
RNA-Seq and Genome-Wide Association Studies Reveal Potential Genes for Rice Seed Shattering

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
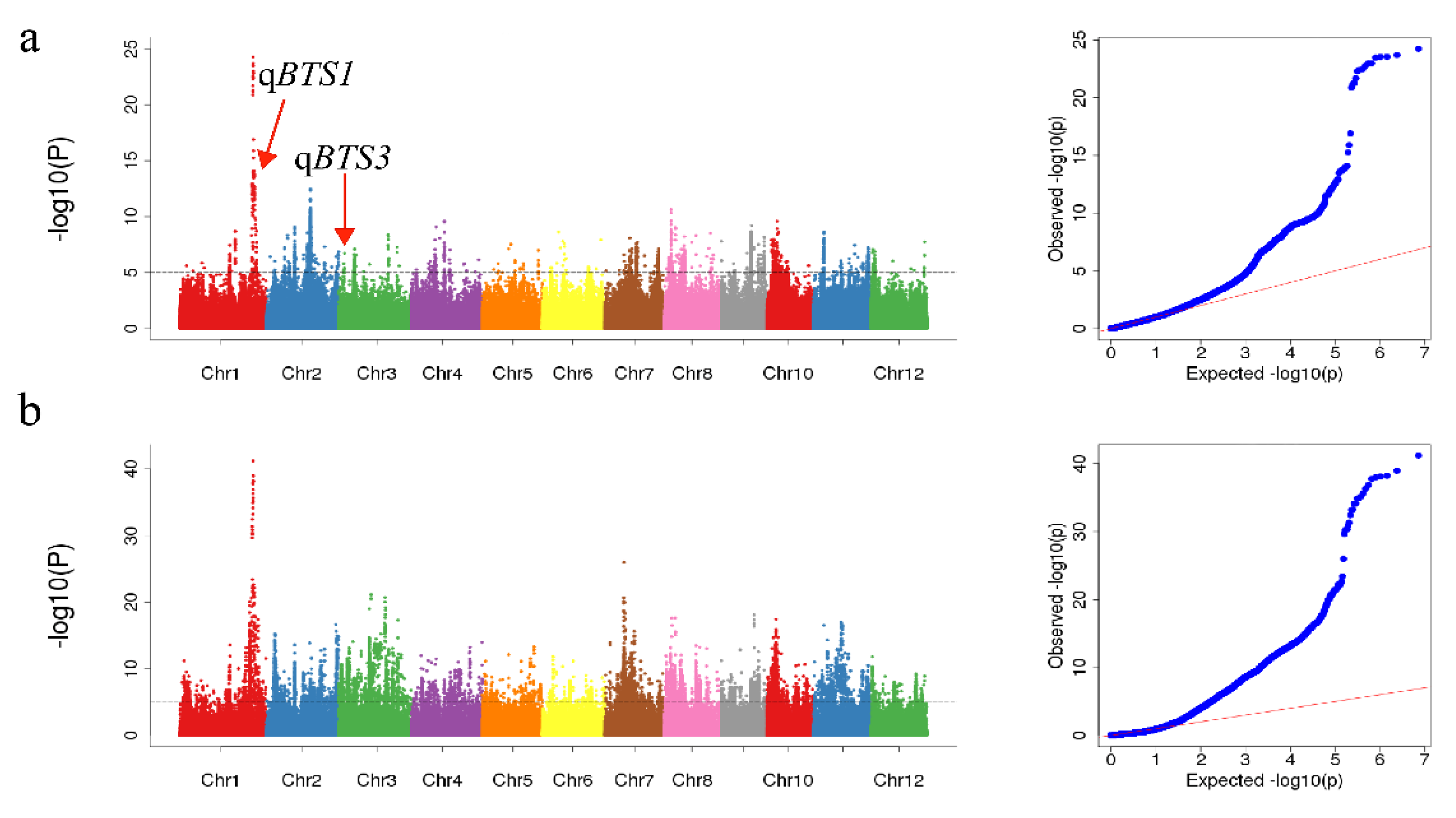
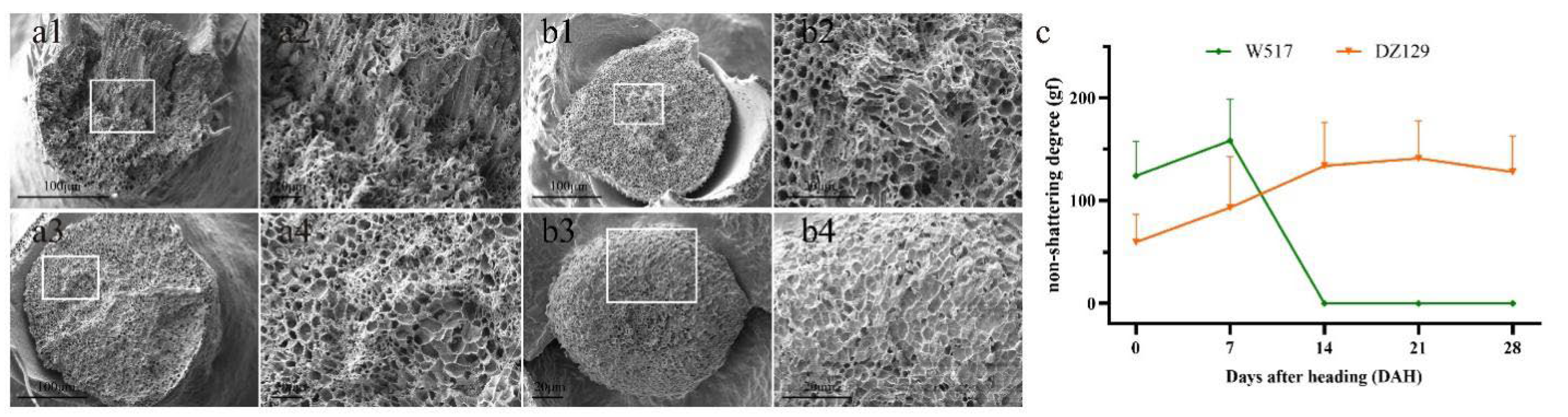
2.1. Phenotypic Evaluations
2.2. Correlation Map or Rice Shattering Traits
2.3. Identification and Histological Observation of Extreme Materials
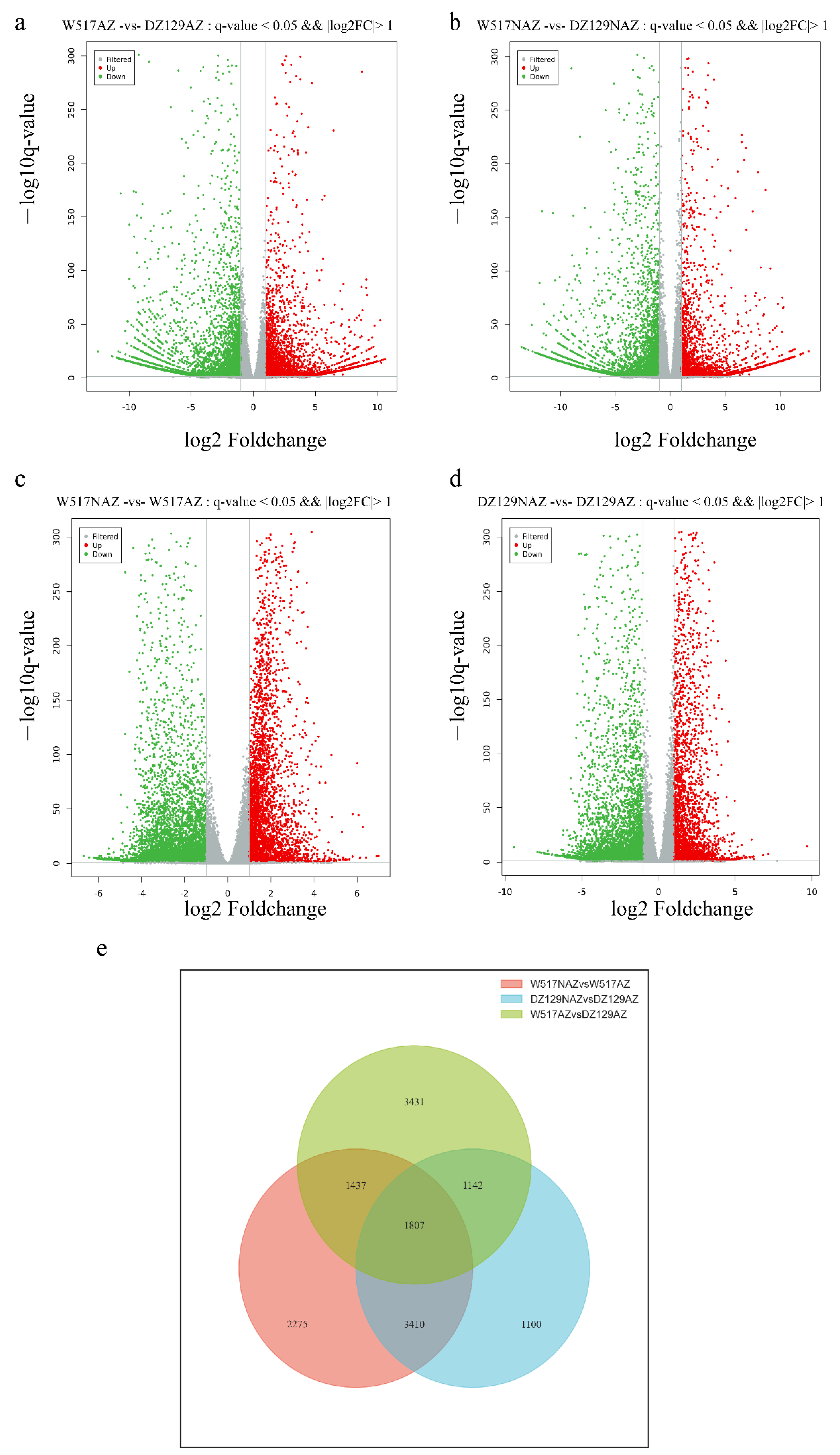
2.4. Differentially Expressed Genes between the AZ and NAZ of Extreme Materials
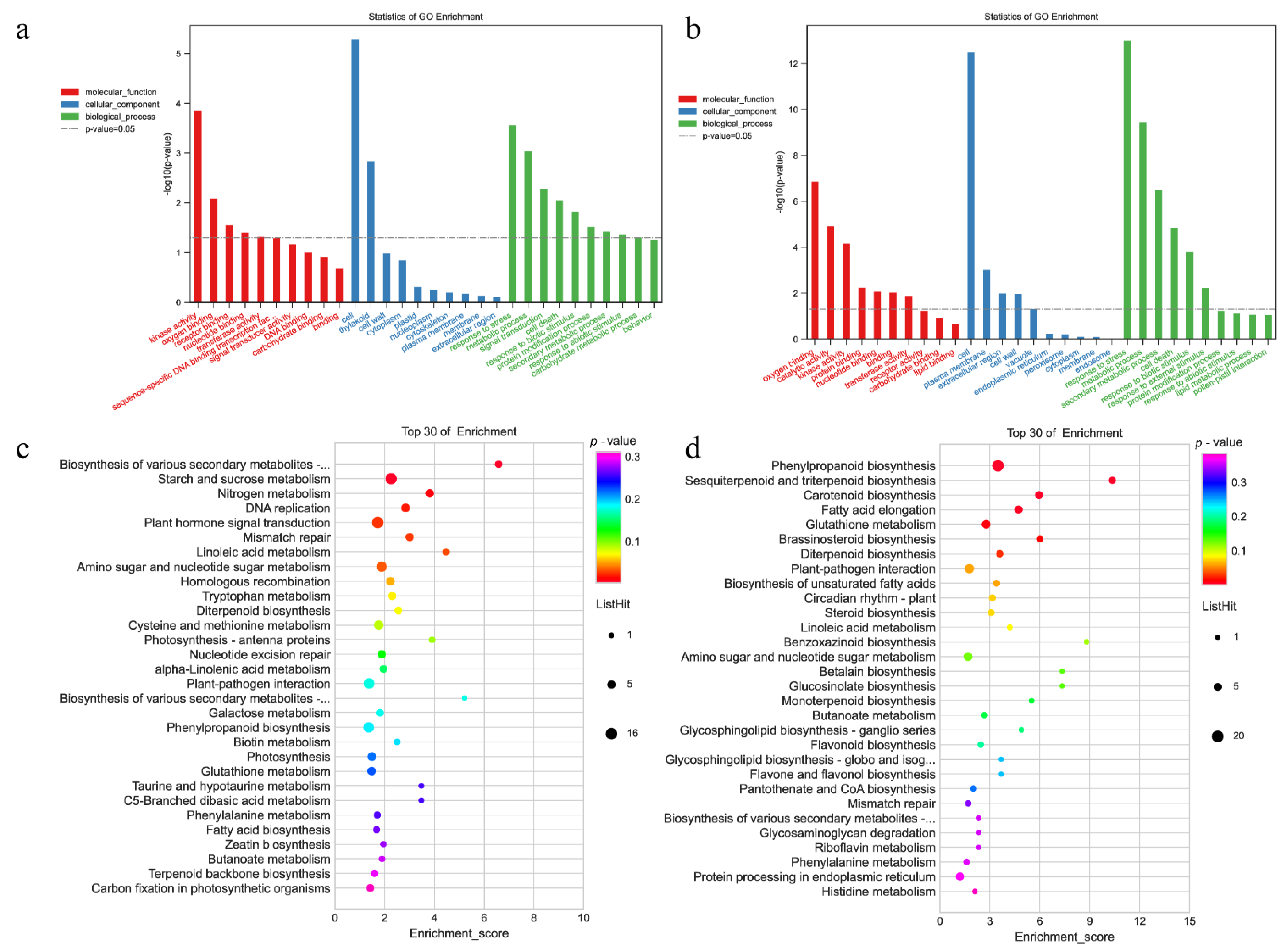
2.5. GO and KEGG Enrichment Analysis of DEGs
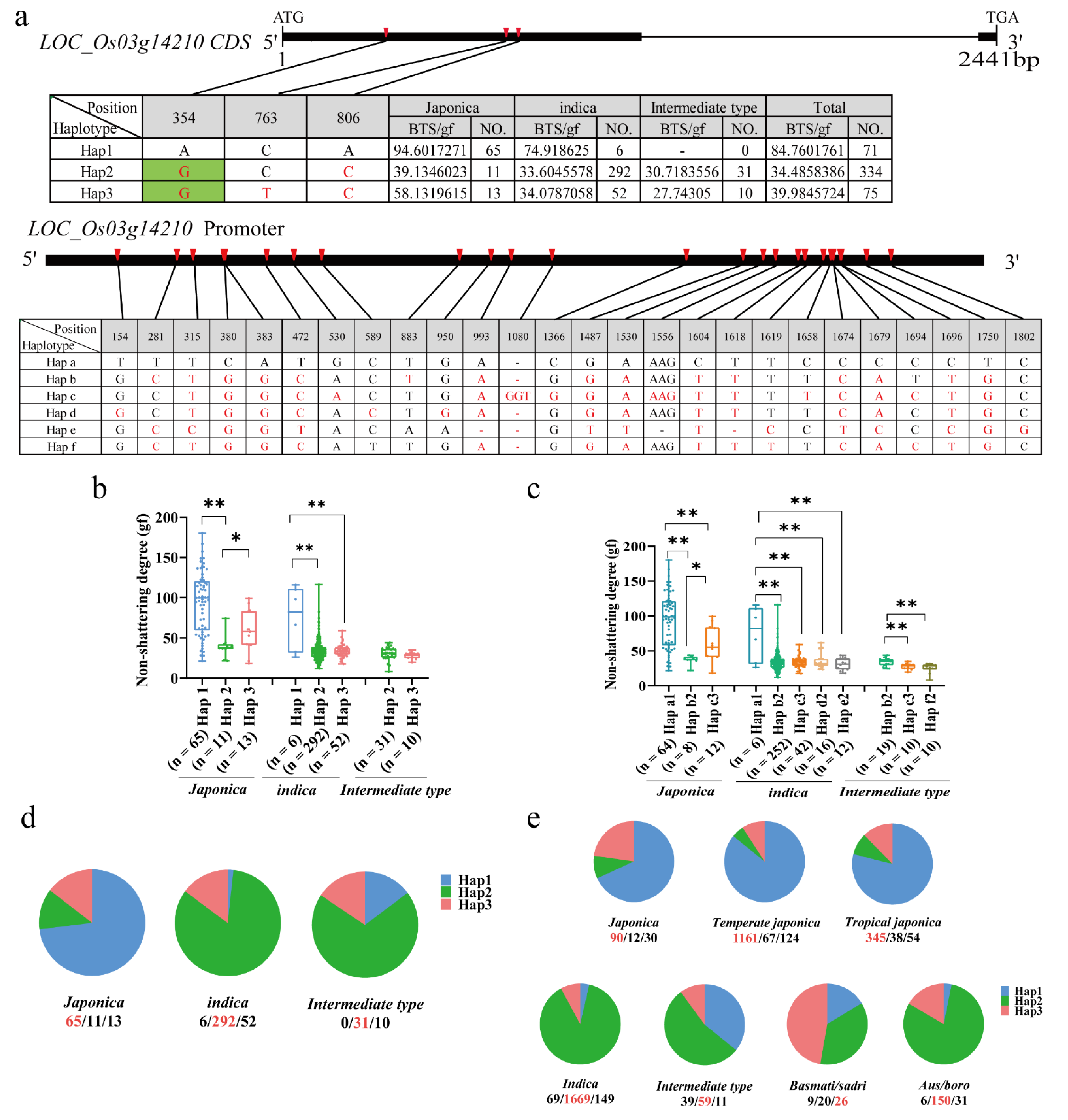
2.6. Candidate Gene Analysis of qBTS1
2.7. Candidate Gene Analysis of qBTS3
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Genome-Wide Association Study
4.3. Phenotypic Evaluation
4.4. RNA Extraction, cDNA Library Construction, and RNA-seq
4.5. Candidate Gene Analysis
4.6. Quantitative Reverse Transcription PCR (qRT–PCR) Analysis
4.7. Scanning Electron Microscopy
4.8. Haplotype Analysis
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, C.; Zhou, A.; Sang, T. Rice domestication by reducing shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, I.-D. On the formation and development of abscission layer in rice plants, Oryza sativa L. Jan. J. Crop. Sci. 1986, 55, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Estornell, L.H.; Agusti, J.; Merelo, P.; Talon, M.; Tadeo, F.R. Elucidating mechanisms underlying organ abscission. Plant Sci. 2013, 199–200, 48–60. [Google Scholar] [CrossRef]
