
DNA Sequence-Dependent Properties of Nucleosome Positioning in Regions of Distinct Chromatin States in Mouse Embryonic Stem Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
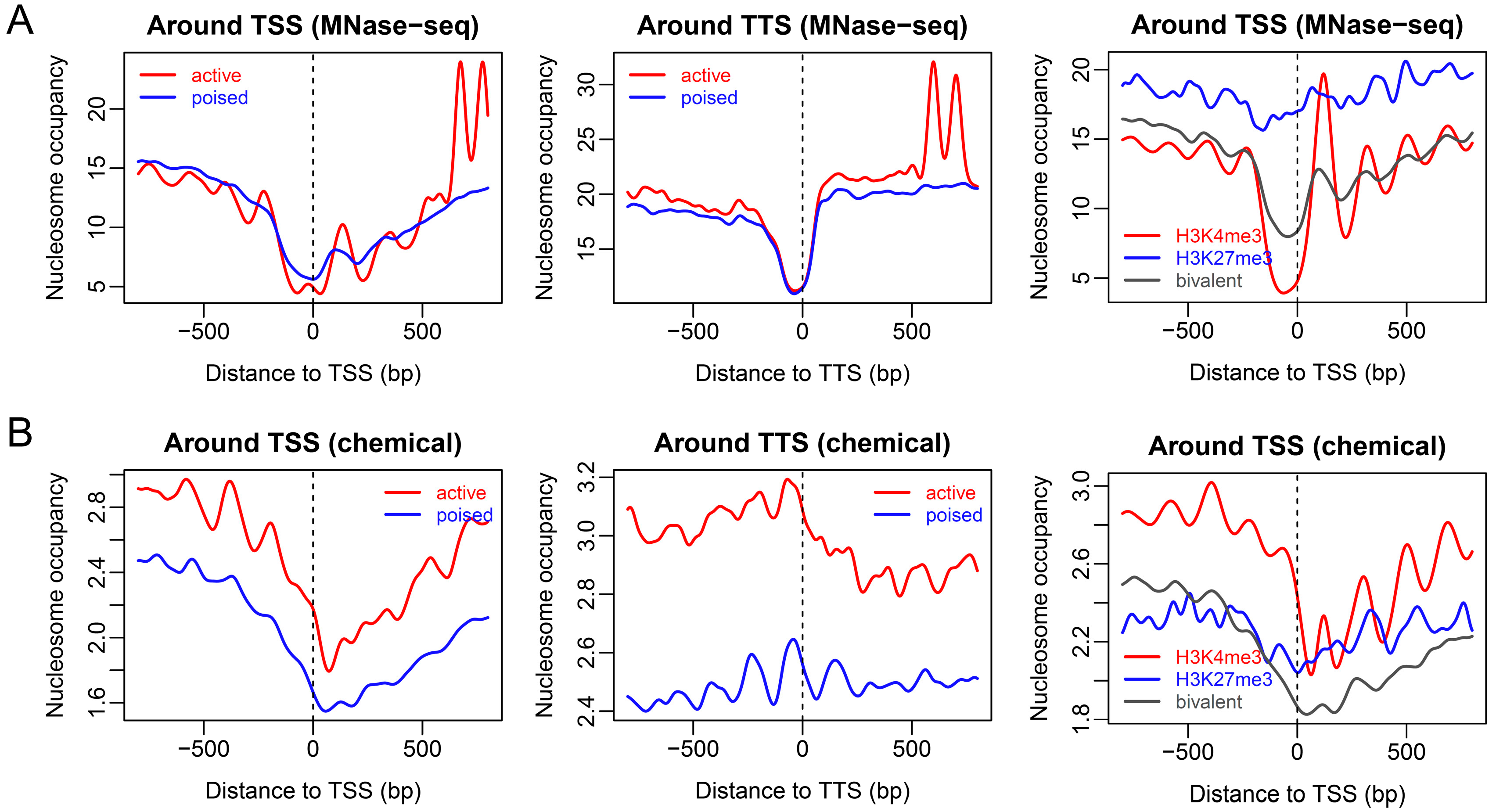
2.1. Nucleosome Organization around Promoters with Distinct Chromatin States in ESCs
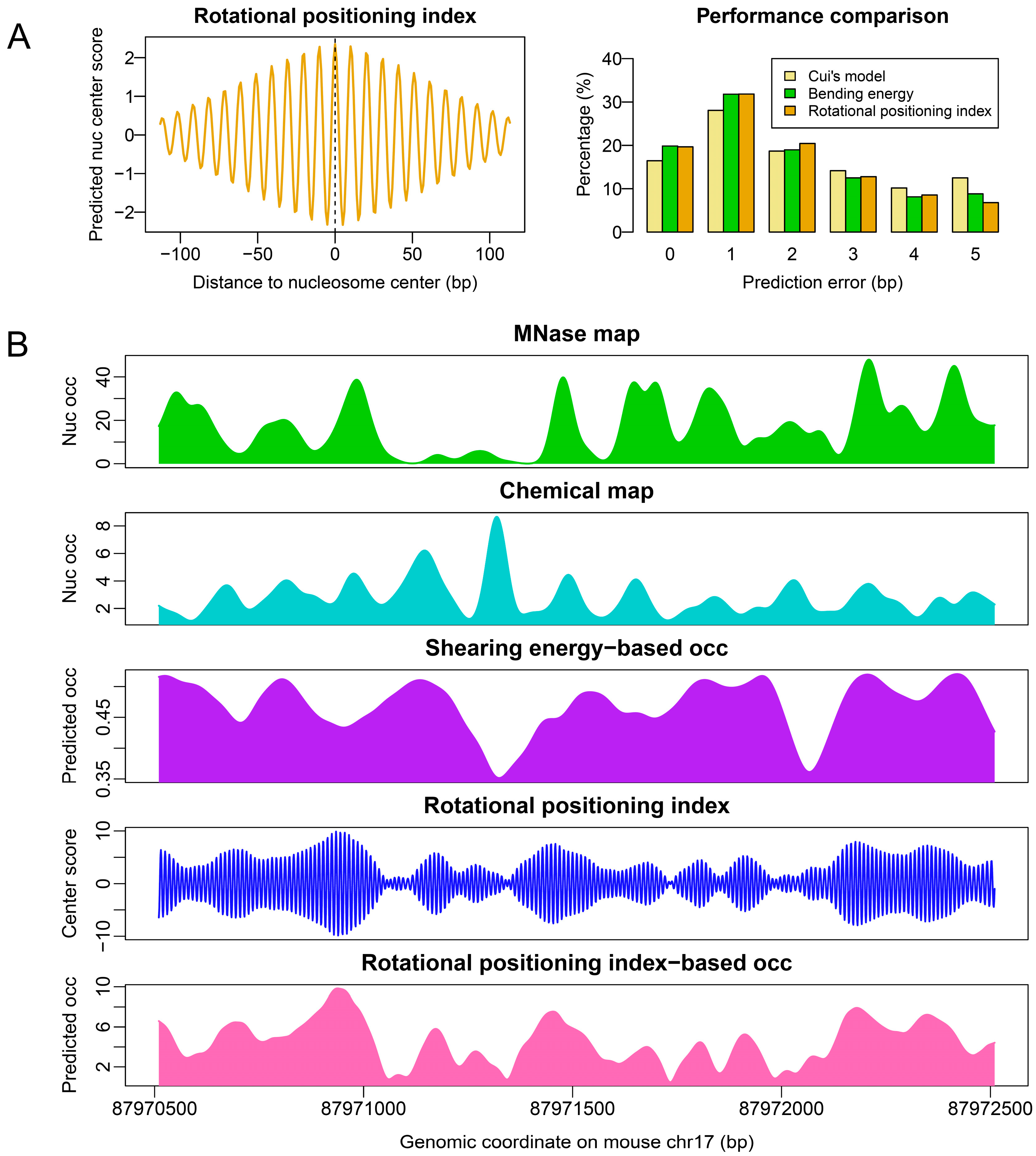
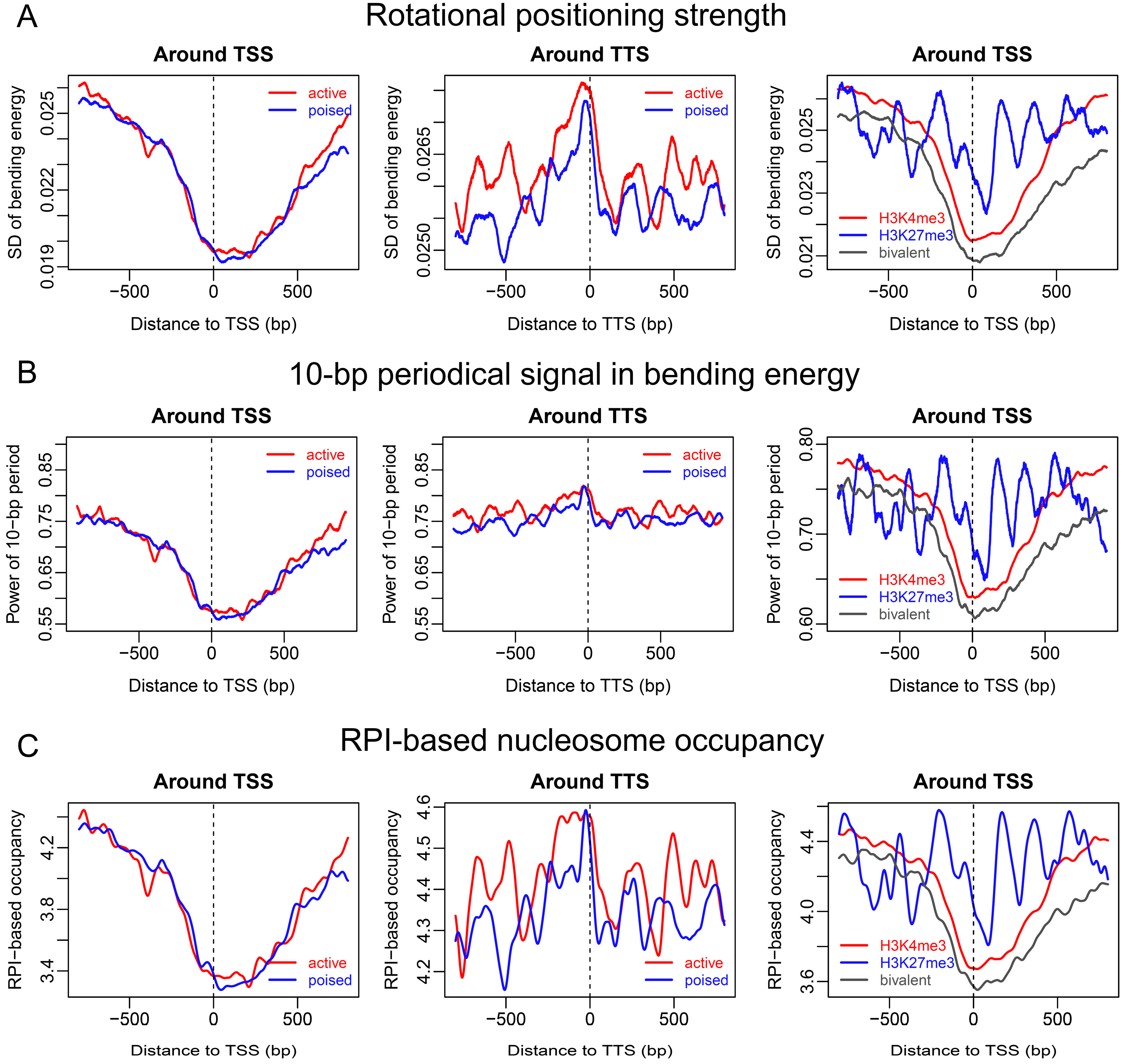
2.2. Sequence-Dependent Nucleosome Positioning around Promoters with Distinct Chromatin States in ESCs
2.3. Nucleosome Organization around Promoters with Different CpG Composition in ESCs
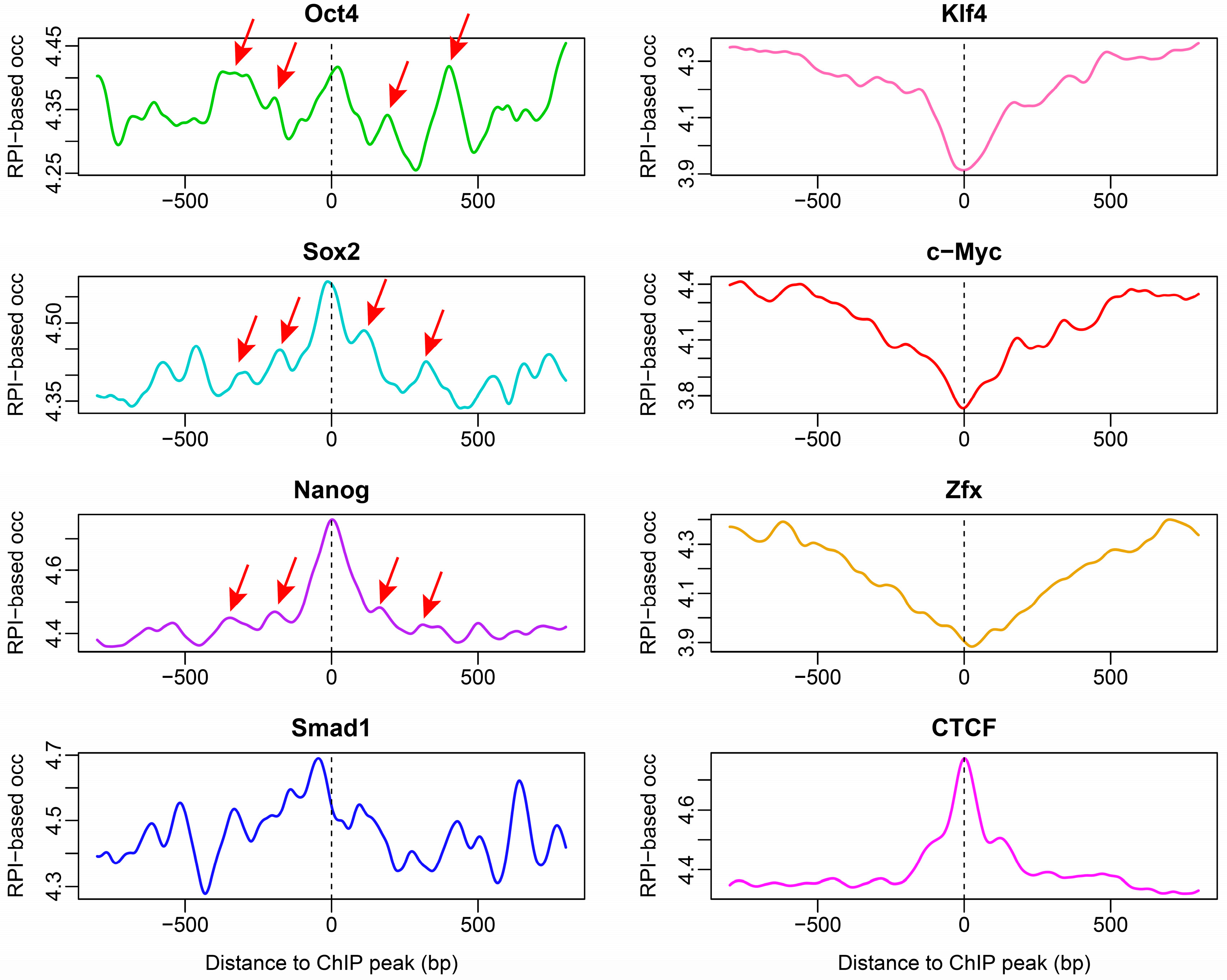
2.4. Nucleosome Organization around Transcription Factor-Binding Sites in ESCs
2.5. Conclusions
3. Materials and Methods
3.1. Genes with Distinct Chromatin States
3.2. Experimental Nucleosome Maps of Mouse ESCs
3.3. DNA Deformation-Energy-Based Descriptors
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ekundayo, B.; Richmond, T.J.; Schalch, T. Capturing structural heterogeneity in chromatin fibers. J. Mol. Biol. 2017, 429, 3031–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoryev, S.A. Chromatin Higher-Order Folding: A Perspective with Linker DNA Angles. Biophys. J. 2018, 114, 2290–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collepardo-Guevara, R.; Schlick, T. Chromatin