
Analysis of QTLs and Candidate Genes for Tassel Symptoms in Maize Infected with Sporisorium reilianum
 , and
, and
Abstract
:1. Introduction
2. Results
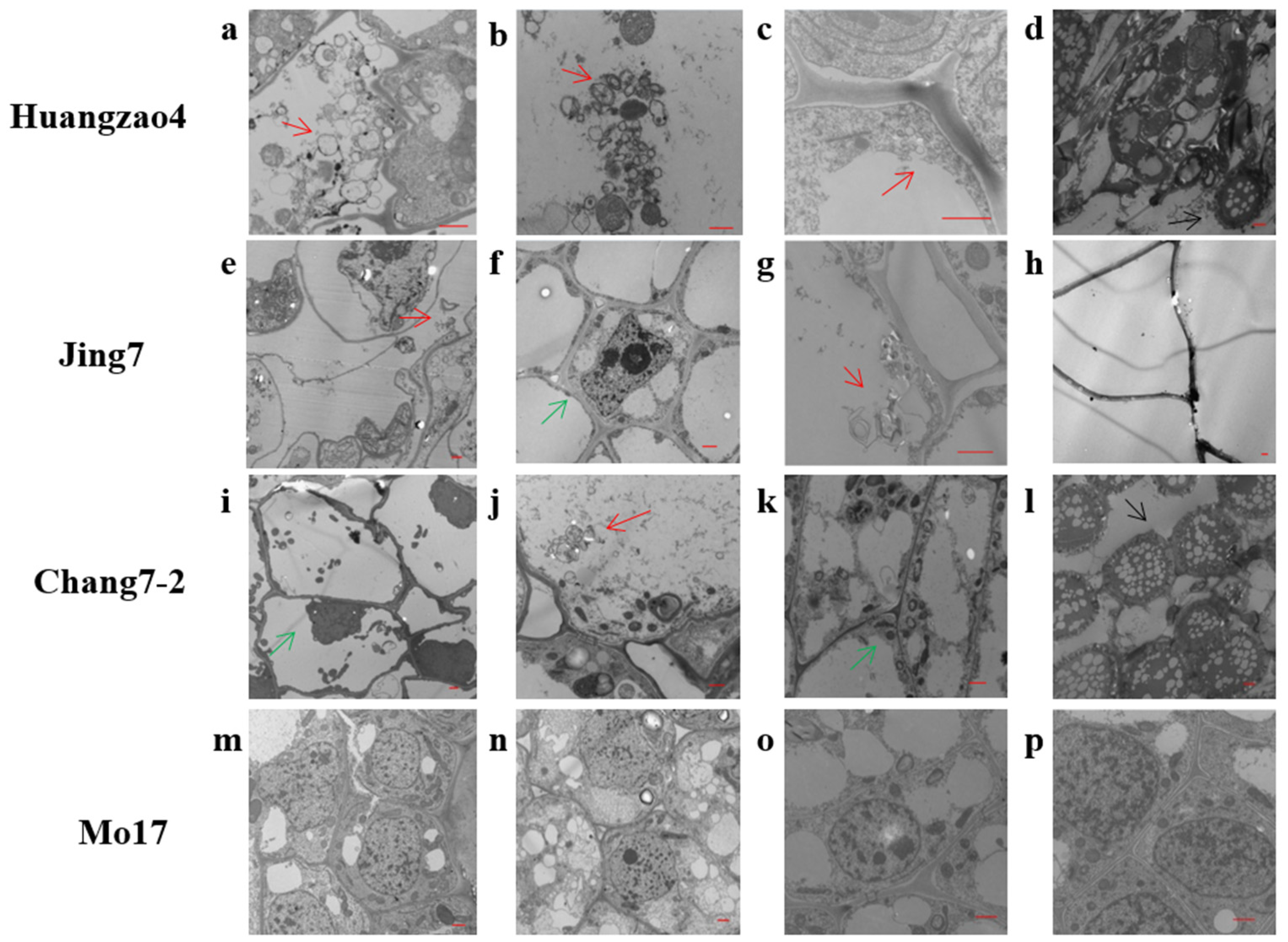
2.1. Sporisorium reilianum Causes Damage to Maize Cell Structure
2.2. Identification of Tassel Symptoms in the Two F2 Populations
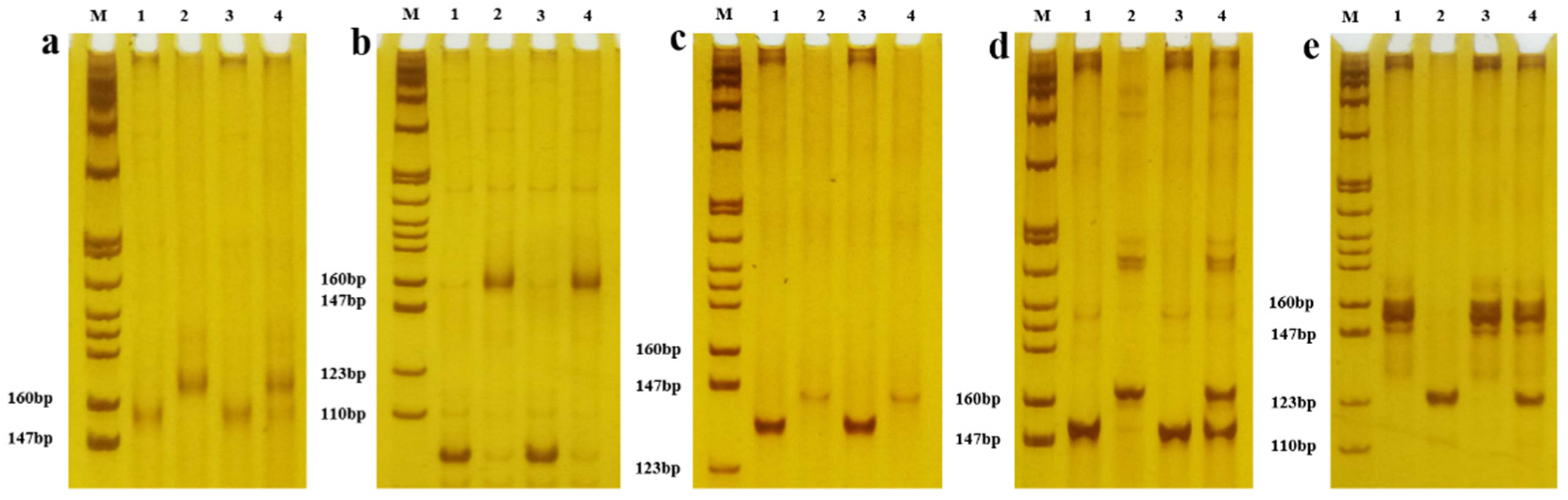
2.3. Identification of Polymorphisms between the Parents
2.4. Construction of Mixed Gene Pools and Selection of Primers
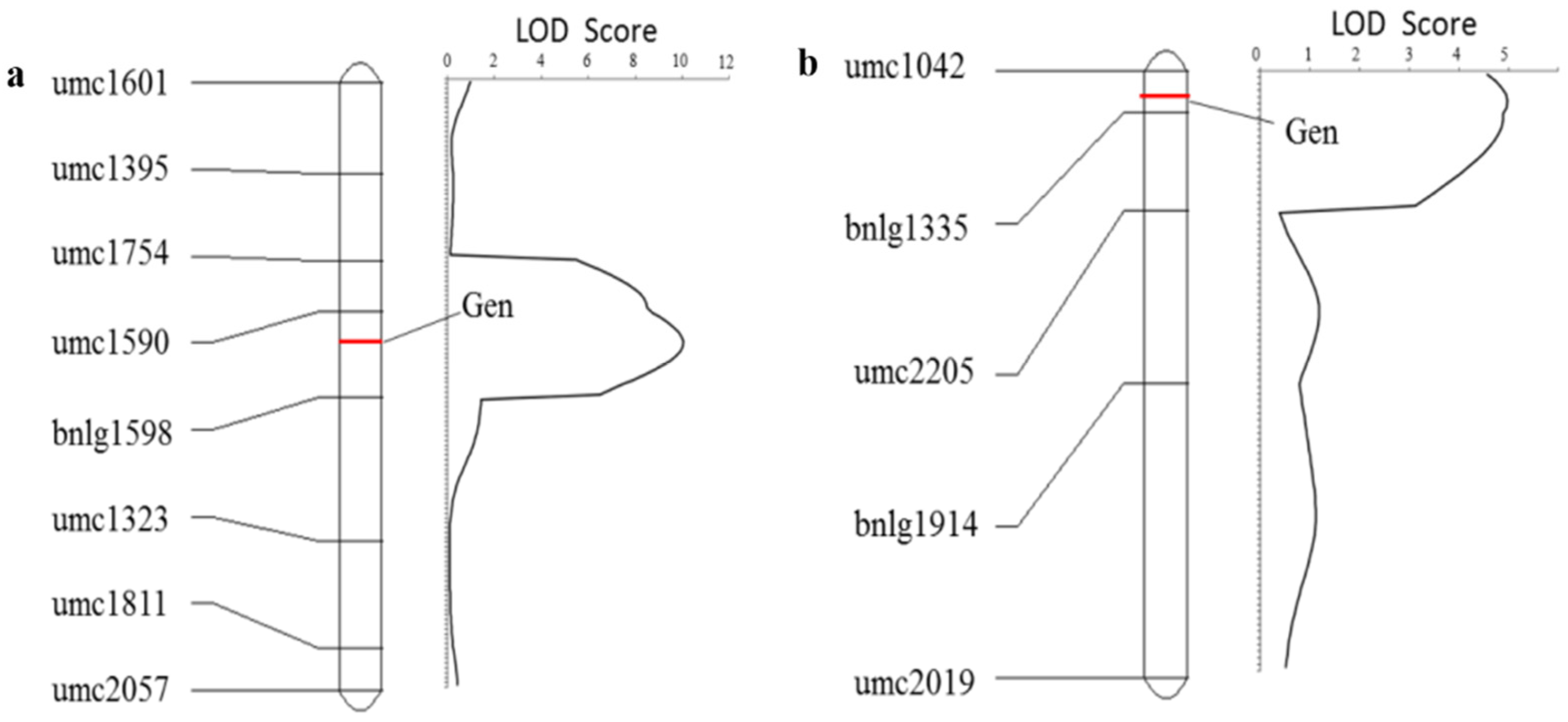
2.5. Preliminary Mapping of QTLs
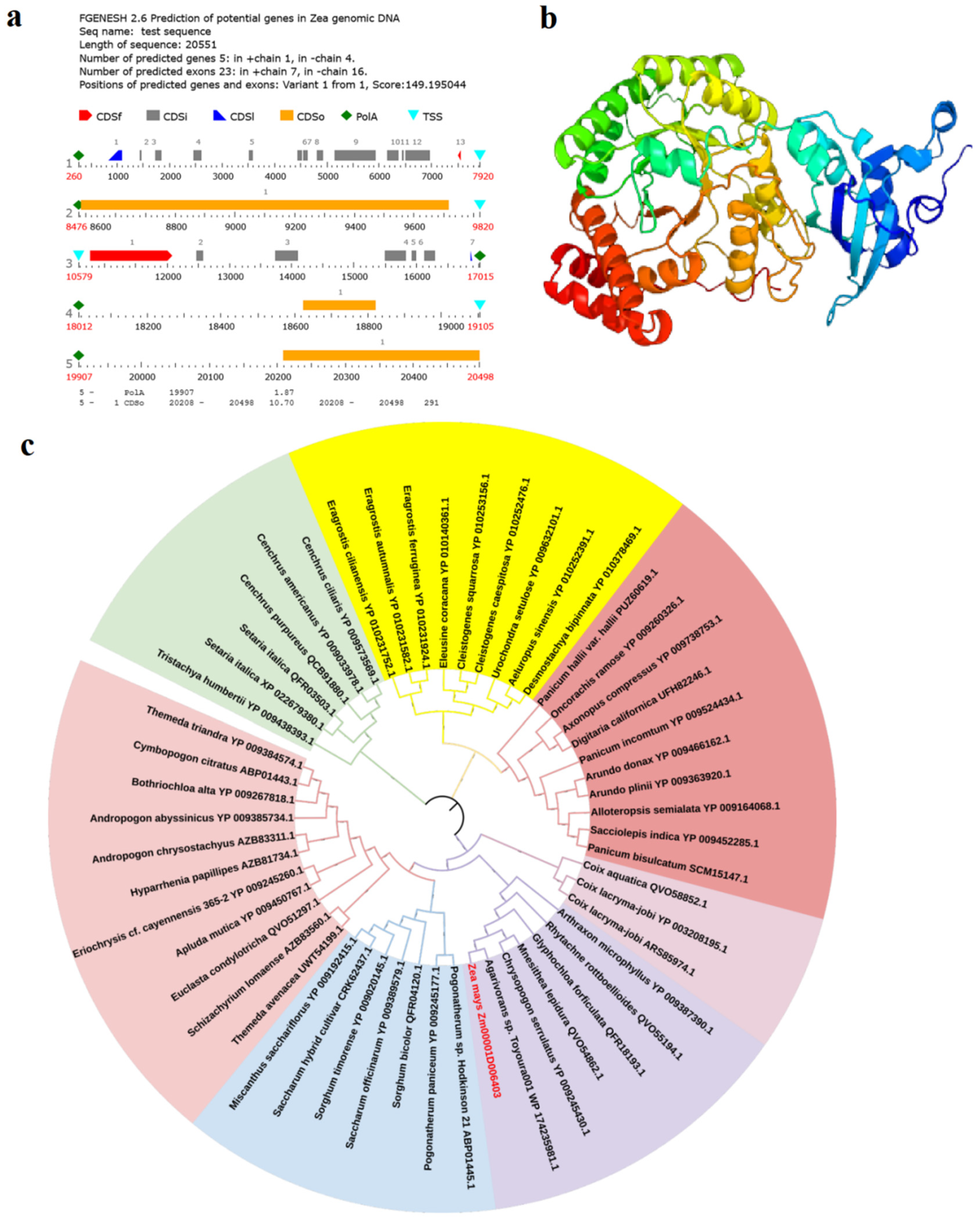
2.6. Predicting Candidate Gene Related to Tassel Symptoms of Maize Infected with Sporisorium reilianum
3. Discussion
3.1. Differences in Tassel Symptoms Infected by Head Smut of Sipingtou Bloodlines of Maize
3.2. Advantage of Using the BSA Method to Analyze Tassel Symptoms in Maize Infected with Sporisorium reilianum
3.3. Genetic Study of Maize Tassels Infected with Sporisorium reilianum
3.4. Function of Two Candidate Genes Related to the Tassel Symptoms of Head Smut
4. Materials and Methods
4.1. Plant and Sporisorium reilianum Materials
4.2. Analysis of Cell Morphological Changes in Tassels Infected with Sporisorium reilianum
4.3. Artificial Inoculation and Phenotypic Investigation
4.4. Construction of a Mixed Gene Pool with Different Symptoms from the F2 Population
4.5. DNA Extraction and Screening of SSR Markers
4.6. Data Processing and Statistical Analysis
4.7. Candidate Genes Prediction and Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Bao, J.; Wei, X.; Wu, S.; Fang, C.; Li, Z.; Qi, Y.; Gao, Y.; Dong, Z.; Wan, X. Genetic structure and molecular mechanisms underlying the formation of tassel, Anther, and pollen in the male inflorescence of maize (Zea Mays L.). Cells 2022, 11, 1753–1783. [Google Scholar] [CrossRef] [PubMed]
- Alana, P.; Schirawski, J. Host specificity in Sporisorium reilianum is determined by distinct mechanisms in maize and sorghum. Mol. Plant Pathol. 2015, 17, 741–754. [Google Scholar]