- Balanza, V.; Roig-Villanova, I.; Di Marzo, M.; Masiero, S.; Colombo, L. Seed abscission and fruit dehiscence required for seed dispersal rely on similar genetic networks. Development 2016, 143, 3372–3381. [Google Scholar] [CrossRef] [Green Version]
- Konishi, S.; Izawa, T.; Lin, S.Y.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP caused loss of seed shattering during rice domestication. Science 2006, 312, 1392–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Griffith, M.E.; Li, X.; Zhu, Z.; Tan, L.; Fu, Y.; Zhang, W.; Wang, X.; Xie, D.; Sun, C. Origin of seed shattering in rice (Oryza sativa L.). Planta 2007, 226, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.B.; Zhu, Q.; Wu, Z.Q.; Ross-Ibarra, J.; Gaut, B.S.; Ge, S.; Sang, T. Selection on grain shattering genes and rates of rice domestication. New Phytol. 2009, 184, 708–720. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, D.; Li, C.; Luo, J.; Zhu, B.-F.; Zhu, J.; Shangguan, Y.; Wang, Z.; Sang, T.; Zhou, B.; et al. Genetic control of seed shattering in rice by the APETALA2 transcription factor SHATTERING ABORTION1. Plant Cell 2012, 24, 1034–1048. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.; Cho, L.H.; Kim, S.L.; Choi, H.; Koh, H.J.; An, G. The BEL1-type homeobox gene SH5 induces seed shattering by enhancing abscission-zone development and inhibiting lignin biosynthesis. Plant J. 2014, 79, 717–728. [Google Scholar] [CrossRef]
- Lewis, M.W.; Leslie, M.E.; Liljegren, S.J. Plant separation: 50 ways to leave your mother. Curr. Opin. Plant Biol. 2006, 9, 59–65. [Google Scholar] [CrossRef]
