
Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Biology of Ketone Bodies
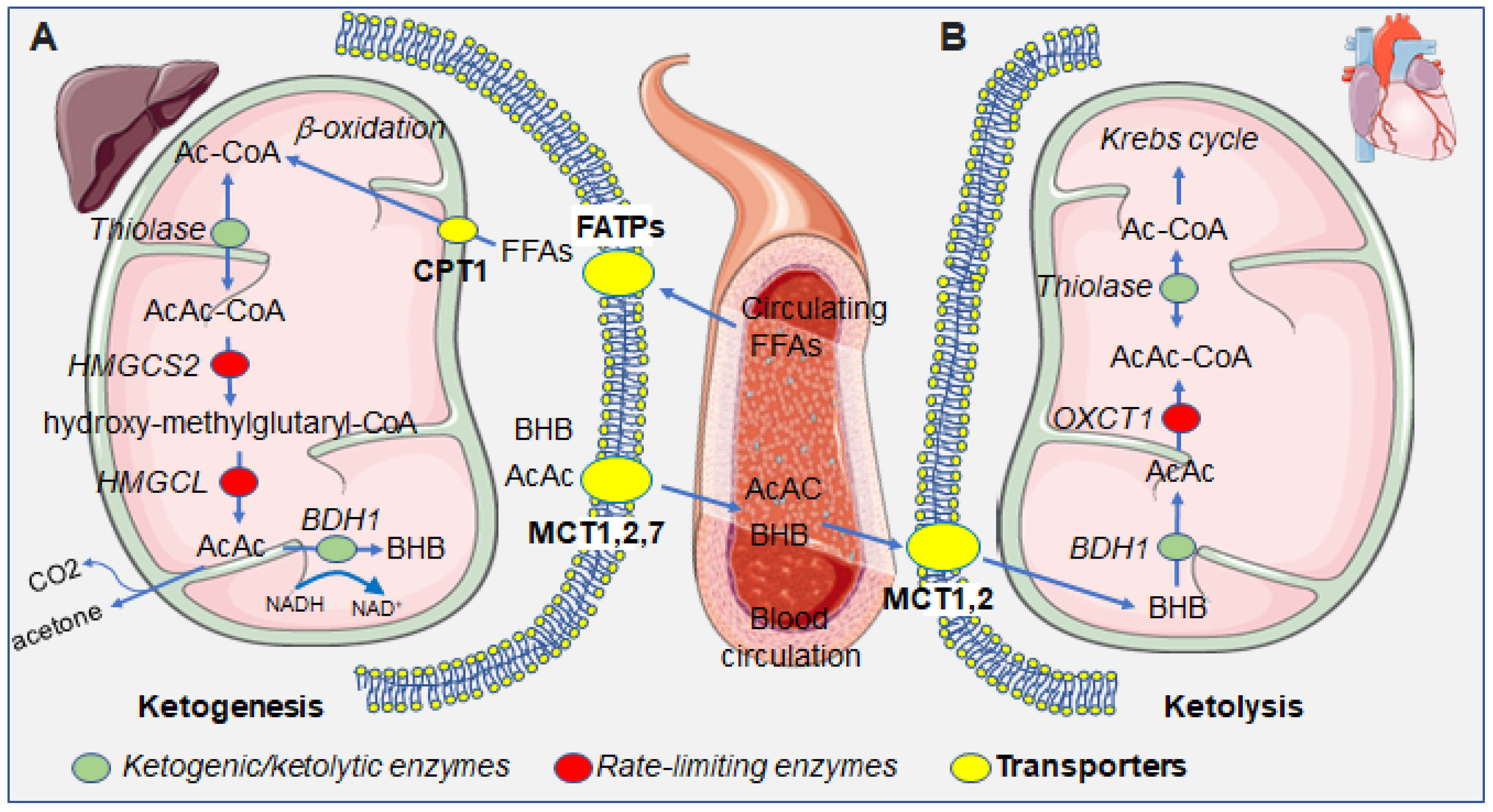
2.1. Ketogenic and Ketolytic Metabolic Pathways
2.2. Ketone Bodies as Epigenetic Modifiers
2.3. The Biochemical Basis of Histone β-Hydroxybutyrylation
2.4. BHB and Ketone Bodies as Signalling Mediators
3. Ketone Bodies as an Alternative Fuel for the Heart
3.1. Cardiovascular Disease and Endothelial Damage Can Be Alleviated by Ketone Bodies
3.2. Potential Therapeutic Actions of the Ketogenic Diet
3.3. The Role of Ketone Bodies in Inflammatory Disease
4. The Effects of a Ketogenic Diet on Inflammation-Dependent Atherosclerosis and Cardiovascular Risk
4.1. The Effects of the Ketogenic Diet on the Inflammasome
4.2. Targeting Cardiac Dysfunction with Ketone Bodies
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Foster, D.W. Studies in the Ketosis of Fasting. J. Clin. Investig. 1967, 46, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Békési, A.; Williamson, D.H. An Explanation for Ketogenesis by the Intestine of the Suckling Rat: The Presence of an Active Hydroxymethylglutaryl-Coenzyme A Pathway. Biol. Neonatol. 1990, 58, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131.e15. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Sylvester, E.; Lian, J.; Deng, C. Kidney Plays an Important Role in Ketogenesis Induced by Risperidone and Voluntary Exercise in Juvenile Female Rats. Psychiatry Res. 2021, 305, 114196. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, K.; Ma, J.; Zhou, L.; Liu, J.; Zeng, L.; Zhu, L.; Xu, P.; Chen, J.; Wei, K.; et al. Ketogenesis-Generated β-Hydroxybutyrate Is an Epigenetic Regulator of CD8+ T-Cell Memory Development. Nat. Cell Biol. 2020, 22, 18–25. [Google Scholar] [CrossRef]
- Auestad, N.; Korsak, R.A.; Morrow, J.W.; Edmond, J. Fatty Acid Oxidation and Ketogenesis by Astrocytes in Primary Culture. J. Neurochem. 1991, 56, 1376–1386. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Puchalska, P.; Martin, S.E.; Huang, X.; Lengfeld, J.E.; Daniel, B.; Graham, M.J.; Han, X.; Nagy, L.; Patti, G.J.; Crawford, P.A. Hepatocyte-Macrophage Acetoacetate Shuttle Protects against Tissue Fibrosis. Cell Metab. 2019, 29, 383–398.e7. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Crawford, M.A. Energetic and Nutritional Constraints on Infant Brain Development: Implications for Brain Expansion during Human Evolution. J. Hum. Evol. 2014, 77, 88–98. [Google Scholar] [CrossRef]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The Ketogenic Diet for the Treatment of Childhood Epilepsy: A Randomised Controlled Trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Tagliabue, A.; Ferraris, C.; Uggeri, F.; Trentani, C.; Bertoli, S.; de Giorgis, V.; Veggiotti, P.; Elli, M. Short-Term Impact of a Classical Ketogenic Diet on Gut Microbiota in GLUT1 Deficiency Syndrome: A 3-Month Prospective Observational Study. Clin. Nutr. ESPEN 2017, 17, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Dąbek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Caprio, M.; Tuccinardi, D.; Moriconi, E.; Di Renzo, L.; Muscogiuri, G.; Colao, A.; Savastano, S. Obesity Programs of nutrition, Education, Research and Assessment (OPERA) group Could Ketogenic Diet “Starve” Cancer? Emerging Evidence. Crit. Rev. Food Sci. Nutr. 2020, 62, 1800–1821. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.A.; Kassovska-Bratinova, S.; Boukaftane, Y.; Robert, M.F.; Wang, S.P.; Ashmarina, L.; Lambert, M.; Lapierre, P.; Potier, E. Medical Aspects of Ketone Body Metabolism. Clin. Invest. Med. 1995, 18, 193–216. [Google Scholar]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and Control of Ketone Body Metabolism: On the Fringe of Lipid Biochemistry. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 243–251. [Google Scholar] [CrossRef]
- Turner, B.M. Defining an Epigenetic Code. Nat. Cell Biol. 2007, 9, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.C.; Allis, C.D. Histone Methylation versus Histone Acetylation: New Insights into Epigenetic Regulation. Curr. Opin. Cell Biol. 2001, 13, 263–273. [Google Scholar] [CrossRef]
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-Methylated Heterochromatin and Its Functions in Tissue Differentiation and Maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Pan, M.-R.; Hsu, M.-C.; Chen, L.-T.; Hung, W.-C. Orchestration of H3K27 Methylation: Mechanisms and Therapeutic Implication. Cell Mol. Life Sci. 2018, 75, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Nasser, S.; Solé, T.; Vega, N.; Thomas, T.; Balcerczyk, A.; Strigini, M.; Pirola, L. Ketogenic Diet Administration to Mice after a High-Fat-Diet Regimen Promotes Weight Loss, Glycemic Normalization and Induces Adaptations of Ketogenic Pathways in Liver and Kidney. Mol. Metab. 2022, 65, 101578. [Google Scholar] [CrossRef] [PubMed]
- Yancy, W.S.; Olsen, M.K.; Guyton, J.R.; Bakst, R.P.; Westman, E.C. A Low-Carbohydrate, Ketogenic Diet versus a Low-Fat Diet to Treat Obesity and Hyperlipidemia: A Randomized, Controlled Trial. Ann. Intern. Med. 2004, 140, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Poplawski, M.M.; Mastaitis, J.W.; Isoda, F.; Grosjean, F.; Zheng, F.; Mobbs, C.V. Reversal of Diabetic Nephropathy by a Ketogenic Diet. PLoS ONE 2011, 6, e18604. [Google Scholar] [CrossRef]
- Sherrier, M.; Li, H. The Impact of Keto-Adaptation on Exercise Performance and the Role of Metabolic-Regulating Cytokines. Am. J. Clin. Nutr. 2019, 110, 562–573. [Google Scholar] [CrossRef]
- Nuttall, F.Q.; Almokayyad, R.M.; Gannon, M.C. Circulating Lipids in Men with Type 2 Diabetes Following 3 Days on a Carbohydrate-Free Diet versus 3 Days of Fasting. Physiol. Rep. 2020, 8, e14569. [Google Scholar] [CrossRef]
- Luzi, L.; Barrett, E.J.; Groop, L.C.; Ferrannini, E.; DeFronzo, R.A. Metabolic Effects of Low-Dose Insulin Therapy on Glucose Metabolism in Diabetic Ketoacidosis. Diabetes 1988, 37, 1470–1477. [Google Scholar] [CrossRef]
- Nasser, S.; Vialichka, V.; Biesiekierska, M.; Balcerczyk, A.; Pirola, L. Effects of Ketogenic Diet and Ketone Bodies on the Cardiovascular System: Concentration Matters. World J. Diabetes 2020, 11, 584–595. [Google Scholar] [CrossRef]
- Qi, H.; Gu, L.; Xu, D.; Liu, K.; Zhou, M.; Wang, Y.; Wang, X.; Li, Y.; Qi, J. β-Hydroxybutyrate Inhibits Cardiac Microvascular Collagen 4 Accumulation by Attenuating Oxidative Stress in Streptozotocin-Induced Diabetic Rats and High Glucose Treated Cells. Eur. J. Pharmacol. 2021, 899, 174012. [Google Scholar] [CrossRef]