- Li, Y.; Wu, X.; Jaqueth, J.; Zhang, D.; Cui, D.; Li, C.; Hu, G.; Dong, H.; Song, Y.; Shi, Y.; et al. The identification of two head smut resistance-related QTL in maize by the joint approach of linkage mapping and association analysis. PLoS ONE 2015, 10, e0145549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berke, T.G.; Rocheford, T.R. Quantitative trait loci for tassel traits in maize. Crop Sci. 1999, 39, 1439–1443. [Google Scholar] [CrossRef]
- Xu, G.; Wang, X.; Huang, C.; Xu, D.; Li, D.; Tian, J.; Chen, Q.; Wang, C.; Liang, Y.; Wu, Y.; et al. Complex genetic architecture underlies maize tassel domestication. New Phytol. 2017, 214, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Z.; Gao, S.; Shi, H.; Zhang, S.; George, M.L.C.; Li, M.; Xie, C. Analysis of QTL for resistance to head smut (Sporisorium Reiliana) in maize. Field Crop Res. 2008, 106, 148–155. [Google Scholar] [CrossRef]
- Chen, Y.; Qing, Q.; Tan, G.; Zhao, J.; Zhang, M. Identification and fine-mapping of a major QTL conferring resistance against head smut in maize. Theor. Appl. Genet. 2008, 117, 1241–1252. [Google Scholar] [CrossRef]
- Weng, J.; Liu, X.; Wang, Z.; Wang, J.; Zhang, L.; Hao, Z.; Xie, C.; Li, M.; Zhang, D.; Li, B.; et al. Molecular mapping of the major resistance quantitative trait locus qhs2.09 with simple sequence repeat and single nucleotide polymorphism markers in maize. Phytopathology 2012, 102, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Zhao, Q.; Zhang, N.; Ye, J.; Tan, G.; Li, B.; Ye, X.; Zhang, B.; Liu, H.; Fengler, K.A.; et al. A maize wall-associated kinase confers quantitative resistance to head smut. Nat. Genet. 2015, 47, 151–157. [Google Scholar] [CrossRef]
- Di, H.; Yu, T.; Deng, Y.; Dong, X.; Li, R.; Zhou, Y.; Wang, Z. Complementary DNA (cDNA) cloning and functional verification of resistance to head smut disease (Sphacelotheca Reiliana) of an NBS–LRR gene ZmNL in maize (Zea Mays). Euphytica 2017, 213, 288–302. [Google Scholar] [CrossRef]
- Lacaze, A.; Joly, D.L. Structural specificity in plant-filamentous pathogen interactions. Mol. Plant Pathol. 2020, 21, 1513–1525. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, N.; Li, C.; Wang, X.; Xu, X.; Chen, W.; Xing, G.; Zheng, W. The early response during the interaction of fungal phytopathogen and host plant. Open Biol. 2017, 7, 170057–170065. [Google Scholar] [CrossRef] [Green Version]