fiber polymorphism triggered by variations of DNA linker lengths. Proc. Natl. Acad. Sci. USA 2014, 111, 8061–8066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcginty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [Green Version]
- Willcockson, M.A.; Healton, S.E.; Weiss, C.N.; Bartholdy, B.A.; Botbol, Y.; Mishra, L.N.; Sidhwani, D.S.; Wilson, T.J.; Pinto, H.B.; Maron, M.I.; et al. H1 histones control the epigenetic landscape by local chromatin compaction. Nature 2021, 589, 293–298. [Google Scholar] [CrossRef]
- Ohno, M.; Ando, T.; Priest, D.G.; Taniguchi, Y. Hi-CO: 3D genome structure analysis with nucleosome resolution. Nat. Protoc. 2021, 16, 3439–3469. [Google Scholar] [CrossRef]
- Ohno, M.; Priest, D.G.; Taniguchi, Y. Nucleosome-level 3D organization of the genome. Biochem. Soc. Trans. 2018, 46, 491–501. [Google Scholar] [CrossRef]
- Hsieh, T.H.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Core, L.; Adelman, K. Promoter-proximal pausing of RNA polymerase II: A nexus of gene regulation. Genes Dev. 2019, 33, 960–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voong, L.N.; Xi, L.; Sebeson, A.C.; Xiong, B.; Wang, J.P.; Wang, X. Insights into nucleosome organization in mouse embryonic stem cells through chemical mapping. Cell 2016, 167, 1555–1570. [Google Scholar] [CrossRef]
