

Obesity-Associated Vitamin D Deficiency Correlates with Adipose Tissue DNA Hypomethylation, Inflammation, and Vascular Dysfunction
 and
and 
Abstract
:1. Introduction
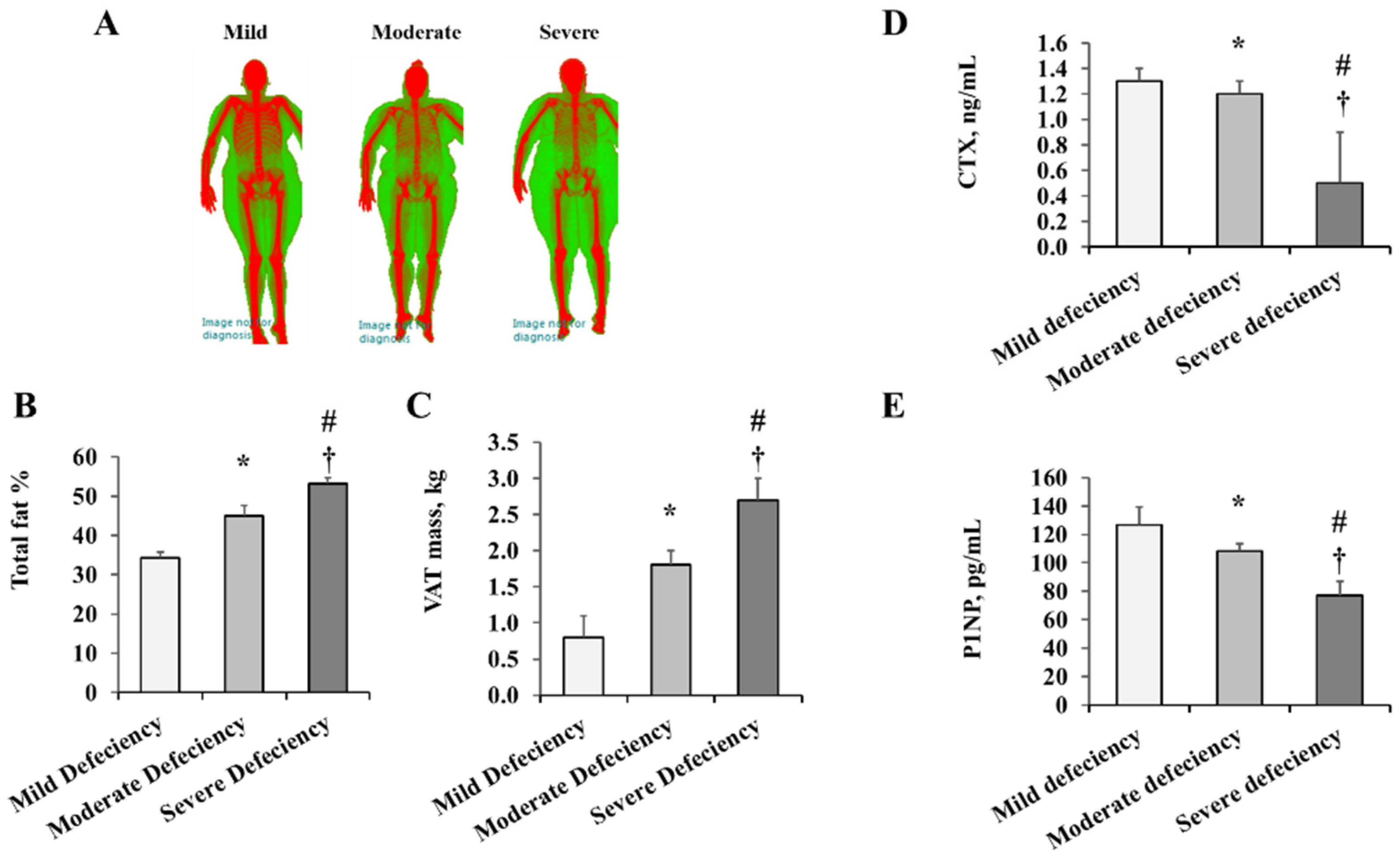
2. Results
2.1. Anthropometric and Cardiometabolic Measures
2.2. Vascular Measurements
2.3. Methylation Modifying Proteins in VAT
2.4. DNA Methylation and Inflammatory Biomarkers
2.5. Vitamin D Deficiency and Cardiometabolic Risk Correlations and Regression Models
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. Body Composition, Cardiovascular, and Metabolic Measures
4.3. Measuring Folate, Vitamin D, Vitamin B12, and Homocysteine (Hcy)
4.4. In Vivo Vasoreactivity Measurements
4.5. Vasoreactivity of AT-Isolated Arterioles
4.6. Global DNA Methylation Analysis
4.7. Methylation-Specific PCR (Polymerase Chain Reaction)
4.8. Quantitative PCR
4.9. Western Blotting
4.10. Immunofluorescent Staining of VAT
4.11. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daniel, D.; Hardigan, P.; Bray, N.; Penzell, D.; Savu, C. The incidence of vitamin D deficiency in the obese: A retrospective chart review. J. Community Hosp. Intern. Med. Perspect. 2015, 5, 26069. [Google Scholar] [CrossRef] [PubMed]
- Compston, J.E.; Vedi, S.; Ledger, J.E.; Webb, A.; Gazet, J.C.; Pilkington, T.R. Vitamin D status and bone histomorphometry in gross obesity. Am. J. Clin. Nutr. 1981, 34, 2359–2363. [Google Scholar] [CrossRef] [PubMed]
- Hengist, A.; Perkin, O.; Gonzalez, J.T.; Betts, J.A.; Hewison, M.; Manolopoulos, K.N.; Jones, K.S.; Koulman, A.; Thompson, D. Mobilising vitamin D from adipose tissue: The potential impact of exercise. Nutr. Bull. 2019, 44, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittas, A.G.; Chung, M.; Trikalinos, T.; Mitri, J.; Brendel, M.; Patel, K.; Lichtenstein, A.H.; Lau, J.; Balk, E.M. Systematic review: Vitamin D and cardiometabolic outcomes. Ann. Intern. Med. 2010, 152, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Mheid, I.; Patel, R.S.; Tangpricha, V.; Quyyumi, A.A. Vitamin D and cardiovascular disease: Is the evidence solid? Eur. Heart J. 2013, 34, 3691–3698. [Google Scholar] [CrossRef] [Green Version]
- Chitalia, N.; Recio-Mayoral, A.; Kaski, J.C.; Banerjee, D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 2012, 220, 265–268. [Google Scholar] [CrossRef]
- Judd, S.E.; Nanes, M.S.; Ziegler, T.R.; Wilson, P.W.; Tangpricha, V. Optimal vitamin D status attenuates the age-associated increase in systolic blood pressure in white Americans: Results from the third National Health and Nutrition Examination Survey. Am. J. Clin. Nutr. 2008, 87, 136–141. [Google Scholar] [CrossRef] [Green Version]
- London, G.M.; Guerin, A.P.; Verbeke, F.H.; Pannier, B.; Boutouyrie, P.; Marchais, S.J.; Metivier, F. Mineral metabolism and arterial functions in end-stage renal disease: Potential role of 25-hydroxyvitamin D deficiency. Am. Soc. Nephrol. 2007, 18, 613–620. [Google Scholar] [CrossRef]