- Rodenburg, S.; Seidl, M.F.; Judelson, H.S.; Vu, A.L.; Govers, F.; de Ridder, D. Metabolic model of the phytophthora infestans-tomato interaction reveals metabolic switches during host colonization. mBio 2019, 10, e00454-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghareeb, H.; Zhao, Y.; Schirawski, J. Sporisorium reilianum possesses a pool of effector proteins that modulate virulence on maize. Mol. Plant Pathol. 2018, 20, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhu, X.; Zhang, Y.; Cai, Y.; Wang, J.; Liu, M.; Wang, J.; Bao, J.; Lin, F. Research on the molecular interaction mechanism between plants and pathogenic fungi. Int. J. Mol. Sci. 2022, 23, 4658–4673. [Google Scholar] [CrossRef]
- Doehlemann, G.; Ökmen, B.; Zhu, W.; Sharon, A. Plant pathogenic fungi. Microbiol. Spectr. 2017, 12, 24–48. [Google Scholar]
- Zhang, S.; Li, C.; Si, J.; Han, Z.; Chen, D. Action mechanisms of effectors in plant-pathogen interaction. Int. J. Mol. Sci. 2022, 23, 6758. [Google Scholar] [CrossRef]
- Pechanova, O.; Pechan, T. Maize-pathogen interactions: An ongoing combat from a proteomics perspective. Int. J. Mol. Sci. 2015, 16, 28429–28448. [Google Scholar] [CrossRef] [Green Version]
- Ghareeb, H.; Becker, A.; Iven, T.; Feussner, I.; Schirawski, J. Sporisorium reilianum infection changes inflorescence and branching architectures of maize. Plant Physiol. 2011, 156, 2037–2052. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yan, J.; Zhao, J.; Song, W.; Zhang, X.; Xiao, Y.; Zheng, Y. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 2012, 196, 125–131. [Google Scholar] [CrossRef]
- Khan, M.; Armin, D. Performing infection assays of Sporisorium reilianum F. Sp. Zeae in maize. Methods Mol. Biol. 2022, 2494, 291–298. [Google Scholar] [PubMed]