- Kaczmarska, Z.; Ortega, E.; Goudarzi, A.; Huang, H.; Kim, S.; Márquez, J.A.; Zhao, Y.; Khochbin, S.; Panne, D. Structure of P300 in Complex with Acyl-CoA Variants. Nat. Chem. Biol. 2017, 13, 21–29. [Google Scholar] [CrossRef]
- Zhou, T.; Cheng, X.; He, Y.; Xie, Y.; Xu, F.; Xu, Y.; Huang, W. Function and Mechanism of Histone β-Hydroxybutyrylation in Health and Disease. Front. Immunol. 2022, 13, 981285. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, D.; Weng, Y.; Delaney, K.; Tang, Z.; Yan, C.; Qi, S.; Peng, C.; Cole, P.A.; Roeder, R.G.; et al. The Regulatory Enzymes and Protein Substrates for the Lysine β-Hydroxybutyrylation Pathway. Sci. Adv. 2021, 7, eabe2771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cao, R.; Niu, J.; Yang, S.; Ma, H.; Zhao, S.; Li, H. Molecular Basis for Hierarchical Histone De-β-Hydroxybutyrylation by SIRT3. Cell Discov. 2019, 5, 35. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent Action of Butyrate over β-Hydroxybutyrate as Histone Deacetylase Inhibitor, Transcriptional Modulator and Anti-Inflammatory Molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Miao, D.; Liu, Z.; Liu, K.; Zhang, B.; Li, J.; Li, Y.; Qi, J. β-Hydroxybutyrate Antagonizes Aortic Endothelial Injury by Promoting Generation of VEGF in Diabetic Rats. Tissue Cell 2020, 64, 101345. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Yu, Y.; Wang, H.; Liu, K.; Wang, Y.; Huang, M.; Xuan, C.; Li, Y.; Qi, J. Up-Regulation of MMP-2 by Histone H3K9 β-Hydroxybutyrylation to Antagonize Glomerulosclerosis in Diabetic Rat. Acta Diabetol. 2020, 57, 1501–1509. [Google Scholar] [CrossRef]
- Diao, M.; Wu, Y.; Yang, J.; Liu, C.; Xu, J.; Jin, H.; Wang, J.; Zhang, J.; Gao, F.; Jin, C.; et al. Identification of Novel Key Molecular Signatures in the Pathogenesis of Experimental Diabetic Kidney Disease. Front. Endocrinol. 2022, 13, 843721. [Google Scholar] [CrossRef]
- Hou, W.; Liu, G.; Ren, X.; Liu, X.; He, L.; Huang, H. Quantitative Proteomics Analysis Expands the Roles of Lysine β-Hydroxybutyrylation Pathway in Response to Environmental β-Hydroxybutyrate. Oxid. Med. Cell Longev. 2022, 2022, 4592170. [Google Scholar] [CrossRef]
- Koronowski, K.B.; Greco, C.M.; Huang, H.; Kim, J.-K.; Fribourgh, J.L.; Crosby, P.; Mathur, L.; Ren, X.; Partch, C.L.; Jang, C.; et al. Ketogenesis Impact on Liver Metabolism Revealed by Proteomics of Lysine β-Hydroxybutyrylation. Cell Rep. 2021, 36, 109487. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Tunaru, S.; Offermanns, S. GPR109A, GPR109B and GPR81, a Family of Hydroxy-Carboxylic Acid Receptors. Trends Pharmacol. Sci. 2009, 30, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The β-Hydroxybutyrate Receptor HCA2 Activates a Neuroprotective Subset of Macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-Chain Fatty Acids and Ketones Directly Regulate Sympathetic Nervous System via G Protein-Coupled Receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- Li, C.; Huang, J.; Chen, X.; Yan, Y.; Li, L.; Zhao, W. Transcriptome Analysis Reveals That NEFA and β-Hydroxybutyrate Induce Oxidative Stress and Inflammatory Response in Bovine Mammary Epithelial Cells. Metabolites 2022, 12, 1060. [Google Scholar] [CrossRef]
