
Duplex DNA Retains the Conformational Features of Single Strands: Perspectives from MD Simulations and Quantum Chemical Computations



Abstract
:1. Introduction
2. Results and Discussion
2.1. MD Simulations
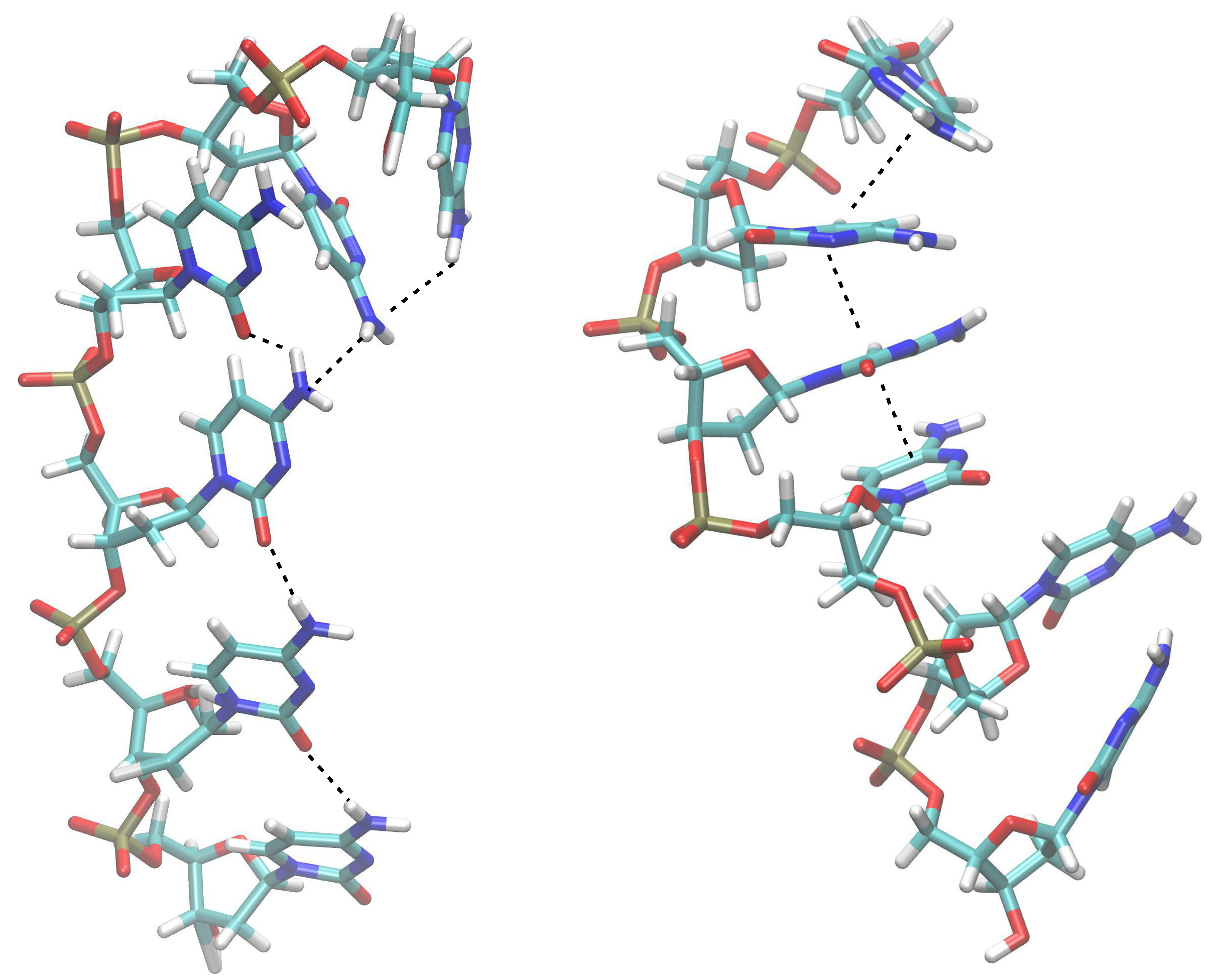
2.2. Results from DFT Geometry Optimizations
2.3. ONIOM Results
2.4. Comparison with Experimental Data
3. Methods and Materials
3.1. Starting Structures and DFT Geometry Optimizations
3.2. MD Simulation of ss-C
3.3. QM/MM ONIOM Computations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bloomfield, A.A.; Crothers, D.M.; Tinoco, J.J. Nucleic Acids: Structures, Properties, and Functions; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]
- Croy, J.E.; Wuttke, D.S. Themes in ssDNA Recognition by Telomere-End Protection Proteins. Trends Biochem. Sci. 2006, 31, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Biagini, P.; Lefeuvre, P.; Golden, M.; Roumagnac, P.; Varsani, A. Recombination in Eukaryotic Single Stranded DNA Viruses. Viruses 2011, 3, 1699–1738. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Transcription as a Source of Genome Instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Rothemund, P. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, N.C. Nanomaterials Based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.N.; Kilchherr, F.; Dietz, H.; Bathe, M. Quantitative Prediction of 3D Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2011, 40, 2862–2868. [Google Scholar] [CrossRef]
- Plumridge, A.; Meisburger, S.P.; Andresen, K.; Pollack, L. The Impact of Base Stacking on the Conformations and Electrostatics of Single-Stranded DNA. Nucleic Acids Res. 2017, 45, 3932–3943. [Google Scholar] [CrossRef]
- Huppert, J.L. Four-Stranded Nucleic Acids: Structure, Function and Targeting of G-Quadruplexes. Chem. Soc. Rev. 2008, 37, 1375–1384. [Google Scholar] [CrossRef]
- McIntosh, D.; Duggan, G.; Gouil, Q.; Saleh, O. Sequence-Dependent Elasticity and Electrostatics of Single-Stranded DNA: Signatures of Base-Stacking. Biophys. J. 2014, 106, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Ke, C.; Humeniuk, M.; S-Gracz, H.; Marszalek, P.E. Direct Measurements of Base Stacking Interactions in DNA by Single-Molecule Atomic-Force Spectroscopy. Phys. Rev. Lett. 2007, 99, 018302. [Google Scholar]
- Ramprakash, J.; Lang, B.; Schwarz, F.P. Thermodynamics of Single Strand DNA Base Stacking. Biopolymers 2008, 89, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Velardo, A.; Peluso, A. Single-Stranded DNA Oligonucleotides Retain Rise Coordinates Characteristic of Double Helices. J. Phys. Chem. B 2018, 122, 7978–7989. [Google Scholar] [CrossRef]
- Zubatiuk, T.A.; Shishkin, O.V.; Gorb, L.; Hovorun, D.M.; Leszczynski, J. B-DNA Characteristics Are Preserved in Double stranded d(A)3·d(T)3 and d(G)3·d(C)3 Mini-Helixes: Conclusions from DFT/M06-2X Study. Phys. Chem. Chem. Phys. 2013, 15, 18155–18166. [Google Scholar] [CrossRef]