- Zhang, S.; Gardiner, J.; Xiao, Y.; Zhao, J.; Wang, F.; Zheng, Y. Floral transition in maize infected with Sporisorium reilianum disrupts compatibility with this biotrophic fungal pathogen. Planta 2013, 237, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Shi, Y.; Song, Y.; Zhang, D.; Li, C.; Buckler, E.S.; Li, Y.; Zhang, Z.; Wang, T. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize. Plant Biotechnol. J. 2016, 14, 1551–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadyayula, N.; da Silva, H.S.; Bohn, M.O.; Rocheford, T.R. Genetic and QTL analysis of maize tassel and ear inflorescence architecture. Theor. Appl. Genet. 2006, 112, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Li, Y.; Duan, S. Study on the resistence to head smut of corn varieties and its inheritance. Chin. Agric. Sci. 1983, 16, 12–17. [Google Scholar]
- Qiu, C.; Lv, W.; Lv, D.; Bai, Y.; Wei, Q. Symptoms of four potato varieties infected with Potato spindle tuber viroid (Pstvd). Plant Prot. 2014, 40, 159–163. [Google Scholar]
- Fu, S.; Zhan, Y.; Zhi, H.; Gai, J.; Yu, D. Mapping of SMV resistance gene Rsc-7 by SSR markers in soybean. Genetica 2006, 128, 63–69. [Google Scholar] [CrossRef]
- Wu, X.; Pang, Z.; Tian, L.; Hu, J. Physiological specialization of Sphacelotheca reiliana (Kuhn) Clint. to sorghum. Acta Phytopathol. Sin. 1982, 1, 13–18. [Google Scholar]
- Maya, H.; Mercado-Flores, Y.; Téllez-Jurado, A.; Pérez-Camarillo, J.; Mejía, O.; Anducho-Reyes, M. Molecular variation of the phytopathogenic fungus Sporisorium reilianum in Valle del mezquital, Hidalgo. Front. Ecol. Evol. 2020, 8, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhou, M.; Lu, W.; Ohm, H. Detection of Fusarium head blight resistance QTL in a wheat population using bulked segregant analysis. Theor. Appl. Genet. 2003, 106, 1041–1047. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Dentification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subudhi, P.K.; Borkakati, R.P.; Virmani, S.S.; Huang, N. Molecular mapping of a thermosensitive genetic male sterility gene in rice using bulked segregant analysis. Genome 1997, 40, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Song, J.; Wen, D.; Xu, L.; Jiang, Y.; Zhang, J.; Xiang, X.; Yu, G. Identification, mapping, and molecular marker development for Rgsr8.1: A new quantitative trait locus conferring resistance to Gibberella stalk rot in maize (Zea Mays L.). Front. Plant Sci. 2017, 8, 1355–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernier, J.; Kumar, A.; Ramaiah, V.; Spaner, D.; Atlin, G. A large-effect QTL for grain yield under reproductive-stage drought stress in upland rice. Crop Sci. 2007, 47, 507–516. [Google Scholar] [CrossRef]