- Yurista, S.R.; Chen, S.; Welsh, A.; Tang, W.H.W.; Nguyen, C.T. Targeting Myocardial Substrate Metabolism in the Failing Heart: Ready for Prime Time? Curr. Heart Fail. Rep. 2022, 19, 180–190. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Price, N.T. The Proton-Linked Monocarboxylate Transporter (MCT) Family: Structure, Function and Regulation. Biochem. J. 1999, 343 Pt 2, 281–299. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The Glucose Fatty-Acid Cycle. Its Role in Insulin Sensitivity and the Metabolic Disturbances of Diabetes Mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Staehr, P.; Hother-Nielsen, O.; Landau, B.R.; Chandramouli, V.; Holst, J.J.; Beck-Nielsen, H. Effects of Free Fatty Acids per Se on Glucose Production, Gluconeogenesis, and Glycogenolysis. Diabetes 2003, 52, 260–267. [Google Scholar] [CrossRef]
- Ross, R. The Pathogenesis of Atherosclerosis: A Perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.S.; Stehouwer, C.D.; Emeis, J.J.; Coppack, S.W. C-Reactive Protein in Healthy Subjects: Associations with Obesity, Insulin Resistance, and Endothelial Dysfunction: A Potential Role for Cytokines Originating from Adipose Tissue? Arterioscler. Thromb. Vasc. Biol. 1999, 19, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y. White Adipose Tissue and Cardiovascular Disease. Best Pr. Res. Clin. Endocrinol. Metab. 2005, 19, 637–647. [Google Scholar] [CrossRef]
- Lyon, C.J.; Hsueh, W.A. Effect of Plasminogen Activator Inhibitor-1 in Diabetes Mellitus and Cardiovascular Disease. Am. J. Med. 2003, 115 (Suppl. 8A), 62S–68S. [Google Scholar] [CrossRef]
- Moller, D.E.; Kaufman, K.D. Metabolic Syndrome: A Clinical and Molecular Perspective. Annu Rev Med 2005, 56, 45–62. [Google Scholar] [CrossRef]
- Carmena, R.; Duriez, P.; Fruchart, J.-C. Atherogenic Lipoprotein Particles in Atherosclerosis. Circulation 2004, 109, III2–III7. [Google Scholar] [CrossRef]
- Sachetelli, S.; Liu, Q.; Zhang, S.-L.; Liu, F.; Hsieh, T.-J.; Brezniceanu, M.-L.; Guo, D.-F.; Filep, J.G.; Ingelfinger, J.R.; Sigmund, C.D.; et al. RAS Blockade Decreases Blood Pressure and Proteinuria in Transgenic Mice Overexpressing Rat Angiotensinogen Gene in the Kidney. Kidney Int. 2006, 69, 1016–1023. [Google Scholar] [CrossRef]
- Caglayan, E.; Blaschke, F.; Takata, Y.; Hsueh, W.A. Metabolic Syndrome-Interdependence of the Cardiovascular and Metabolic Pathways. Curr. Opin. Pharmacol. 2005, 5, 135–142. [Google Scholar] [CrossRef]
- Oram, J.F.; Bornfeldt, K.E. Direct Effects of Long-Chain Non-Esterified Fatty Acids on Vascular Cells and Their Relevance to Macrovascular Complications of Diabetes. Front. Biosci. 2004, 9, 1240–1253. [Google Scholar] [CrossRef]
- François, L.L.; Manel, V.; Rousselle, C.; David, M. Ketogenic regime as anti-epileptic treatment: Its use in 29 epileptic children. Arch. Pediatr. 2003, 10, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Bodenant, M.; Moreau, C.; Sejourné, C.; Auvin, S.; Delval, A.; Cuisset, J.-M.; Derambure, P.; Destée, A.; Defebvre, L. Interest of the ketogenic diet in a refractory status epilepticus in adults. Rev. Neurol. 2008, 164, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Winesett, S.P.; Bessone, S.K.; Kossoff, E.H.W. The Ketogenic Diet in Pharmacoresistant Childhood Epilepsy. Expert Rev. Neurother. 2015, 15, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Grochowska, K.; Przeliorz, A. The Effect of the Ketogenic Diet on the Therapy of Neurodegenerative Diseases and Its Impact on Improving Cognitive Functions. Dement. Geriatr. Cogn. Dis. Extra 2022, 12, 100–106. [Google Scholar] [CrossRef]