- Šponer, J.; Bussi, G.; Krepl, M.; Banáš, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, H.; Mladek, A.; Gkionis, K.; Hansen, A.; Grimme, S.; Šponer, J. Quantum Chemical Benchmark Study on 46 RNA Backbone Families Using a Dinucleotide Unit. J. Chem. Theory Comput. 2015, 11, 2615–2629. [Google Scholar] [CrossRef]
- Pérez, A.; Marchán, I.; Svozil, D.; Šponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbovic, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E., III. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Liebl, K.; Zacharias, M. Tumuc1: A New Accurate DNA Force Field Consistent with High-Level Quantum Chemistry. J. Chem. Theory Comput. 2021, 17, 7096–7105. [Google Scholar] [CrossRef]
- Tucker, M.R.; Piana, S.; Tan, D.; LeVine, M.V.; Shaw, D.E. Development of Force Field Parameters for the Simulation of Single- and Double-Stranded DNA Molecules and DNA-Protein Complexes. J. Phys. Chem. B 2022, 126, 4442–4457. [Google Scholar] [CrossRef]
- Chakraborty, K.; Mantha, S.R.; Bandyopadhyay, S. Molecular Dynamics Simulation of a Single-Stranded DNA with Heterogeneous Distribution of Nucleobases in Aqueous Medium. J. Chem. Phys. 2013, 139, 075103. [Google Scholar] [CrossRef] [PubMed]
- Norberg, J.; Nilsson, L. Potential of Mean Force Calculations of the Stacking-Unstacking Process in Single Stranded Deoxyribonucleoside Monophosphates. Biophys. J. 1995, 69, 2277–2285. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Nilsson, L. MD Simulations of Homomorphous PNA, DNA, and RNA Single Strands: Characterization and Comparison of Conformations and Dynamics. J. Am. Chem. Soc. 2001, 123, 7414–7422. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- Jafilan, S.; Klein, L.; Hyun, C.; Florián, J. Intramolecular Base Stacking of Dinucleoside Monophosphate Anions in Aqueous Solution. J. Phys. Chem. B 2012, 116, 3613–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.F.; Andrews, C.T.; Elcock, A.H. Stacking Free Energies of All DNA and RNA Nucleoside Pairs and Dinucleoside-Monophosphates Computed Using Recently Revised AMBER Parameters and Compared with Experiment. J. Chem. Theory Comput. 2015, 11, 2315–2328. [Google Scholar] [CrossRef] [Green Version]
- Hayatshahi, H.S.; Henriksen, N.M.; Cheatham, T.E. Consensus Conformations of Dinucleoside Monophosphates Described with Well-Converged Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 1456–1470. [Google Scholar] [CrossRef]
- Kruse, H.; Havrila, M.; Šponer, J. QM Computations on Complete Nucleic Acids Building Blocks: Analysis of the Sarcin-Ricin RNA Motif Using DFT-D3, HF-3c, PM6-D3H, and MM Approaches. J. Chem. Theory Comput. 2014, 10, 2615–2629. [Google Scholar] [CrossRef]
- Sponer, J.; Riley, K.E.; Hobza, P. Nature and Magnitude of Aromatic Stacking of Nucleic Acid Bases. Phys. Chem. Chem. Phys. 2008, 10, 2595–2610. [Google Scholar] [CrossRef]