- Motiwala, S.R.; Wang, T.J. Vitamin D and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 345–353. [Google Scholar] [CrossRef]
- Scragg, R.; Sowers, M.; Bell, C. Serum 25-hydroxyvitamin D, ethnicity, and blood pressure in the Third National Health and Nutrition Examination Survey. Am. J. Hypertens. 2007, 20, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Yiu, Y.F.; Chan, Y.H.; Yiu, K.H.; Siu, C.W.; Li, S.W.; Wong, L.Y.; Lee, S.W.; Tam, S.; Wong, E.W.; Cheung, B.M.; et al. Vitamin D deficiency is associated with depletion of circulating endothelial progenitor cells and endothelial dysfunction in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, E830–E835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, L.M.; Simhan, H.N.; Catov, J.M.; Roberts, J.M.; Platt, R.W.; Diesel, J.C.; Klebanoff, M.A. Maternal vitamin D status and the risk of mild and severe preeclampsia. Epidemiology 2014, 25, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Al-Dujaili, E.A.; Munir, N.; Iniesta, R.R. Effect of vitamin D supplementation on cardiovascular disease risk factors and exercise performance in healthy participants: A randomized placebo-controlled preliminary study. Ther. Adv. Endocrinol. Metab. 2016, 7, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, A.; Sun, B.; Larson, C.; Forman, J.P.; Williams, J.S. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin ii akin to the action of a converting enzyme inhibitor in obese hypertensives: An interventional study. J. Clin. Endocrinol. Metab. 2012, 97, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, N.; Campodonico, J.; Milazzo, V.; De Metrio, M.; Brambilla, M.; Camera, M.; Marenzi, G. Vitamin D and Cardiovascular Disease: Current Evidence and Future Perspectives. Nutrients 2021, 13, 3603. [Google Scholar] [CrossRef]
- Judd, S.E.; Tangpricha, V. Vitamin D deficiency and risk for cardiovascular disease. Am. J. Med. Sci. 2009, 338, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Roffe-Vazquez, D.N.; Huerta-Delgado, A.S.; Castillo, E.C.; Villarreal-Calderon, J.R.; Gonzalez-Gil, A.M.; Enriquez, C.; Garcia-Rivas, G.; Elizondo-Montemayor, L. Correlation of Vitamin D with Inflammatory Cytokines, Atherosclerotic Parameters, and Lifestyle Factors in the Setting of Heart Failure: A 12-Month Follow-Up Study. Int. J. Mol. Sci. 2019, 20, 5811. [Google Scholar] [CrossRef] [Green Version]
- Yin, K.; Agrawal, D.K. Vitamin D and inflammatory diseases. J. Inflamm. Res. 2014, 7, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Barbarawi, M.; Kheiri, B.; Zayed, Y.; Barbarawi, O.; Dhillon, H.; Swaid, B.; Yelangi, A.; Sundus, S.; Bachuwa, G.; Alkotob, M.L.; et al. Vitamin D Supplementation and Cardiovascular Disease Risks in More Than 83,000 Individuals in 21 Randomized Clinical Trials: A Meta-analysis. JAMA Cardiol. 2019, 4, 765–776. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Szczurek, M.; Hassan, C.; Masrur, M.; Gangemi, A.; Phillips, S.A. Vitamin D Improves Nitric Oxide-Dependent Vasodilation in Adipose Tissue Arterioles from Bariatric Surgery Patients. Nutrients 2019, 11, 2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.M.; Naquiallah, D.; Qureshi, M.; Mirza, M.I.; Hassan, C.; Masrur, M.; Bianco, F.M.; Frederick, P.; Cristoforo, G.P.; Gangemi, A.; et al. DNA methylation profile of genes involved in inflammation and autoimmunity correlates with vascular function in morbidly obese adults. Epigenetics 2022, 17, 93–109. [Google Scholar] [CrossRef]