- Ji, H.; Kim, S.R.; Kim, Y.H.; Kim, H.; Eun, M.Y.; Jin, I.D.; Cha, Y.S.; Yun, D.W.; Ahn, B.O.; Lee, M.C.; et al. Inactivation of the CTD phosphatase-like gene OsCPL1 enhances the development of the abscission layer and seed shattering in rice. Plant J. 2010, 61, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ma, X.; Zhao, S.; Tang, Y.; Liu, F.; Gu, P.; Fu, Y.; Zhu, Z.; Cai, H.; Sun, C.; et al. The APETALA2-Like Transcription Factor SUPERNUMERARY BRACT Controls Rice Seed Shattering and Seed Size. Plant Cell 2019, 31, 17–36. [Google Scholar] [CrossRef] [Green Version]
- Budiman, M.; Chang, S.; Lee, S.; Yang, T.; Zhang, H.; De Jong, H.; Wing, R. Localization of jointless-2 gene in the centromeric region of tomato chromosome 12 based on high resolution genetic and physical mapping. TAG. Theor. Appl. Genet. Theor. Und Angew. Genet. 2004, 108, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Cho, L.H.; Antt, H.W.; Koh, H.J.; An, G. KNOX Protein OSH15 Induces Grain Shattering by Repressing Lignin Biosynthesis Genes. Plant Physiol. 2017, 174, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.H.; Zhao, Y.; Wei, X.H.; Li, C.Y.; Wang, A.; Zhao, Q.; Li, W.J.; Guo, Y.L.; Deng, L.W.; Zhu, C.R.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32–39. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, K.; Morinaka, Y.; Wang, F.; Huang, P.; Takehara, S.; Hirai, T.; Ito, A.; Koketsu, E.; Kawamura, M.; Kotake, K.; et al. GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proc. Natl. Acad. Sci. USA 2019, 116, 21262–21267. [Google Scholar] [CrossRef] [PubMed]
- Nayyeripasand, L.; Garoosi, G.A.; Ahmadikhah, A. Genome-Wide Association Study (GWAS) to Identify Salt-Tolerance QTLs Carrying Novel Candidate Genes in Rice During Early Vegetative Stage. Rice 2021, 14, 9. [Google Scholar] [CrossRef]