- Zubatiuk, T.; Kukuev, M.A.; Korolyova, A.S.; Gorb, L.; Nyporko, A.; Hovorun, D.; Leszczynski, J. Structure and Binding Energy of Double-Stranded A-DNA Mini-helices: Quantum-Chemical Study. J. Phys. Chem. B 2015, 119, 12741–12749. [Google Scholar] [CrossRef]
- Barone, G.; Fonseca Guerra, C.; Bickelhaupt, F.M. B-DNA Structure and Stability as Function of Nucleic Acid Composition: Dispersion-Corrected DFT Study of Dinucleoside Monophosphate Single and Double Strands. ChemistryOpen 2013, 2, 186–193. [Google Scholar] [CrossRef]
- Šponer, J.; Mladek, A.; Špackova, N.; Cheatham, T.E.; Grimme, S. Relative Stability of Different DNA Guanine Quadruplex Stem Topologies Derived Using Large-Scale Quantum-Chemical Computations. J. Am. Chem. Soc. 2013, 135, 9785–9796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, C.D.M.; Wetmore, S.D. Developing a Computational Model that Accurately Reproduces the Structural Features of a Dinucleoside Monophosphate Unit within B-DNA. Phys. Chem. Chem. Phys. 2011, 13, 16373–16383. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Caruso, T.; D’Ursi, A.M.; Fusco, S.; Masi, A.; Scrima, M.; Chatgilialoglu, C.; Peluso, A. Delocalized Hole Domains in Guanine-Rich DNA Oligonucleotides. J. Phys. Chem. B 2015, 119, 5462–5466. [Google Scholar] [CrossRef] [PubMed]
- Calladine, C.R.; Drew, H.R.; Luisi, B.F.; Travers, A.A. Understanding DNA, 3rd ed.; Elsevier Academic Press: Oxford, UK, 2004; Chapter 3. [Google Scholar]
- Dickerson, R.E. Nucleic Acids. In International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules; Rossmann, R.G., Arnold, E., Eds.; Kluwer Academic Press: Dordrecht, The Netherlands, 2001; pp. 588–622. [Google Scholar]
- Isaksson, J.; Acharya, S.; Barman, J.; Cheruku, P.; Chattopadhyaya, J. Single-Stranded Adenine-Rich DNA and RNA Retain Structural Characteristics of Their Respective Double-Stranded Conformations and Show Directional Differences in Stacking Pattern. Biochemistry 2004, 43, 15996–16010. [Google Scholar] [CrossRef]
- Mills, J.B.; Vacano, E.; Hagerman, P.J. Flexibility of Single-Stranded DNA: Use of Gapped Duplex Helices to Determine the Persistence Lengths of Poly(dT) and Poly(dA). J. Mol. Biol. 1999, 285, 245–257. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Celentano, M.; D’Ursi, A.M.; Scrima, M.; Peluso, A. Stacking Interactions between Adenines in Oxidized Oligonucleotides. J. Phys. Chem. B 2013, 117, 8947–8953. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [Green Version]
