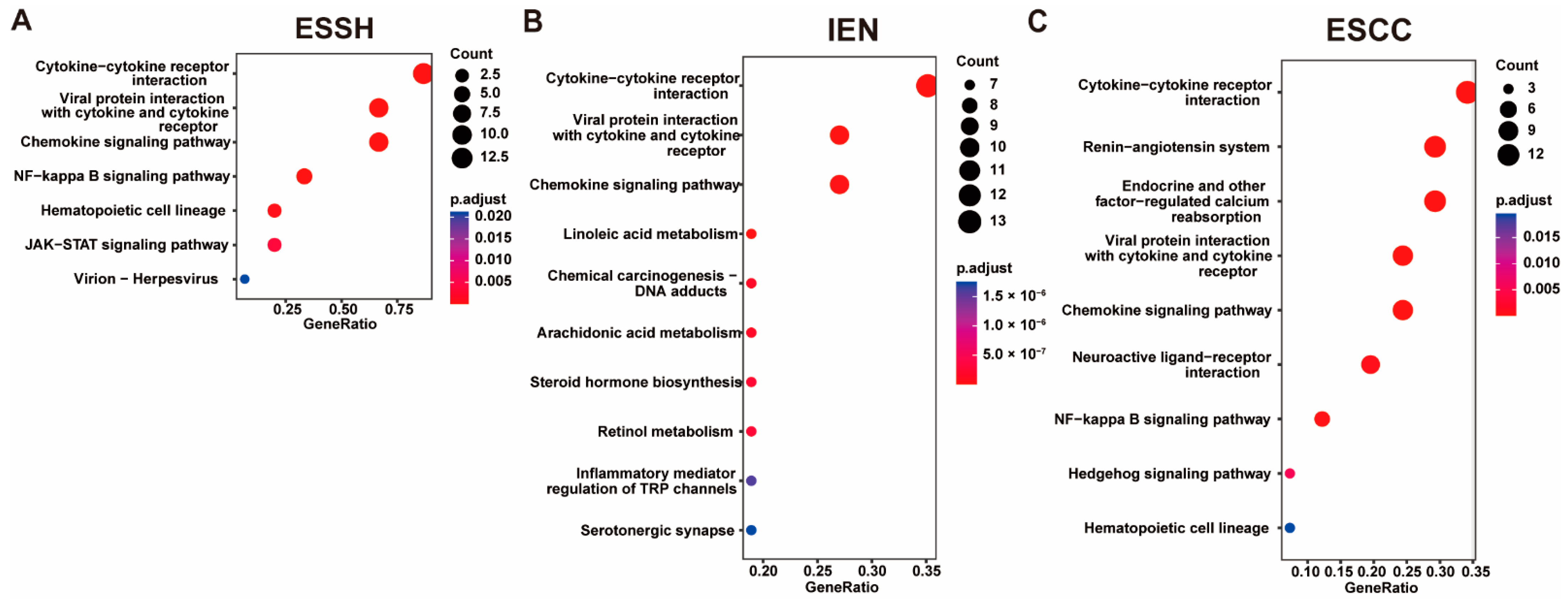
2.4. Copy Number Variations
3. Discussion
4. Materials and Methods
4.1. Cell Line and Culture Conditions
4.2. Animals and Carcinogen Treatment
4.3. Histopathological Analysis
4.4. Immunohistochemistry (IHC)
4.5. Immunofluorescence Staining
4.6. Isolation of Genomic DNA
4.7. Whole-Exome Sequencing (WES)
4.8. Bioinformatics Data Analysis
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abnet, C.C.; Arnold, M.; Wei, W.Q. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology 2018, 154, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichenbach, Z.W.; Murray, M.G.; Saxena, R.; Farkas, D.; Karassik, E.G.; Klochkova, A.; Patel, K.; Tice, C.; Hall, T.M.; Gang, J.; et al. Clinical and translational advances in esophageal squamous cell carcinoma. Adv. Cancer Res. 2019, 144, 95–135. [Google Scholar]
- Codipilly, D.C.; Qin, Y.; Dawsey, S.M.; Kisiel, J.; Topazian, M.; Ahlquist, D.; Iyer, P.G. Screening for esophageal squamous cell carcinoma: Recent advances. Gastrointest Endosc. 2018, 88, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 390, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.K. Histopathological Assessment for Esophageal Squamous Cell Carcinoma. Methods Mol. Biol. 2020, 2129, 7–18. [Google Scholar] [PubMed]
- Lee, N.P.; Chan, C.M.; Tung, L.N.; Wang, H.K.; Law, S. Tumor xenograft animal models for esophageal squamous cell carcinoma. J. Biomed. Sci. 2018, 25, 66. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, R.A.; Farshchian, M.; Abbaszadegan, M.R. Genetically engineered mouse models of esophageal cancer. Exp. Cell Res. 2021, 406, 112757. [Google Scholar] [CrossRef]
- Tang, X.H.; Knudsen, B.; Bemis, D.; Tickoo, S.; Gudas, L.J. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin. Cancer Res. 2004, 10, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Cui, Q.; Fan, W.; Ma, Y.; Chen, Y.; Liu, T.; Zhang, X.; Xi, Y.; Wang, C.; Peng, L.; et al. Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer. Nat. Commun. 2020, 11, 3715. [Google Scholar] [CrossRef]
- Ribeiro, D.A.; Fracalossi, A.C.; Uatari, S.A.; Oshima, C.T.; Salvadori, D.M. Imbalance of tumor suppression genes expression following rat tongue carcinogenesis induced by 4-nitroquinoline 1-oxide. In Vivo 2009, 23, 937–942. [Google Scholar] [PubMed]
- Booth, D.R. A relationship found between intra-oral sites of 4NQO reductase activity and chemical carcinogenesis. Cell Tissue Kinet 1990, 23, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Fronza, G.; Campomenosi, P.; Iannone, R.; Abbondandolo, A. The 4-nitroquinoline 1-oxide mutational spectrum in single stranded DNA is characterized by guanine to pyrimidine transversions. Nucleic Acids Res. 1992, 20, 1283–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramotar, D.; Belanger, E.; Brodeur, I.; Masson, J.Y.; Drobetsky, E.A. A yeast homologue of the human phosphotyrosyl phosphatase activator PTPA is implicated in protection against oxidative DNA damage induced by the model carcinogen 4-nitroquinoline 1-oxide. J. Biol. Chem. 1998, 273, 21489–21496. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Hetrick, K.M.; Foster, P.L. Interplay of DNA repair, homologous recombination, and DNA polymerases in resistance to the DNA damaging agent 4-nitroquinoline-1-oxide in Escherichia coli. DNA Repair 2010, 9, 1090–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahsan, A.; Liu, Z.; Su, R.; Liu, C.; Liao, X.; Su, M. Potential Chemotherapeutic Effect of Selenium for Improved Canceration of Esophageal Cancer. Int. J. Mol. Sci. 2022, 23, 5509. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Killcoyne, S.; Yusuf, A.; Fitzgerald, R.C. Genomic instability signals offer diagnostic possibility in early cancer detection. Trends Genet. 2021, 37, 966–972. [Google Scholar] [CrossRef]
- Li, X.; Tian, D.; Guo, Y.; Qiu, S.; Xu, Z.; Deng, W.; Su, M. Genomic characterization of a newly established esophageal squamous cell carcinoma cell line from China and published esophageal squamous cell carcinoma cell lines. Cancer Cell Int. 2020, 20, 184. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zheng, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic Alterations in Esophageal Tissues From Squamous Dysplasia to Carcinoma. Gastroenterology 2017, 153, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; The WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Rice, T.W.; Ishwaran, H.; Ferguson, M.K.; Blackstone, E.H.; Goldstraw, P. Cancer of the Esophagus and Esophagogastric Junction: An Eighth Edition Staging Primer. J. Thorac. Oncol. 2017, 12, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Niestroj, L.M.; Perez-Palma, E.; Howrigan, D.P.; Zhou, Y.; Cheng, F.; Saarentaus, E.; Nurnberg, P.; Stevelink, R.; Daly, M.J.; Palotie, A.; et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain 2020, 143, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feizi, N.; Chi, C.; Hu, P. Association Analysis of Somatic Copy Number Alteration Burden With Breast Cancer Survival. Front Genet. 2018, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Xiao, D.; Guo, Y.; Tian, D.; Yun, H.; Chen, D.; Su, M. Chronic inflammation-related DNA damage response: A driving force of gastric cardia carcinogenesis. Oncotarget 2015, 6, 2856–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Tan, W.; Ling, Z.; Xi, R.; Shao, M.; Chen, M.; Luo, Y.; Zhao, Y.; Liu, Y.; Huang, X.; et al. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun. 2017, 8, 15290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Weng, X.; Ye, J.; Zhou, D.; Liu, Y.; Zhao, K. Identification of the Germline Mutation Profile in Esophageal Squamous Cell Carcinoma by Whole Exome Sequencing. Front Genet. 2019, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yao, N.; Zhang, S.; Song, Y.; Shao, Q.; Gu, H.; Ma, J.; Chen, B.; Zhao, H.; Tian, Y. Identification of critical radioresistance genes in esophageal squamous cell carcinoma by whole-exome sequencing. Ann. Transl. Med. 2020, 8, 998. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Tian, D.; Guo, J.; Liu, M.; Chen, Z.; Hamdy, F.C.; Helleday, T.; Su, M.; Ying, S. DNA damage response in peritumoral regions of oesophageal cancer microenvironment. Carcinogenesis 2013, 34, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.C.; Wang, M.R.; Koeffler, H.P. Genomic and Epigenomic Aberrations in Esophageal Squamous Cell Carcinoma and Implications for Patients. Gastroenterology 2018, 154, 374–389. [Google Scholar] [CrossRef] [Green Version]