- Ali, M.M.; Hassan, C.; Masrur, M.; Bianco, F.M.; Naquiallah, D.; Mirza, I.; Frederick, P.; Fernandes, E.T.; Giulianotti, C.P.; Gangemi, A.; et al. Adipose Tissue Hypoxia Correlates with Adipokine Hypomethylation and Vascular Dysfunction. Biomedicines 2021, 9, 1034. [Google Scholar] [CrossRef]
- Pike, J.W.; Meyer, M.B. The vitamin D receptor: New paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3). Endocrinol. Metab. Clin. N. Am. 2010, 39, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Fetahu, I.S.; Hobaus, J.; Kallay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5, 164. [Google Scholar] [CrossRef] [Green Version]
- Cruz, M.A.D.; Ulfenborg, B.; Blomstrand, P.; Faresjo, M.; Stahl, F.; Karlsson, S. Characterization of methylation patterns associated with lifestyle factors and vitamin D supplementation in a healthy elderly cohort from Southwest Sweden. Sci. Rep. 2022, 12, 12670. [Google Scholar] [CrossRef]
- Ong, L.T.C.; Booth, D.R.; Parnell, G.P. Vitamin D and its Effects on DNA Methylation in Development, Aging, and Disease. Mol. Nutr. Food Res. 2020, 64, e2000437. [Google Scholar] [CrossRef]
- Haloul, M.; Vinjamuri, S.J.; Naquiallah, D.; Mirza, M.I.; Qureshi, M.; Hassan, C.; Masrur, M.; Bianco, F.M.; Frederick, P.; Cristoforo, G.P.; et al. Hyperhomocysteinemia and Low Folate and Vitamin B12 Are Associated with Vascular Dysfunction and Impaired Nitric Oxide Sensitivity in Morbidly Obese Patients. Nutrients 2020, 12, 2014. [Google Scholar] [CrossRef]
- Selhub, J. Folate, vitamin B12 and vitamin B6 and one carbon metabolism. J. Nutr. Health Aging 2002, 6, 39–42. [Google Scholar]
- Wierzbicki, A.S. Homocysteine and cardiovascular disease: A review of the evidence. Diabetes Vasc. Dis. Res. 2007, 4, 143–150. [Google Scholar] [CrossRef]
- Humphrey, L.L.; Fu, R.; Rogers, K.; Freeman, M.; Helfand, M. Homocysteine level and coronary heart disease incidence: A systematic review and meta-analysis. Mayo Clin. Proc. 2008, 83, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Jonas, M.I.; Kurylowicz, A.; Bartoszewicz, Z.; Lisik, W.; Jonas, M.; Kozniewski, K.; Puzianowska-Kuznicka, M. Vitamin D Receptor Gene Expression in Adipose Tissue of Obese Individuals is Regulated by miRNA and Correlates with the Pro-Inflammatory Cytokine Level. Int. J. Mol. Sci. 2019, 20, 5272. [Google Scholar] [CrossRef] [Green Version]