- Heddi, B.; Oguey, C.; Lavelle, C.; Foloppe, N.; Hartmann, B. Intrinsic Flexibility of B-DNA: The Experimental TRX Scale. Nucleic Acids Res. 2010, 38, 1034–1047. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, J.D.; Condon, D.E.; Kennedy, S.D.; Hauser, M.; Bevilacqua, P.C.; Turner, D.H. The Nuclear Magnetic Resonance of CCCC RNA Reveals a Right-Handed Helix, and Revised Parameters for AMBER Force Field Torsions Improve Structural Predictions from Molecular Dynamics. Biochemistry 2013, 52, 996–1010. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Lewis, F.D.; Berlin, Y.A.; Ratner, M.A.; Siebbeles, L.D.A. Absolute Rates of Hole Transfer in DNA. J. Am. Chem. Soc. 2005, 127, 14894–14903. [Google Scholar] [CrossRef]
- Sugiyama, H.; Saito, I. Theoretical Studies of GG-Specific Photocleavage of DNA via Electron Transfer: Significant Lowering of Ionization Potential and 5′-Localization of HOMO of Stacked GG Bases in B-form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar] [CrossRef]
- Capobianco, A.; Landi, A.; Peluso, A. Modeling DNA Oxidation in Water. Phys. Chem. Chem. Phys. 2017, 19, 13571–13578. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, R.; Capobianco, A.; Landi, A.; Peluso, A. Vibronic Couplings and Coherent Electron Transfer in Bridged Systems. Phys. Chem. Chem. Phys. 2015, 17, 30937–30945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubař, T.; Woiczikowski, P.B.; Cuniberti, G.; Elstner, M. Efficient Calculation of Charge-Transfer Matrix Elements for Hole Transfer in DNA. J. Phys. Chem. B 2008, 112, 7937–7947. [Google Scholar] [CrossRef]
- Landi, A.; Borrelli, R.; Capobianco, A.; Peluso, A. Transient and Enduring Electronic Resonances Drive Coherent Long Distance Charge Transport in Molecular Wires. J. Phys. Chem. Lett. 2019, 10, 1845–1851. [Google Scholar] [CrossRef]
- Landi, A.; Capobianco, A.; Peluso, A. Coherent Effects in Charge Transport in Molecular Wires: Toward a Unifying Picture of Long-Range Hole Transfer in DNA. J. Phys. Chem. Lett. 2020, 11, 7769–7775. [Google Scholar] [CrossRef]
- Landi, A.; Capobianco, A.; Peluso, A. The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study. Molecules 2021, 26, 5497. [Google Scholar] [CrossRef]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef] [Green Version]
- Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528. [Google Scholar] [CrossRef]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Kanvah, S.; Joseph, J.; Schuster, G.B.; Barnett, R.N.; Cleveland, C.L.; Landman, U. Oxidation of DNA: Damage to Nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Fu, Y.; Wang, D.; Wei, Y.; Kobayashi, N.; Minari, T. DNA as Functional Material in Organic-Based Electronics. Appl. Sci. 2018, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Etheve, L.; Martin, J.; Lavery, R. Decomposing Protein-DNA Binding and Recognition Using simplified Protein Models. Nucleic Acids Res. 2017, 45, 10270–10283. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.A.; Holroyd, L.F.; van Mourik, T.; Jones, A.C. A DFT Study of 2-Aminopurine-Containing Dinucleotides: Prediction of Stacked Conformations with B-DNA Structure. Phys. Chem. Chem. Phys. 2016, 18, 14691–14700. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.S.; van Mourik, T. DNA Base Stacking: The Stacked Uracil/Uracil and Thymine/Thymine Minima. J. Comput. Chem. 2012, 33, 2161–2172. [Google Scholar] [CrossRef]