- Dingwall, C.; Lomonossoff, G.P.; Laskey, R.A. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981, 9, 2659–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.R.; Dunkel, I.; Heise, F.; Linke, C.; Krobitsch, S.; Ehrenhofer-Murray, A.E.; Sperling, S.R.; Vingron, M. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS ONE 2010, 5, e15754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mieczkowski, J.; Cook, A.; Bowman, S.K.; Mueller, B.; Alver, B.H.; Kundu, S.; Deaton, A.M.; Urban, J.A.; Larschan, E.; Park, P.J.; et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016, 7, 11485. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Ramachandran, S.; Bryson, T.D.; Henikoff, S. Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol. 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogaard, K.; Xi, L.; Wang, J.P.; Widom, J. A map of nucleosome positions in yeast at base-pair resolution. Nature 2012, 486, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thåström, A.; Field, Y.; Moore, I.K.; Wang, J.P.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Cui, F.; Chen, L.; LoVerso, P.R.; Zhurkin, V.B. Prediction of nucleosome rotational positioning in yeast and human genomes based on sequence-dependent DNA anisotropy. BMC Bioinform. 2014, 15, 313. [Google Scholar] [CrossRef] [Green Version]
- Tolstorukov, M.Y.; Colasanti, A.V.; McCandlish, D.M.; Olson, W.K.; Zhurkin, V.B. A novel roll-and-slide mechanism of DNA folding in chromatin: Implications for nucleosome positioning. J. Mol. Biol. 2007, 371, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Bishop, T.C. Geometry of the nucleosomal DNA superhelix. Biophys. J. 2008, 95, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Morozov, A.V.; Fortney, K.; Gaykalova, D.A.; Studitsky, V.M.; Widom, J.; Siggia, E.D. Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res. 2009, 37, 4707–4722. [Google Scholar] [CrossRef]
- Liu, G.; Xing, Y.; Zhao, H.; Wang, J.; Shang, Y.; Cai, L. A deformation energy-based model for predicting nucleosome dyads and occupancy. Sci. Rep. 2016, 6, 24133. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zhao, H.; Meng, H.; Xing, Y.; Cai, L. A deformation energy model reveals sequence-dependent property of nucleosome positioning. Chromosoma 2021, 130, 27–40. [Google Scholar] [CrossRef]
- Wang, J.Y.; Wang, J.; Liu, G. Calculation of nucleosomal DNA deformation energy: Its implication for nucleosome positioning. Chromosome Res. 2012, 20, 889–902. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, H.; Meng, H.; Xing, Y.; Yang, H.; Lin, H. Deform-nu: A DNA Deformation Energy-Based Predictor for Nucleosome Positioning. Front. Cell Dev. Biol. 2020, 8, 596341. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Teif, V.B.; Vainshtein, Y.; Caudron-Herger, M.; Mallm, J.P.; Marth, C.; Höfer, T.; Rippe, K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012, 19, 1185–1192. [Google Scholar] [CrossRef]
- Yu, J.; Xiong, C.; Zhuo, B.; Wen, Z.; Shen, J.; Liu, C.; Chang, L.; Wang, K.; Wang, M.; Wu, C.; et al. Analysis of local chromatin states reveals gene transcription potential during mouse neural progenitor cell differentiation. Cell Rep. 2020, 32, 107953. [Google Scholar] [CrossRef]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef]
- Li, K.; Carroll, M.; Vafabakhsh, R.; Wang, X.A.; Wang, J.P. DNAcycP: A deep learning tool for DNA cyclizability prediction. Nucleic Acids Res. 2022, 50, 3142–3154. [Google Scholar] [CrossRef] [PubMed]
- Dreos, R.; Ambrosini, G.; Bucher, P. Influence of Rotational Nucleosome Positioning on Transcription Start Site Selection in Animal Promoters. PLoS Comput. Biol. 2016, 12, e1005144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Farnung, L.; Kaasinen, E.; Sahu, B.; Yin, Y.; Wei, B.; Dodonova, S.O.; Nitta, K.R.; Morgunova, E.; Taipale, M.; et al. The interaction