- Vranic, L.; Mikolasevic, I.; Milic, S. Vitamin D Deficiency: Consequence or Cause of Obesity? Medicina (Kaunas) 2019, 55, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F.; Chen, T.C. Vitamin D deficiency: A worldwide problem with health consequences. Am. J. Clin. Nutr. 2008, 87, 1080S–1086S. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Han, L.; Liu, Q.; Zhao, Y.; Wang, L.; Wang, Y. Effects of Vitamin D Supplementation on General and Central Obesity: Results from 20 Randomized Controlled Trials Involving Apparently Healthy Populations. Ann. Nutr. Metab. 2020, 76, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Theik, N.W.Y.; Raji, O.E.; Shenwai, P.; Shah, R.; Kalluri, S.R.; Bhutta, T.H.; Hannoodee, H.; Al Khalili, M.; Khan, S. Relationship and Effects of Vitamin D on Metabolic Syndrome: A Systematic Review. Cureus 2021, 13, e17419. [Google Scholar] [CrossRef]
- Zakharova, I.; Klimov, L.; Kuryaninova, V.; Nikitina, I.; Malyavskaya, S.; Dolbnya, S.; Kasyanova, A.; Atanesyan, R.; Stoyan, M.; Todieva, A.; et al. Vitamin D Insufficiency in Overweight and Obese Children and Adolescents. Front. Endocrinol. (Lausanne) 2019, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- McGill, A.T.; Stewart, J.M.; Lithander, F.E.; Strik, C.M.; Poppitt, S.D. Relationships of low serum vitamin D3 with anthropometry and markers of the metabolic syndrome and diabetes in overweight and obesity. Nutr. J. 2008, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Alkhatatbeh, M.J.; Abdul-Razzak, K.K.; Khasawneh, L.Q.; Saadeh, N.A. High Prevalence of Vitamin D Deficiency and Correlation of Serum Vitamin D with Cardiovascular Risk in Patients with Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2017, 15, 213–219. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Insufficiency Exacerbates Adipose Tissue Macrophage Infiltration and Decreases AMPK/SIRT1 Activity in Obese Rats. Nutrients 2017, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Trayhurn, P.; Bing, C. 1,25-Dihydroxyvitamin D3 inhibits the cytokine-induced secretion of MCP-1 and reduces monocyte recruitment by human preadipocytes. Int. J. Obes. 2013, 37, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lotfi-Dizaji, L.; Mahboob, S.; Aliashrafi, S.; Vaghef-Mehrabany, E.; Ebrahimi-Mameghani, M.; Morovati, A. Effect of vitamin D supplementation along with weight loss diet on meta-inflammation and fat mass in obese subjects with vitamin D deficiency: A double-blind placebo-controlled randomized clinical trial. Clin. Endocrinol. 2019, 90, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Han, S.N. The Role of Vitamin D in Adipose Tissue Biology: Adipocyte Differentiation, Energy Metabolism, and Inflammation. J. Lipid Atheroscler. 2021, 10, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bhagatwala, J.; Huang, Y.; Pollock, N.K.; Parikh, S.; Raed, A.; Gutin, B.; Harshfield, G.A.; Dong, Y. Race/Ethnicity-Specific Association of Vitamin D and Global DNA Methylation: Cross-Sectional and Interventional Findings. PLoS ONE 2016, 11, e0152849. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Witkowska-Sedek, E.; Ruminska, M.; Stelmaszczyk-Emmel, A.; Sobol, M.; Majcher, A.; Pyrzak, B. Vitamin D Effects on Selected Anti-Inflammatory and Pro-Inflammatory Markers of Obesity-Related Chronic Inflammation. Front. Endocrinol. 2022, 13, 920340. [Google Scholar] [CrossRef]