- Hanafi, R.; Anestopoulos, I.; Voulgaridou, G.P.; Franco, R.; Georgakilas, A.G.; Ziech, D.; Malamou-Mitsi, V.; Pappa, A.; Panayiotidis, M.I. Oxidative stress based-biomarkers in oral carcinogenesis: How far have we gone? Curr. Mol. Med. 2012, 12, 698–703. [Google Scholar] [CrossRef]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumor microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef]
- Arima, Y.; Nishigori, C.; Takeuchi, T.; Oka, S.; Morimoto, K.; Utani, A.; Miyachi, Y. 4-Nitroquinoline 1-oxide forms 8-hydroxydeoxyguanosine in human fibroblasts through reactive oxygen species. Toxicol. Sci. 2006, 91, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L.; Theodoropoulos, G.; Kozloski, G.A.; Carothers Carraway, C.A. Muc4/MUC4 functions and regulation in cancer. Future Oncol. 2009, 5, 1631–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, P.; Singh, A.P.; Batra, S.K. Structure, evolution, and biology of the MUC4 mucin. FASEB J. 2008, 22, 966–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, C.A.; VanderVorst, K.; Free, S.; Rowson-Hodel, A.; Carraway, K.L. The role of membrane mucin MUC4 in breast cancer metastasis. Endocr. Relat. Cancer 2021, 29, R17–R32. [Google Scholar] [CrossRef]
- Gao, X.P.; Dong, J.J.; Xie, T.; Guan, X. Integrative Analysis of MUC4 to Prognosis and Immune Infiltration in Pan-Cancer: Friend or Foe? Front Cell Dev. Biol. 2021, 9, 695544. [Google Scholar] [CrossRef]
- Gautam, S.K.; Kumar, S.; Dam, V.; Ghersi, D.; Jain, M.; Batra, S.K. MUCIN-4 (MUC4) is a novel tumor antigen in pancreatic cancer immunotherapy. Semin Immunol. 2020, 47, 101391. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Li, A.; Liu, X.; Miao, Y.; Wang, Z. Analysis of the Relationship between Bladder Cancer Gene Mutation and Clinical Prognosis by High-Throughput Sequencing. Lab. Med. 2022; Advance online publication. lmac083. [Google Scholar] [CrossRef]
- Peng, L.; Li, Y.; Gu, H.; Xiang, L.; Xiong, Y.; Wang, R.; Zhou, H.; Wang, J. Mucin 4 mutation is associated with tumor mutation burden and promotes antitumor immunity in colon cancer patients. Aging 2021, 13, 9043–9055. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, X.; Fu, Y.; Li, B.; Xu, J.; Wang, H.; Huang, Z.; Xu, H.; Qiu, Y.; Shi, Y.; et al. Targeted next-generation sequencing for cancer-associated gene mutation and copy number detection in 206 patients with non-small-cell lung cancer. Bioengineered 2021, 12, 791–802. [Google Scholar] [CrossRef]