- An, H.; Liu, K.; Wang, B.; Tian, Y.; Ge, Y.; Zhang, Y.; Tang, W.; Chen, G.; Yu, J.; Wu, W.; et al. Genome-wide association study identifies QTLs conferring salt tolerance in rice. Plant Breed. 2019, 139, 73–82. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, X.; Zhang, B.; Yuan, X.; Xing, Y.; Liu, H.; Luo, L.; Chen, G.; Xiong, L. Genetic analyses of lodging resistance and yield provide insights into post-Green-Revolution breeding in rice. Plant Biotechnol. J. 2021, 19, 814–829. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Yuan, Q.; Lin, H.; Li, X.; Zhang, C.; Gao, H.; Zhang, B.; He, H.; Liu, T.; Jie, Z.; et al. Linkage analysis, GWAS, transcriptome analysis to identify candidate genes for rice seedlings in response to high temperature stress. BMC Plant Biol. 2021, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, C.; Zhu, D.; He, H.; Wei, Z.; Yuan, Q.; Li, X.; Gao, X.; Zhang, B.; Gao, H. Identifying candidate genes and patterns of heat-stress response in rice using a genome-wide association study and transcriptome analyses. Crop J. 2022; in press. [Google Scholar] [CrossRef]
- Li, Q.; Lu, X.; Wang, C.; Shen, L.; Dai, L.; He, J.; Yang, L.; Li, P.; Hong, Y.; Zhang, Q. Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen-deficiency tolerance in rice. Crop J. 2022, 10, 942–951. [Google Scholar] [CrossRef]
- Lin, Z.; Li, X.; Shannon, L.M.; Yeh, C.T.; Wang, M.L.; Bai, G.; Peng, Z.; Li, J.; Trick, H.N.; Clemente, T.E.; et al. Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 2012, 44, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.S.; Chu, S.H.; Jiang, W.Z.; Cho, Y.I.; Hahn, J.H.; Eun, M.Y.; McCouch, S.R.; Koh, H.J. Characterization and mapping of a shattering mutant in rice that corresponds to a block of domestication genes. Genetics 2006, 173, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Numaguchi, K.; Miura, K.; Yoshida, K.; Thanh, P.T.; Htun, T.M.; Yamasaki, M.; Komeda, N.; Matsumoto, T.; Terauchi, R.; et al. OsLG1 regulates a closed panicle trait in domesticated rice. Nat. Genet. 2013, 45, 462–465. [Google Scholar] [CrossRef]
- Li, F.; Numa, H.; Hara, N.; Sentoku, N.; Ishii, T.; Fukuta, Y.; Nishimura, N.; Kato, H. Identification of a locus for seed shattering in rice (Oryza sativa L.) by combining bulked segregant analysis with whole-genome sequencing. Mol. Breed. 2019, 39, 36. [Google Scholar] [CrossRef]
- Li, F.; Komatsu, A.; Ohtake, M.; Eun, H.; Shimizu, A.; Kato, H. Direct identification of a mutation in OsSh1 causing non-shattering in a rice (Oryza sativa L.) mutant cultivar using whole-genome resequencing. Sci. Rep. 2020, 10, 14936. [Google Scholar] [CrossRef]
- Peng, L.; Sun, S.; Yang, B.; Zhao, J.; Li, W.; Huang, Z.; Li, Z.; He, Y.; Wang, Z. Genome-wide association study reveals that the cupin domain protein OsCDP3. 10 regulates seed vigour in rice. Plant Biotechnol. J. 2022, 20, 485. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.; He, Y.T.; Li, F.C.; Ma, D.R.; Sun, J. Integrative hormone and transcriptome analysis underline the role of abscisic acid in seed shattering of weedy rice. Plant Growth Regul. 2021, 94, 261–273. [Google Scholar] [CrossRef]