- Hall, E.; Volkov, P.; Dayeh, T.; Esguerra, J.L.; Salo, S.; Eliasson, L.; Ronn, T.; Bacos, K.; Ling, C. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 2014, 15, 522. [Google Scholar] [CrossRef] [Green Version]
- Aroke, E.N.; Joseph, P.V.; Roy, A.; Overstreet, D.S.; Tollefsbol, T.O.; Vance, D.E.; Goodin, B.R. Could epigenetics help explain racial disparities in chronic pain? J. Pain Res. 2019, 12, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Pheiffer, C.; Willmer, T.; Dias, S.; Abrahams, Y.; Louw, J.; Goedecke, J.H. Ethnic and Adipose Depot Specific Associations Between DNA Methylation and Metabolic Risk. Front. Genet. 2020, 11, 967. [Google Scholar] [CrossRef]
- Do, W.L.; Nguyen, S.; Yao, J.; Guo, X.; Whitsel, E.A.; Demerath, E.; Rotter, J.I.; Rich, S.S.; Lange, L.; Ding, J.; et al. Associations between DNA methylation and BMI vary by metabolic health status: A potential link to disparate cardiovascular outcomes. Clin. Epigenetics 2021, 13, 230. [Google Scholar] [CrossRef]
- Park, Y.M.; Myers, M.; Vieira-Potter, V.J. Adipose tissue inflammation and metabolic dysfunction: Role of exercise. Mo. Med. 2014, 111, 65–72. [Google Scholar]
- Mousa, A.; Naderpoor, N.; Teede, H.; Scragg, R.; de Courten, B. Vitamin D supplementation for improvement of chronic low-grade inflammation in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Nutr. Rev. 2018, 76, 380–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.J.; Mousa, A.; Ebeling, P.R.; Scott, D.; de Courten, B. Effects of vitamin D supplementation on inflammatory markers in heart failure: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 1169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Hsieh, C.C.; Kuo, H.F.; Tsai, M.K.; Yang, S.N.; Kuo, C.H.; Lee, M.S.; Hung, C.H. Effect of Vitamin D3 on Monocyte Chemoattractant Protein 1 Production in Monocytes and Macrophages. Acta Cardiol. Sin. 2014, 30, 144–150. [Google Scholar] [PubMed]
- Szymczak-Pajor, I.; Miazek, K.; Selmi, A.; Balcerczyk, A.; Sliwinska, A. The Action of Vitamin D in Adipose Tissue: Is There the Link between Vitamin D Deficiency and Adipose Tissue-Related Metabolic Disorders? Int. J. Mol. Sci. 2022, 23, 956. [Google Scholar] [CrossRef] [PubMed]
- Elkhwanky, M.S.; Kummu, O.; Piltonen, T.T.; Laru, J.; Morin-Papunen, L.; Mutikainen, M.; Tavi, P.; Hakkola, J. Obesity Represses CYP2R1, the Vitamin D 25-Hydroxylase, in the Liver and Extrahepatic Tissues. JBMR Plus 2020, 4, e10397. [Google Scholar] [CrossRef] [PubMed]
- Wamberg, L.; Cullberg, K.B.; Rejnmark, L.; Richelsen, B.; Pedersen, S.B. Investigations of the anti-inflammatory effects of vitamin D in adipose tissue: Results from an in vitro study and a randomized controlled trial. Horm. Metab. Res. 2013, 45, 456–462. [Google Scholar] [CrossRef]