- Salunkhe, A.S.; Poornima, R.; Prince, K.S.; Kanagaraj, P.; Sheeba, J.A.; Amudha, K.; Suji, K.K.; Senthil, A.; Babu, R.C. Fine mapping QTL for drought resistance traits in rice (Oryza sativa L.) using bulk segregant analysis. Mol. Biotechnol. 2011, 49, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Altinkut, A.; Gozukirmizi, N. Search for microsatellite markers associated with water-stress tolerance in wheat through bulked segregant analysis. Mol. Biotechnol. 2003, 23, 97–106. [Google Scholar] [CrossRef]
- Lu, H.; Lin, T.; Klein, J.; Wang, S.; Qi, J.; Zhou, Q.; Sun, J.; Zhang, Z.; Weng, Y.; Huang, S. QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor. Appl. Genet. 2014, 127, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yao, J.; Chu, L.; Yuan, Z.; Li, Y.; Zhang, Y. Genetic mapping of the nulliplex-branch gene (gb_nb1) in cotton using next-generation sequencing. Theor. Appl. Genet. 2015, 128, 539–547. [Google Scholar] [CrossRef]
- Trick, M.; Adamski, N.M.; Mugford, S.G.; Jiang, C.C.; Febrer, M.; Uauy, C. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol. 2012, 12, 14–31. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Gonzalez, R.H.; Segovia, V.; Bird, N.; Fenwick, P.; Holdgate, S.; Berry, S.; Jack, P.; Caccamo, M.; Uauy, C. RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. J. 2015, 13, 613–624. [Google Scholar] [CrossRef]
- Veldboom, L.R.; Lee, M.; Woodman, W.L. Molecular marker-facilitated studies in an elite maize population: I. Linkage analysis and determination of QTL for morphological traits. Theor. Appl. Genet. 1994, 88, 7–16. [Google Scholar] [CrossRef]
- Ji, H.; Li, X.; Xie, C.; Hao, Z.; Lü, X.; Shi, L.; Zhang, S. Comparative QTL mapping of resistance to Sporisorium reiliana in maize based on meta-analysis of QTL locations. J. Plant Res. 2007, 8, 132–139. [Google Scholar]
- Lu, X.; Brewbaker, J.L. Molecular mapping of qtls conferring resistance to Sphacelotheca reiliana (Kühn) Clint. Maize Genet. Coop. Newsl. 1999, 73, 36. [Google Scholar]