- Kannan, S.; Kohlhoff, K.; Zacharias, M. B-DNA Under Stress: Over- and Untwisting of DNA during Molecular Dynamics Simulations. Biophys. J. 2006, 91, 2956–2965. [Google Scholar] [CrossRef] [Green Version]
- El Hassan, M.A.; Calladine, C.R. Conformational Characteristics of DNA: Empirical Classifications and a Hypothesis for the Conformational Behaviour of Dinucleotide Steps. Philos. Trans. R. Soc. A 1997, 355, 43–100. [Google Scholar] [CrossRef]
- Stellwagen, E.; Stellwagen, N.C. Electrophoretic Mobility of DNA in Solutions of High Ionic Strength. Byophys. J. 2020, 118, 2783. [Google Scholar] [CrossRef]
- Chen, H.; Meisburger, S.P.; Pabit, S.A.; Sutton, J.L.; Webb, W.W.; Pollack, L. Ionic Strength-Dependent Persistence Lengths of Single-Stranded RNA and DNA. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Peluso, A. The Oxidization Potential of AA Steps in Single Strand DNA Oligomers. RSC Adv. 2014, 4, 47887–47893. [Google Scholar] [CrossRef]
- Capobianco, A.; Caruso, T.; Peluso, A. Hole Delocalization over Adenine Tracts in Single Stranded DNA Oligonucleotides. Phys. Chem. Chem. Phys. 2015, 17, 4750–4756. [Google Scholar] [CrossRef] [PubMed]
- González-Olvera, J.C.; Zamorano-Carrillo, A.; Arreola-Jardón, G.; Pless, R.C. Residue Interactions Affecting the Deprotonation of Internal Guanine Moieties in Oligodeoxyribonucleotides, Calculated by FMO Methods. J. Mol. Model. 2022, 28, 43. [Google Scholar] [CrossRef]
- Han, X.; Wang, E.; Cui, Y.; Lin, Y.; Chen, H.; An, R.; Liang, X.; Komiyama, M. The Staining Efficiency of Cyanine Dyes for Single-Stranded DNA is Enormously Dependent on Nucleotide Composition. Electrophoresis 2019, 40, 1708–1714. [Google Scholar] [CrossRef]
- Norberg, J.; Nilsson, L. Influence of Adjacent Bases on the Stacking-Unstacking Process of Single-stranded Oligonucleotides. Biopolymers 1996, 39, 765–768. [Google Scholar] [CrossRef]
- Erie, D.A.; Breslauer, K.J.; Olson, W.K. A Monte Carlo Method for Generating Structures of Short Single-Stranded DNA Sequences. Biopolymers 1993, 33, 75–105. [Google Scholar] [CrossRef]
- Vokáčová, Z.; Buděšínský, M.; Rosenberg, I.; Schneider, B.; Šponer, J.; Sychrovský, V. Structure and Dynamics of the ApA, ApC, CpA, and CpC RNA Dinucleoside Monophosphates Resolved with NMR Scalar Spin-Spin Couplings. J. Phys. Chem. B 2009, 113, 1182–1191. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Kim, S.H. Conformational Studies of Nucleic Acids. V. Sequence Specificities in the Conformational Energetics of Oligonucleotides: The Homo-Tetramers. Biopolymers 1988, 27, 59–77. [Google Scholar] [CrossRef]