- Yuzbashian, E.; Asghari, G.; Hedayati, M.; Zarkesh, M.; Mirmiran, P.; Khalaj, A. Determinants of vitamin D receptor gene expression in visceral and subcutaneous adipose tissue in non-obese, obese, and morbidly obese subjects. J. Steroid Biochem. Mol. Biol. 2019, 187, 82–87. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Munoz-Garach, A.; Serrano, M.; Garrido-Sanchez, L.; Bernal-Lopez, M.R.; Fernandez-Garcia, D.; Moreno-Santos, I.; Garriga, N.; Castellano-Castillo, D.; Camargo, A.; et al. Serum 25-hydroxyvitamin D and adipose tissue vitamin D receptor gene expression: Relationship with obesity and type 2 diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E591–E595. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.M. Vitamin D as A Protector of Arterial Health: Potential Role in Peripheral Arterial Disease Formation. Int. J. Mol. Sci. 2019, 20, 4907. [Google Scholar] [CrossRef] [Green Version]
- Hiemstra, T.; Lim, K.; Thadhani, R.; Manson, J.E. Vitamin D and Atherosclerotic Cardiovascular Disease. J. Clin. Endocrinol. Metab. 2019, 104, 4033–4050. [Google Scholar] [CrossRef]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, F.E.L.; Dos Santos, L.; Tenorio, A.P.O.; Lopes, M.R.; Barbosa, R.H.A. Supplementation with vitamin D and its analogs for treatment of endothelial dysfunction and cardiovascular disease. J. Vasc. Bras. 2020, 19, e20190150. [Google Scholar] [CrossRef] [PubMed]
- Gkastaris, K.; Goulis, D.G.; Potoupnis, M.; Anastasilakis, A.D.; Kapetanos, G. Obesity, osteoporosis and bone metabolism. J. Musculoskelet. Neuronal Interact. 2020, 20, 372–381. [Google Scholar] [PubMed]
- Fassio, A.; Idolazzi, L.; Rossini, M.; Gatti, D.; Adami, G.; Giollo, A.; Viapiana, O. The obesity paradox and osteoporosis. Eat. Weight Disord. 2018, 23, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Purnamasari, D.; Puspitasari, M.D.; Setiyohadi, B.; Nugroho, P.; Isbagio, H. Low bone turnover in premenopausal women with type 2 diabetes mellitus as an early process of diabetes-associated bone alterations: A cross-sectional study. BMC Endocr. Disord. 2017, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, A.M.; Szczurek, M.R.; Blackburn, B.K.; Mey, J.T.; Chen, Z.; Robinson, A.T.; Bian, J.T.; Unterman, T.G.; Minshall, R.D.; Brown, M.D.; et al. Hyperinsulinemia augments endothelin-1 protein expression and impairs vasodilation of human skeletal muscle arterioles. Physiol. Rep. 2016, 4, e12895. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Hwang, C.L.; Szczurek, M.R.; Bian, J.T.; Ranieri, C.; Gutterman, D.D.; Phillips, S.A. Low-Fat Diet Designed for Weight Loss But Not Weight Maintenance Improves Nitric Oxide-Dependent Arteriolar Vasodilation in Obese Adults. Nutrients 2019, 11, 1339. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.M.; Naquiallah, D.; Hassan, C.; Masrur, M.; Bianco, F.M.; Frederick, P.; Cristoforo, G.; Gangemi, A.; Phillips, S.A.; Mahmoud, A.M. Obesity-associated Hypoxia Contributes to Aberrant Methylation of Genes Implicated in Inflammation and vascular Function. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Solomon, T.P.J.; Phillips, S.A.; Kirwan, J.P.; Haus, J.M. Aerobic Exercise Reduces NOX2 in Skeletal Muscle of Obese Insulin-resistant Adults Via Interfering with RAGE/p-IkB-α Axis. FASEB J. 2016, 30, lb762. [Google Scholar] [CrossRef]