- Lübberstedt, T.; Xia, X.; Tan, G.; Liu, X.; Melchinger, A.E. QTL mapping of resistance to Sporisorium reiliana in maize. Theor. Appl. Genet. 1999, 99, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Jiang, Y.; Wang, Z.; Li, X.; Zhang, S. QTL identification of resistance to head smut in maize (in Chinese, English abstract). Acta Agron Sin. 2005, 31, 1449–1454. [Google Scholar]
- Di, H.; Liu, X.; Wang, Q.; Weng, J.; Zhang, L.; Li, X.; Wang, Z. Development of SNP-based dCAPS markers linked to major head smut resistance quantitative trait locus qhs2.09 in maize. Euphytica 2015, 202, 69–79. [Google Scholar] [CrossRef]
- Wu, Y.; Vicente, F.S.; Huang, K.; Dhliwayo, T.; Costich, D.E.; Semagn, K.; Sudha, N.; Olsen, M.; Prasanna, B.M.; Zhang, X.; et al. Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theor. Appl. Genet. 2016, 129, 753–765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, B.; Zuo, W.; Xing, Y.; Konlasuk, S.; Tan, G.; Zhang, Q.; Ye, J.; Xu, M. Cytological and molecular characterization of ZmWAK-mediated head-smut resistance in maize. Mol. Plant Microbe Interact. 2017, 30, 455–465. [Google Scholar] [CrossRef]
- Gao, S. Inheritance and quantitative trait loci mapping of resistance to Head Smut caused by Sphacelotheca reiliana(Kühn) in Maize. Ph.D. Thesis, Jilin University, Changchun, China, 2005. [Google Scholar]
- Huang, J.; Sun, W.; Ren, J.; Yang, R.; Fan, J.; Li, Y.; Wang, X.; Joseph, S.; Deng, W.; Zhai, L. Genome-wide identification and characterization of actin-depolymerizing factor (ADF) family genes and expression analysis of responses to various stresses in Zea Mays L. Int. J. Mol. Sci. 2020, 21, 1751–1768. [Google Scholar] [CrossRef] [Green Version]
- Burgos-Rivera, B.; Ruzicka, D.R.; Deal, R.B.; McKinney, E.C.; King-Reid, L.; Meagher, R.B. Actin depolymerizing factor9 controls development and gene expression in Arabidopsis. Plant Mol. Biol. 2008, 68, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Lopez, I.; Anthony, R.G.; Maciver, S.K.; Jiang, C.J.; Khan, S.; Weeds, A.G.; Hussey, P.J. Pollen specific expression of maize genes encoding