- Lee, Y.; Yoon, T.H.; Lee, J.; Jeon, S.Y.; Lee, J.H.; Lee, M.K.; Chen, H.; Yun, J.; Oh, S.Y.; Wen, X.; et al. A lignin molecular brace controls precision processing of cell walls critical for surface integrity in Arabidopsis. Cell 2018, 173, 1468–1480.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, G.; Ori, N.; Sato, Y.; Hake, S.J. The knotted1-like homeobox gene BREVIPEDICELLUS regulates cell differentiation by modulating metabolic pathways. Genes Dev. 2003, 17, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Fooyontphanich, K.; Morcillo, F.; Joet, T.; Dussert, S.; Serret, J.; Collin, M.; Amblard, P.; Tangphatsornruang, S.; Roongsattham, P.; Jantasuriyarat, C.; et al. Multi-scale comparative transcriptome analysis reveals key genes and metabolic reprogramming processes associated with oil palm fruit abscission. BMC Plant Biol. 2021, 21, 92. [Google Scholar] [CrossRef]
- Wang, J.; Feng, J.; Jia, W.; Fan, P.; Bao, H.; Li, S.; Li, Y. Genome-Wide Identification of Sorghum bicolor Laccases Reveals Potential Targets for Lignin Modification. Front. Plant Sci. 2017, 8, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeto, J.; Tsutsumi, Y. Diverse functions and reactions of class III peroxidases. New Phytol. 2016, 209, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Chin, H.-S.; Wu, Y.-P.; Hour, A.-L.; Hong, C.-Y.; Lin, Y.-R. Genetic and evolutionary analysis of purple leaf sheath in rice. Rice 2016, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, J.; Zhang, Z.; Xie, W. Elymus nutans genes for seed shattering and candidate gene-derived EST-SSR markers for germplasm evaluation. BMC Plant Biol. 2019, 19, 102. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Elliott, K.A.; Gonzalez-Carranza, Z.H. Abscission, dehiscence, and other cell separation processes. Annu. Rev. Plant Biol. 2002, 53, 131–158. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Leung, H. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Guo, Z.; Huang, C.; Duan, L.; Chen, G.; Jiang, N.; Fang, W.; Feng, H.; Xie, W.; Lian, X.J. Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat. Commun. 2014, 5, 5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level—The DESeq Package; European Molecular Biology Laboratory: Heidelberg, Germany, 2012; Volume 10, p. f1000research. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wang, K.; Chen, Z.; Cao, Y.; Gao, Q.; Li, Y.; Li, X.; Lu, H.; Du, H.; Lu, M.; et al. MBKbase for rice: An integrated omics knowledgebase for molecular breeding in rice. Nucleic Acids Res. 2020, 48, D1085–D1092. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Yue, J.; Wang, J.; Lu, W.; Huang, M.; Guo, T.; Wang, H. RNA-Seq and Genome-Wide Association Studies Reveal Potential Genes for Rice Seed Shattering. Int. J. Mol. Sci. 2022, 23, 14633. https://doi.org/10.3390/ijms232314633
Wu L, Yue J, Wang J, Lu W, Huang M, Guo T, Wang H. RNA-Seq and Genome-Wide Association Studies Reveal Potential Genes for Rice Seed Shattering. International Journal of Molecular Sciences. 2022; 23(23):14633. https://doi.org/10.3390/ijms232314633
Chicago/Turabian StyleWu, Linxuan, Jicheng Yue, Jiafeng Wang, Wenyu Lu, Ming Huang, Tao Guo, and Hui Wang. 2022. "RNA-Seq and Genome-Wide Association Studies Reveal Potential Genes for Rice Seed Shattering" International Journal of Molecular Sciences 23, no. 23: 14633. https://doi.org/10.3390/ijms232314633