- Sharun, K.; Pawde, A.M.; Kumar, R.; Kalaiselvan, E.; Kinjavdekar, P.; Dhama, K.; Pal, A. Standardization and characterization of adipose-derived stromal vascular fraction from New Zealand white rabbits for bone tissue engineering. Vet. World 2021, 14, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Carswell, K.A.; Lee, M.J.; Fried, S.K. Culture of isolated human adipocytes and isolated adipose tissue. Methods Mol. Biol. 2012, 806, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Mild Deficiency (n = 21) | Moderate Deficiency (n = 30) | Severe Deficiency (n = 26) | p-Value |
|---|---|---|---|---|
| Age, y | 38.9 ± 2.3 | 36.4 ± 1.3 | 34.3 ± 0.2 | 0.1473 |
| Gender (♀) | 15 | 17 | 14 | 0.4303 ¥ |
| Race/ethnicity (AA) | 10 | 16 | 19 | 0.1629 ¥ |
| Vitamin D and Ca2+ measurements | ||||
| 25-hydroxyvitamin D, ng/mL | 13.5 ± 0.4 | 9.9 ± 0.1 * | 4.7 ± 0.2 *† | <0.0001 |
| Calcium, mg/dL | 9.8 ± 0.1 | 9.4 ± 0.1 * | 9.2 ± 0.1 * | 0.0006 |
| Phosphorous, mg/dL | 3.4 ± 0.1 | 3.3 ± 0.2 | 3.1 ± 0.1 | 0.4050 |
| Anthropometric DEXA measurements | ||||
| Weight, kg | 94.9 ± 5.2 | 101.2 ± 11.5 | 148.6 ± 6.0 *† | <0.0001 |
| WC, cm | 82.1 ± 7.7 | 106.7 ± 6.2 * | 134.9 ± 4.1 *† | <0.0001 |
| BMI, kg/m2 | 34.1 ± 4.2 | 34.5 ± 1.9 | 52.9 ± 1.7 *† | <0.0001 |
| BSA | 2.1 ± 0.1 | 2.2 ± 0.1 | 2.6 ± 0.1 *† | 0.0022 |
| Fat % | 34.3 ± 1.5 | 45.0 ± 2.6 * | 53.1 ± 1.6 *† | <0.0001 |
| Lean % | 63.1 ± 3.7 | 53.4 ± 2.4 * | 45.9 ± 1.5 * | 0.0001 |
| Android fat % | 34.3 ± 1.9 | 47.1 ± 2.7 * | 56.2 ± 1.5 *† | <0.0001 |
| VAT mass, kg | 0.8 ± 0.3 | 1.8 ± 0.2 * | 2.7 ± 0.3 *† | 0.0003 |
| Bone density measurements | ||||
| BMC, cm2 | 2801.3 ± 91.0 | 3172.9 ± 100.5 | 3578.0 ± 163.7 *† | 0.0004 |
| BMD, gm/cm2 | 1.3 ± 0.1 | 1.3 ± 0.03 | 1.5 ± 0.03 *† | 0.0134 |
| T-score | 1.8 ± 0.4 | 3.0 ± 0.2 * | 3.6 ± 0.3 * | 0.0004 |
| Z-score | 0.6 ± 0.1 | 1.5 ± 0.3 | 1.6 ± 0.3 * | 0.0314 |
| Variable | Mild Deficiency (n = 21) | Moderate Deficiency (n = 30) | Severe Deficiency (n = 26) | p-Value |
|---|---|---|---|---|
| Cardiometabolic measurements | ||||
| FPI, µU/mL | 10.1 ± 1.2 | 11.9 ± 0.9 | 15.2 ± 0.8 *† | 0.0018 |
| FPG, mg/dL | 93.5 ± 3.7 | 97.7 ± 3.9 | 101.2 ± 3.2 | 0.3749 |
| HOMA-IR | 2.4 ± 0.4 | 3.4 ± 0.7 | 3.9 ± 0.4 * | 0.0233 |
| HbA1c, % | 5.4 ± 0.1 | 5.5 ± 0.2 | 5.9 ± 0.3 | 0.2737 |
| Chol, mg/dL | 159.0 ± 4.9 | 163.8 ± 5.3 | 179.1 ± 4.8 * | 0.0213 |
| LDL, mg/dL | 90.9 ± 5.8 | 93.0 ± 5.3 | 113.2 ± 4.8 *† | 0.0070 |
| HDL, mg/dL | 48.7 ± 1.9 | 47.2 ± 1.8 | 43.2 ± 1.2 | 0.0690 |
| Trig, mg/dL | 97.7 ± 2.8 | 112.2 ± 4.1 | 116.3 ± 4.5 *† | 0.0079 |
| HR, bpm | 76 ± 4 | 78 ± 2 | 82 ± 2 | 0.2866 |
| Systolic BP, mmHg | 123 ± 2 | 125 ± 2 | 132 ± 3 * | 0.0292 |
| Diastolic BP, mmHg | 77 ± 2 | 78 ± 2 | 84 ± 1 *† | 0.0132 |
| Other measurements | ||||
| Folate, ng/mL | 16.6 ± 1.4 | 18.6 ± 0.7 | 13.5 ± 0.9 *† | 0.0010 |