actin depolymerizing factor-like proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 7415–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurepa, J.; Smalle, J.A. Oxidative stress-induced formation of covalently linked ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit dimer in tobacco plants. BMC Res. Notes 2019, 12, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Doron, L.; Segal, N.; Gibori, H.; Shapira, M. The BSD2 ortholog in Chlamydomonas reinhardtii is a polysome-associated chaperone that co-migrates on sucrose gradients with the rbcL transcript encoding the Rubisco large subunit. Plant J. 2014, 80, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.; Guo, Z.; Zarzycki, J.; Steinchen, W.; Schuller, J.M.; Heimerl, T.; Prinz, S.; Mueller-Cajar, O.; Erb, T.J.; Hochberg, G. Evolution of increased complexity and specificity at the dawn of form I Rubiscos. Science 2022, 378, 155–160. [Google Scholar] [CrossRef]
- Grabsztunowicz, M.; Górski, Z.; Luciński, R.; Jackowski, G. A reversible decrease in ribulose 1,5-bisphosphate carboxylase/oxygenase carboxylation activity caused by the aggregation of the enzyme’s large subunit is triggered in response to the exposure of moderate irradiance-grown plants to low irradiance. Physiol. Plant. 2015, 154, 591–608. [Google Scholar] [CrossRef]
- Qu, Y.; Sakoda, K.; Fukayama, H.; Kondo, E.; Suzuki, Y.; Makino, A.; Terashima, I.; Yamori, W. Overexpression of both rubisco and rubisco activase rescues rice photosynthesis and biomass under heat stress. Plant Cell Environ. 2021, 44, 2308–2320. [Google Scholar] [CrossRef]
- Feng, Z.; Yuan, M.; Zou, J.; Wu, L.; Wei, L.; Chen, T.; Zhou, N.; Xue, W.; Zhang, Y.; Chen, Z.; et al. Development of marker-free rice with stable and high resistance to rice black-streaked dwarf virus disease through RNA interference. Plant Biotechnol. J. 2021, 19, 212–214. [Google Scholar] [CrossRef]
- Xu, M.L.; Melchinger, A.E.; Lübberstedt, T. Species-specific detection of the maize pathogens Sporisorium reiliana and Ustilago maydis by dot blot hybridization and PCR-based assays. Plant Dis. 1999, 83, 390–395. [Google Scholar] [CrossRef]