landscape between transcription factors and the nucleosome. Nature 2018, 562, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Xi, L.; Brogaard, K.; Zhang, Q.; Lindsay, B.; Widom, J.; Wang, J.P. A locally convoluted cluster model for nucleosome positioning signals in chemical map. J. Am. Stat. Assoc. 2014, 109, 48–62. [Google Scholar] [CrossRef]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.J.; Neidle, S.; Shakked, Z.; et al. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Zhang, Z.; Dong, B.; Liu, J. DNA Sequence-Dependent Properties of Nucleosome Positioning in Regions of Distinct Chromatin States in Mouse Embryonic Stem Cells. Int. J. Mol. Sci. 2022, 23, 14488. https://doi.org/10.3390/ijms232214488
Liu G, Zhang Z, Dong B, Liu J. DNA Sequence-Dependent Properties of Nucleosome Positioning in Regions of Distinct Chromatin States in Mouse Embryonic Stem Cells. International Journal of Molecular Sciences. 2022; 23(22):14488. https://doi.org/10.3390/ijms232214488
Chicago/Turabian StyleLiu, Guoqing, Zhi Zhang, Biyu Dong, and Jia Liu. 2022. "DNA Sequence-Dependent Properties of Nucleosome Positioning in Regions of Distinct Chromatin States in Mouse Embryonic Stem Cells" International Journal of Molecular Sciences 23, no. 22: 14488. https://doi.org/10.3390/ijms232214488