- Kabeláč, M.; Hobza, P. Potential Energy and Free Energy Surfaces of all Ten Canonical and Methylated Nucleic Acid Base Pairs: Molecular Dynamics and Quantum Chemical ab initio Studies. J. Phys. Chem. B 2001, 105, 5804–5817. [Google Scholar] [CrossRef]
- Kypr, J.; Kejnovská, I.; Renčiuk, D.; Vorlíčková, M. Circular Dichroism and Conformational Polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.S.; Lamm, G.; Pack, G.R. The Triplex-Hairpin Transition in Cytosine-Rich DNA. Biophys. J. 2004, 87, 3954–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnott, S.; Chandrasekaran, R.; Leslie, A.G.W. Structure of the Single-Stranded Polyribonucleotide Polycytidylic Acid. J. Mol. Biol. 1976, 106, 735–748. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisné, C.; Hantz, E.; Hartmann, B.; Delepierre, M. Solution Structure of a Non-Palindromic 16 Base-Pair DNA Related to the HIV-1 κb site: Evidence for BI-BII Equilibrium Inducing a Global Dynamic Curvature of the Duplex. J. Mol. Biol. 1998, 279, 127–142. [Google Scholar] [CrossRef]
- DiGabriele, A.D.; Steitz, T.A. A DNA Dodecamer Containing an Adenine Tract Crystallizes in a Unique Lattice and Exhibits a New Bend. J. Mol. Biol. 1993, 231, 1024–1039. [Google Scholar] [CrossRef]
- MacDonald, D.; Herbert, K.; Zhang, X.; Polgruto, T.; Lu, P. Solution Structure of an A-Tract DNA Bend. J. Mol. Biol. 2001, 306, 1081–1098. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A Software Package for the Analysis, Rebuilding and Visualization of Three-Dimensional Nucleic Acid Structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.J.; Olson, W.K. 3DNA: A Versatile, Integrated Software System for the Analysis, Rebuilding and Visualization of Three-dimensional Nucleic-Acid Structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Singh, A.; Singh, N. Effect of Salt Concentration on the Stability of Heterogeneous DNA. Physica A 2015, 419, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Schreck, J.S.; Šulc, P. Understanding DNA Interactions in Crowded Environments with a Coarse-Grained Model. Nucleic Acids Res. 2020, 48, 10726–10738. [Google Scholar] [CrossRef]
- World, T.; Fund, W.; Archipelago, S. Spartan’04; Wavefunction, Inc.: Irvine, CA, USA, 2007. [Google Scholar]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham III, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Price, D.J.; Brooks, C.L. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Toukmaji, A.; Sagui, C.; Board, J.; Darden, T. Efficient Particle-Mesh Ewald Based Approach to Fixed and Induced Dipolar Interactions. J. Chem. Phys. 2000, 113, 10913–10927. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Clemente, F.R.; Vreven, T.; Frisch, M.J. Getting the Most out of ONIOM: Guidelines and Pitfalls. In Quantum Biochemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; Chapter 2; pp. 61–83. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3LYP-D3 | M06-2X | |||||||
|---|---|---|---|---|---|---|---|---|