- Steven, S.; Taylor, R. Restoring Normoglycaemia by Use of a Very Low Calorie Diet in Long- and Short-Duration Type 2 Diabetes. Diabet. Med. 2015, 32, 1149–1155. [Google Scholar] [CrossRef]
- Goday, A.; Bellido, D.; Sajoux, I.; Crujeiras, A.B.; Burguera, B.; García-Luna, P.P.; Oleaga, A.; Moreno, B.; Casanueva, F.F. Short-Term Safety, Tolerability and Efficacy of a Very Low-Calorie-Ketogenic Diet Interventional Weight Loss Program versus Hypocaloric Diet in Patients with Type 2 Diabetes Mellitus. Nutr. Diabetes 2016, 6, e230. [Google Scholar] [CrossRef]
- Schneider, S.; Biggerstaff, D.L.; Barber, T.M. Helpful or Harmful? The Impact of the Ketogenic Diet on Eating Disorder Outcomes in Type 1 Diabetes Mellitus. Expert. Rev. Endocrinol. Metab. 2022, 17, 319–331. [Google Scholar] [CrossRef]
- Morris, C.G.; Low, J. Metabolic Acidosis in the Critically Ill: Part 2. Causes and Treatment. Anaesthesia 2008, 63, 396–411. [Google Scholar] [CrossRef]
- Yancy, W.S.; Foy, M.; Chalecki, A.M.; Vernon, M.C.; Westman, E.C. A Low-Carbohydrate, Ketogenic Diet to Treat Type 2 Diabetes. Nutr. Metab. 2005, 2, 34. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, J.; Du, D.; Niu, N.; Liu, S.; Yang, X.; Lu, P.; Shen, X.; Shi, N.; Yao, L.; et al. Ketogenesis Acts as an Endogenous Protective Programme to Restrain Inflammatory Macrophage Activation during Acute Pancreatitis. EBioMedicine 2022, 78, 103959. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome-Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Nayor, M.; Brown, K.J.; Vasan, R.S. The Molecular Basis of Predicting Atherosclerotic Cardiovascular Disease Risk. Circ. Res. 2021, 128, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte Subsets Differentially Employ CCR2, CCR5, and CX3CR1 to Accumulate within Atherosclerotic Plaques. J. Clin. Invest. 2007, 117, 185–194. [Google Scholar] [CrossRef]
- Tall, A.R.; Westerterp, M. Inflammasomes, Neutrophil Extracellular Traps, and Cholesterol. J. Lipid Res. 2019, 60, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Björkbacka, H.; Kunjathoor, V.V.; Moore, K.J.; Koehn, S.; Ordija, C.M.; Lee, M.A.; Means, T.; Halmen, K.; Luster, A.D.; Golenbock, D.T.; et al. Reduced Atherosclerosis in MyD88-Null Mice Links Elevated Serum Cholesterol Levels to Activation of Innate Immunity Signaling Pathways. Nat. Med. 2004, 10, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; St Hilaire, C.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β Has Atheroprotective Effects in Advanced Atherosclerotic Lesions of Mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Tabas, I. Consequences and Therapeutic Implications of Macrophage Apoptosis in Atherosclerosis: The Importance of Lesion Stage and Phagocytic Efficiency. Arter. Thromb. Vasc. Biol. 2005, 25, 2255–2264. [Google Scholar] [CrossRef]