- Shen, L.Y.; Wang, H.; Dong, B.; Yan, W.P.; Lin, Y.; Shi, Q.; Chen, K.N. Possible prediction of the response of esophageal squamous cell carcinoma to neoadjuvant chemotherapy based on gene expression profiling. Oncotarget 2016, 7, 4531–4541. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.S.; Liu, W.; Zheng, S.Y.; Cai, H.Y.; Luo, H.H.; Feng, Y.F.; Lei, Y.Y. A Novel Ras--Related Signature Improves Prognostic Capacity in Oesophageal Squamous Cell Carcinoma. Front Genet. 2022, 13, 822966. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, M.A.; Kwok, S.; Montgomery, K.D.; Lowe, A.W.; Pai, R.K. Immunohistochemical panel for distinguishing esophageal adenocarcinoma from squamous cell carcinoma: A combination of p63, cytokeratin 5/6, MUC5AC, and anterior gradient homolog 2 allows optimal subtyping. Hum. Pathol. 2012, 43, 1799–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niv, Y.; Fass, R. The role of mucin in GERD and its complications. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killcoyne, S.; Gregson, E.; Wedge, D.C.; Woodcock, D.J.; Eldridge, M.D.; de la Rue, R.; Miremadi, A.; Abbas, S.; Blasko, A.; Kosmidou, C.; et al. Genomic copy number predicts esophageal cancer years before transformation. Nat. Med. 2020, 26, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, G.; Harutyunyan, T.; Aroutiounian, R.; Liehr, T. DNA Copy Number Variations as Markers of Mutagenic Impact. Int. J. Mol. Sci. 2019, 20, 4723. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.P.; Ligtenberg, M.J.; Hoogerbrugge, N.; Geurts van Kessel, A. Germline copy number variation and cancer risk. Curr. Opin. Genet. Dev. 2010, 20, 282–289. [Google Scholar] [CrossRef]
- Pos, O.; Radvanszky, J.; Buglyo, G.; Pos, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef]
- Li, L.; Song, D.; Qi, L.; Jiang, M.; Wu, Y.; Gan, J.; Cao, K.; Li, Y.; Bai, Y.; Zheng, T. Photodynamic therapy induces human esophageal carcinoma cell pyroptosis by targeting the PKM2/caspase-8/caspase-3/GSDME axis. Cancer Lett. 2021, 520, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.B.; Fang, J.Y. The role of pyroptosis in gastrointestinal cancer and immune responses to intestinal microbial infection. Biochim Biophys Acta Rev. Cancer 2019, 1872, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Erickson, A.; He, M.; Berglund, E.; Marklund, M.; Mirzazadeh, R.; Schultz, N.; Kvastad, L.; Andersson, A.; Bergenstrahle, L.; Bergenstrahle, J.; et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature 2022, 608, 360–367. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Su, R.; Ahsan, A.; Liu, C.; Liao, X.; Tian, D.; Su, M. Esophageal Squamous Cancer from 4NQO-Induced Mice Model: CNV Alterations. Int. J. Mol. Sci. 2022, 23, 14304. https://doi.org/10.3390/ijms232214304
Liu Z, Su R, Ahsan A, Liu C, Liao X, Tian D, Su M. Esophageal Squamous Cancer from 4NQO-Induced Mice Model: CNV Alterations. International Journal of Molecular Sciences. 2022; 23(22):14304. https://doi.org/10.3390/ijms232214304
Chicago/Turabian StyleLiu, Zhiwei, Ruibing Su, Anil Ahsan, Chencai Liu, Xiaoqi Liao, Dongping Tian, and Min Su. 2022. "Esophageal Squamous Cancer from 4NQO-Induced Mice Model: CNV Alterations" International Journal of Molecular Sciences 23, no. 22: 14304. https://doi.org/10.3390/ijms232214304