| 6-31G(d,p) | TZVP | 6-31G(d,p) | TZVP | |||||
| Rise | h-Rise | Rise | h-Rise | Rise | h-Rise | Rise | h-Rise | |
| A | 3.14 | 3.25 | 3.30 | 3.34 | 2.96 | 3.09 | 3.18 | 3.15 |
| C | 2.74 | 2.56 | 3.24 | 2.94 | 2.53 | 2.20 | 3.42 | 3.31 |
| G | 3.05 | 3.12 | 3.14 | 3.21 | 2.94 | 3.03 | 3.02 | 3.09 |
| T | 2.55 | 2.34 | 2.66 | 2.43 | 2.33 | 2.00 | 2.40 | 1.93 |
| ACA | 2.92 | 3.02 | 3.12 | 3.08 | 2.75 | 2.73 | 3.18 | 3.15 |
| ds-G | 3.43 | 3.25 | 3.56 | 3.33 | 3.25 | 3.09 | 3.40 | 3.19 |
| ds-A | 3.25 | 3.03 | 3.26 | 2.95 | 2.98 | 2.96 | 3.12 | 2.81 |
| B3LYP-D3 | M06-2X | |||||||
|---|---|---|---|---|---|---|---|---|
| 6-31G(d,p) | TZVP | 6-31G(d,p) | TZVP | |||||
| Twist | h-Twist | Twist | h-Twist | Twist | h-Twist | Twist | h-Twist | |
| ss-A | 37.51 | 38.23 | 43.21 | 44.25 | 37.21 | 38.53 | 43.90 | 44.17 |
| ss-C | 33.12 | 35.11 | 34.53 | 36.23 | 33.26 | 36.54 | 38.69 | 39.51 |
| ss-G | 44.04 | 45.32 | 45.85 | 46.77 | 44.26 | 45.03 | 47.21 | 47.68 |
| ss-T | 33.24 | 36.36 | 32.01 | 34.50 | 33.62 | 38.26 | 31.60 | 37.29 |
| ss-ACA | 36.23 | 37.66 | 39.39 | 39.95 | 36.91 | 38.58 | 42.27 | 42.78 |
| ds-G | 41.74 | 43.18 | 45.76 | 46.66 | 44.42 | 45.57 | 45.95 | 47.12 |
| ds-A | 43.56 | 44.37 | 46.48 | 47.39 | 40.22 | 40.49 | 46.56 | 47.70 |
| ME | EE | ||||
|---|---|---|---|---|---|
| 6-31G(d,p) | TZVP | 6-31G(d,p) | TZVP | ||
| Rise | |||||
| B3LYP-D3 | 2.49 | 2.75 | 3.10 | 3.17 | |
| M06-2X | 2.41 | 2.77 | 3.03 | 3.10 | |
| Twist | |||||
| B3LYP-D3 | 29.50 | 32.60 | 36.84 | 36.54 | |
| M06-2X | 30.22 | 33.02 | 37.19 | 38.22 | |
| ME | EE | |
|---|---|---|
| B3LYP-D3/6-31G(d,p) | 0.74 | 0.59 |
| B3LYP-D3/TZVP | 0.66 | 0.61 |
| M06-2X/6-31G(d,p) | 0.82 | 0.59 |
| M06-2X/TZVP | 0.70 | 0.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capobianco, A.; Landi, A.; Peluso, A. Duplex DNA Retains the Conformational Features of Single Strands: Perspectives from MD Simulations and Quantum Chemical Computations. Int. J. Mol. Sci. 2022, 23, 14452. https://doi.org/10.3390/ijms232214452
Capobianco A, Landi A, Peluso A. Duplex DNA Retains the Conformational Features of Single Strands: Perspectives from MD Simulations and Quantum Chemical Computations. International Journal of Molecular Sciences. 2022; 23(22):14452. https://doi.org/10.3390/ijms232214452
Chicago/Turabian StyleCapobianco, Amedeo, Alessandro Landi, and Andrea Peluso. 2022. "Duplex DNA Retains the Conformational Features of Single Strands: Perspectives from MD Simulations and Quantum Chemical Computations" International Journal of Molecular Sciences 23, no. 22: 14452. https://doi.org/10.3390/ijms232214452
APA StyleCapobianco, A., Landi, A., & Peluso, A. (2022). Duplex DNA Retains the Conformational Features of Single Strands: Perspectives from MD Simulations and Quantum Chemical Computations. International Journal of Molecular Sciences, 23(22), 14452. https://doi.org/10.3390/ijms232214452