- Gonzalez, L.; Trigatti, B.L. Macrophage Apoptosis and Necrotic Core Development in Atherosclerosis: A Rapidly Advancing Field with Clinical Relevance to Imaging and Therapy. Can. J. Cardiol. 2017, 33, 303–312. [Google Scholar] [CrossRef]
- Sharman, M.J.; Kraemer, W.J.; Love, D.M.; Avery, N.G.; Gómez, A.L.; Scheett, T.P.; Volek, J.S. A Ketogenic Diet Favorably Affects Serum Biomarkers for Cardiovascular Disease in Normal-Weight Men. J. Nutr. 2002, 132, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Sharman, M.J.; Forsythe, C.E. Modification of Lipoproteins by Very Low-Carbohydrate Diets. J. Nutr. 2005, 135, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The Failing Heart Utilizes 3-Hydroxybutyrate as a Metabolic Stress Defense. JCI Insight 2019, 4, 124079. [Google Scholar] [CrossRef] [PubMed]
- Bedi, K.C.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H. On the Inability of Ketone Bodies to Serve as the Only Energy Providing Substrate for Rat Heart at Physiological Work Load. Basic Res. Cardiol. 1983, 78, 435–450. [Google Scholar] [CrossRef]
- Russell, R.R.; Taegtmeyer, H. Coenzyme A Sequestration in Rat Hearts Oxidizing Ketone Bodies. J. Clin. Invest. 1992, 89, 968–973. [Google Scholar] [CrossRef][Green Version]
- Newman, J.C.; Verdin, E. Ketone Bodies as Signaling Metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Di Mauro, E. Role of Histone Acetylation in the Control of Gene Expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Lee, A.K.; Kim, D.H.; Bang, E.; Choi, Y.J.; Chung, H.Y. β-Hydroxybutyrate Suppresses Lipid Accumulation in Aged Liver through GPR109A-Mediated Signaling. Aging Dis. 2020, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef]
- Yamanashi, T.; Iwata, M.; Kamiya, N.; Tsunetomi, K.; Kajitani, N.; Wada, N.; Iitsuka, T.; Yamauchi, T.; Miura, A.; Pu, S.; et al. Beta-Hydroxybutyrate, an Endogenic NLRP3 Inflammasome Inhibitor, Attenuates Stress-Induced Behavioral and Inflammatory Responses. Sci. Rep. 2017, 7, 7677. [Google Scholar] [CrossRef] [PubMed]
- Mohammadifard, N.; Haghighatdoost, F.; Rahimlou, M.; Rodrigues, A.P.S.; Gaskarei, M.K.; Okhovat, P.; de Oliveira, C.; Silveira, E.A.; Sarrafzadegan, N. The Effect of Ketogenic Diet on Shared Risk Factors of Cardiovascular Disease and Cancer. Nutrients 2022, 14, 3499. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bendridi, N.; Selmi, A.; Balcerczyk, A.; Pirola, L. Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders. Int. J. Mol. Sci. 2022, 23, 14564. https://doi.org/10.3390/ijms232314564
Bendridi N, Selmi A, Balcerczyk A, Pirola L. Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders. International Journal of Molecular Sciences. 2022; 23(23):14564. https://doi.org/10.3390/ijms232314564
Chicago/Turabian StyleBendridi, Nadia, Anna Selmi, Aneta Balcerczyk, and Luciano Pirola. 2022. "Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders" International Journal of Molecular Sciences 23, no. 23: 14564. https://doi.org/10.3390/ijms232314564
APA StyleBendridi, N., Selmi, A., Balcerczyk, A., & Pirola, L. (2022). Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders. International Journal of Molecular Sciences, 23(23), 14564. https://doi.org/10.3390/ijms232314564