- Qi, F.; Zhang, L.; Dong, X.; Di, H.; Zhang, J.; Yao, M.; Dong, L.; Zeng, X.; Liu, X.; Wang, Z.; et al. Analysis of cytology and expression of resistance genes in maize infected with Sporisorium Reilianum. Plant Dis. 2019, 103, 2100–2107. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, P.; Li, N.; Di, H.; Wang, Y.; Liu, X.; Zhang, L.; Yu, T. Indoors infecting factors optimization of maize head smut under seedling stage. J. Northeast Agric. Univ. 2015, 46, 1–5. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Huangzao4×Jing7 F2 (Numbers) | Jing7×Chang7-2 F2 (Numbers) |
|---|---|---|
| A | 265 | - |
| B | 234 | 146 |
| C | - | 183 |
| D | 890 | 263 |
| Inbred Lines | Types of Tassel Symptoms | Blood and Pedigree | Average Incidence | Tassel Symptoms |
|---|---|---|---|---|
| Huangzao4 | A | Sipingtou group, widely used in China | 92.31% | Exhibits severe carbonization of tassels and shows black filament with many spores |
| Jing7 | B | Sipingtou group, selfing of Jingzao7 (Huangzao4×luoxi3) | 89.58% | Tassels expand without spores |
| Chang7-2 | C | Sipingtou group, single-cross hybrid Changdan7 (Huangzao4×Wei95) | 73.81% | Base of its tassel is afflicted, whereas the upper part develops normally |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Yao, M.; Wang, Q.; Zhang, X.; Di, H.; Zhang, L.; Dong, L.; Xu, Q.; Liu, X.; Zeng, X.; et al. Analysis of QTLs and Candidate Genes for Tassel Symptoms in Maize Infected with Sporisorium reilianum. Int. J. Mol. Sci. 2022, 23, 14416. https://doi.org/10.3390/ijms232214416
Zhou Y, Yao M, Wang Q, Zhang X, Di H, Zhang L, Dong L, Xu Q, Liu X, Zeng X, et al. Analysis of QTLs and Candidate Genes for Tassel Symptoms in Maize Infected with Sporisorium reilianum. International Journal of Molecular Sciences. 2022; 23(22):14416. https://doi.org/10.3390/ijms232214416
Chicago/Turabian StyleZhou, Yu, Minhao Yao, Qian Wang, Xiaoming Zhang, Hong Di, Lin Zhang, Ling Dong, Qingyu Xu, Xianjun Liu, Xing Zeng, and et al. 2022. "Analysis of QTLs and Candidate Genes for Tassel Symptoms in Maize Infected with Sporisorium reilianum" International Journal of Molecular Sciences 23, no. 22: 14416. https://doi.org/10.3390/ijms232214416