| Vitamin B12, ng/L | 564.5 ± 36.4 | 524.5 ± 26.5 | 323.8 ± 18.2 *† | <0.0001 |
| Hcy, µmol/L | 10.2 ± 1.2 | 12.3 ± 1.3 | 23.5 ± 3.1 *† | <0.0001 |
| Variable | Mild Deficiency (n = 21) | Moderate Deficiency (n = 30) | Severe Deficiency (n = 26) | p-Value |
|---|---|---|---|---|
| Circulating biomarkers of inflammation and vascular function | ||||
| CRP, mg/dL | 1.8 ± 0.8 | 1.9 ± 0.3 | 4.4 ± 0.3 *† | 0.0001 |
| CXCL10, pg/mL | 82.2 ± 7.7 | 135.2 ± 13.1 * | 211.6 ± 16.8 *† | <0.0001 |
| IL1β, pg/mL | 11.5 ± 1.4 | 15.6 ± 2.1 | 19.0 ± 1.7 | 0.0295 |
| IL1RA, pg/mL | 61.8 ± 7.4 | 81.2 ± 6.5 | 84.1 ± 8.1 | 0.0962 |
| IL6, pg/mL | 9.1 ± 0.3 | 13.0 ± 0.7 * | 20.3 ± 1.5 *† | <0.0001 |
| IL8, pg/mL | 40.2 ± 3.8 | 38.7 ± 2.2 | 53.6 ± 3.9 *† | 0.0025 |
| IL10, pg/mL | 39.0 ± 1.2 | 42.4 ± 3.3 | 38.3 ± 3.1 | 0.5471 |
| IL17, pg/mL | 69.6 ± 3.2 | 86.3 ± 3.0 * | 126.4 ± 7.0 *† | <0.0001 |
| TNFα, pg/mL | 19.7 ± 1.1 | 23.1 ± 2.0 | 30.7 ± 2.4 *† | 0.0012 |
| MCP-1, pg/mL | 307.0 ± 12.3 | 332.0 ± 16.4 | 370.5 ± 13.5 * | 0.0166 |
| Active PAI, ng/mL | 21.7 ± 2.0 | 27.9 ± 1.3 * | 29.3 ± 1.7 * | 0.0061 |
| sCD40L, pg/mL | 346.8 ± 19.1 | 401.4 ± 21.0 | 448.7 ± 25.5 | 0.0125 |
| sICAM-1, ng/mL | 116.8 ± 4.8 | 124.9 ± 6.7 | 220.3 ± 5.4 *† | <0.0001 |
| sVCAM-1, ng/mL | 776.6 ± 11.7 | 851.1 ± 12.2 * | 920.7 ± 13.5 *† | <0.0001 |
| NO, µmol/L | 7.9 ± 0.9 | 5.9 ± 0.4 * | 4.1 ± 0.4 *† | 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirza, I.; Mohamed, A.; Deen, H.; Balaji, S.; Elsabbahi, D.; Munasser, A.; Naquiallah, D.; Abdulbaseer, U.; Hassan, C.; Masrur, M.; et al. Obesity-Associated Vitamin D Deficiency Correlates with Adipose Tissue DNA Hypomethylation, Inflammation, and Vascular Dysfunction. Int. J. Mol. Sci. 2022, 23, 14377. https://doi.org/10.3390/ijms232214377
Mirza I, Mohamed A, Deen H, Balaji S, Elsabbahi D, Munasser A, Naquiallah D, Abdulbaseer U, Hassan C, Masrur M, et al. Obesity-Associated Vitamin D Deficiency Correlates with Adipose Tissue DNA Hypomethylation, Inflammation, and Vascular Dysfunction. International Journal of Molecular Sciences. 2022; 23(22):14377. https://doi.org/10.3390/ijms232214377
Chicago/Turabian StyleMirza, Imaduddin, Ariej Mohamed, Hania Deen, Swetha Balaji, Duaa Elsabbahi, Amier Munasser, Dina Naquiallah, Uzma Abdulbaseer, Chandra Hassan, Mario Masrur, and et al. 2022. "Obesity-Associated Vitamin D Deficiency Correlates with Adipose Tissue DNA Hypomethylation, Inflammation, and Vascular Dysfunction" International Journal of Molecular Sciences 23, no. 22: 14377. https://doi.org/10.3390/ijms232214377
APA StyleMirza, I., Mohamed, A., Deen, H., Balaji, S., Elsabbahi, D., Munasser, A., Naquiallah, D., Abdulbaseer, U., Hassan, C., Masrur, M., Bianco, F. M., Ali, M. M., & Mahmoud, A. M. (2022). Obesity-Associated Vitamin D Deficiency Correlates with Adipose Tissue DNA Hypomethylation, Inflammation, and Vascular Dysfunction. International Journal of Molecular Sciences, 23(22), 14377. https://doi.org/10.3390/ijms232214377