1. Introduction
Carbenes are undoubtedly one of the most important groups of organic compounds [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18]. They are characterized by having only two valence carbon atoms, thanks to which it generally forms two single covalent bonds to the substituents, CR
R
. As a consequence, the carbon atom retains two unbound electrons, which in a singlet state occupy the same
p orbital, creating a lone electron pair, which is the cause of nucleophilic properties of carbenes. In other words, singlet carbenes are good Lewis bases. It is therefore not surprising that carbenes participate in various kinds of intermolecular interactions, such as hydrogen bonds [
19,
20,
21,
22,
23,
24,
25,
26,
27], lithium bonds [
16,
18,
28,
29,
30], beryllium bonds [
16,
18,
31,
32,
33,
34,
35], magnesium bonds [
16,
18,
34,
35,
36,
37,
38], triel bonds [
18,
39,
40,
41,
42], tetrel bonds [
18,
43,
44,
45], pnictogen bonds [
18,
46,
47,
48], chalcogen bonds [
18,
49], halogen bonds [
18,
50,
51,
52,
53] (in particular to the iodine atom [
50,
51]), and aerogen bonds [
54]. However, carbenes in particular are known for their great ease of bonding with transition metal atoms [
8,
12,
16,
36,
55,
56,
57,
58,
59,
60,
61] to form various adducts, which is the goal of numerous studies in organometallic chemistry. In this case, the N-heterocyclic carbenes (NHCs), especially those derived from imidazol-2-ylidene, are of particular importance [
4,
8,
9,
11,
12,
15,
16,
17,
18,
27]. In turn, within the group of the NHC–M complexes, those involving zinc are of increasing practical importance [
35,
36,
38,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78], due in part to the easy availability and relatively low cost of precursors of such complexes. Some practical applications of NHC–Zn complexes in organometallic catalysis have recently been extensively described [
62,
63,
64,
65]. It is worth mentioning, however, that apart from the lone electron pair which gives singlet carbenes nucleophilic properties, singlet carbenes also feature electrophilic properties [
7,
79,
80,
81,
82,
83,
84] resulting from the presence of an empty
p orbital perpendicular to the plane formed by CR
R
.
Very recently, one of us (M. J.) has investigated [
35] the beryllium [
85,
86,
87,
88,
89,
90,
91,
92,
93], magnesium [
85,
92,
93,
94,
95,
96] and zinc (spodium) [
78,
97,
98,
99,
100] bonds in a large group of dimers formed by carbenes ((NH
)
C, imidazol-2-ylidene, imidazolidin-2-ylidene, tetrahydropyrymid-2-ylidene, and cyclopropenylidene) or carbodiphosphoranes ((PH
)
C and (NH
)
C) and MX
(M = Be, Mg, Zn and X = H, F, Cl, Br, Me) molecules. This was the first study of beryllium, magnesium, and zinc bonds of the C⋯M type. Moreover, in the vast majority of cases, in earlier systems with the beryllium or magnesium bond, the Lewis base was a small molecule involving either atom being good electron donors [
85,
86,
87,
90,
92,
93,
95] or
-electron bonds [
88]. On the other hand, theoretical research on zinc bonds is rather sporadic [
35,
78,
97,
100].
Among many important findings was the demonstration that carbene⋯MX
dimers are characterized by a very high charge transfer from carbene to the MX
molecule [
35]. Namely, it was from two to almost four times greater than in the case of HOH⋯OH
and HOH⋯NH
dimers. This result showed that the carbene⋯MX
(M = Be, Mg, Zn; X = H, F, Cl, Br, Me) dimers are undoubtedly systems in which the charge transfer effect plays a large role, especially in the presence of very polarizable bromine atoms as X. This, in turn, proves the strong interorbital interactions between the carbene molecule and MX
. Importantly, it has been shown that in terms of the electron density and the total electronic energy density values computed at the bond critical point [
101] of C⋯M the C⋯Zn zinc bond is similar to the C⋯Be beryllium bond. Treating the value of electron density as a measure of bond strength [
101], this result has shown that the zinc bonds should be of similar strength to the beryllium bonds, while the magnesium bonds should be significantly weaker than them [
35]. Moreover, together with positive values of the Laplacian of the electron density, negative values of the total electronic energy density obtained for the beryllium and zinc bonds have indicated a significant degree of electron sharing, in turn reflecting a high degree of covalence [
102].
A somewhat surprisingly different picture of the similarity of these three types of bonding has been obtained on the basis of the delocalization index,
(C,M), which describes the exchange of the electrons in the basins of atoms C and M [
103,
104,
105,
106]. Namely, the
(C,M) values for the zinc bond were large, while those for the beryllium and magnesium bond were much lower and similar to each other [
35]. Thus, taking into account the relationship of
(A,B) to the exchange energy [
107] and therefore treating
(A,B) as a measure of the covalent character of an A–B bond [
108], this result has shown that the zinc bond is much more covalent than the beryllium and magnesium bond. On the other hand, the NCI-based (the abbreviation NCI is derived from the Non-Covalent Interactions index [
109,
110]) analysis has shown [
35] that the zinc bond is the strongest; however, the beryllium bond is only slightly weaker.
In light of the previously obtained results [
35] briefly recalled here, it is necessary to investigate comparatively the physical nature of the beryllium, magnesium and TM-type zinc bonds, not only qualitatively but also quantitatively. This is the main goal of this article, and this nature will be analyzed using complementary energy decomposition methods, IQA (i.e., the Interacting Quantum Atoms approach [
111,
112]), which provides a local picture of bonding and ETS-NOCV (i.e., the combination of the Extended Transition State (ETS) method [
113] with the Natural Orbitals for Chemical Valence (NOCV) method [
114,
115,
116,
117,
118]), which is more suited for the description of bonding between molecular fragments. Results will also be compared with the state-of-the-art DLPNO-CCSD(T) (i.e., the Domain-based Localized Pair-Natural Orbital Singles and Doubles Coupled Cluster with perturbative Triples) [
119,
120,
121,
122,
123] calculations and the Local Energy Decomposition (LED) scheme [
124,
125].
2. Results and Discussion
As already mentioned in the Introduction section, one of us (M. J.) has recently investigated [
35] beryllium, magnesium and zinc (spodium) bonds in a large group of dimers formed by either a carbene ((NH
)
C, imidazol-2-ylidene, imidazolidin-2-ylidene, tetrahydropyrymid-2-ylidene, and cyclopropenylidene) or a carbodiphosphorane ((PH
)
C and (NH
)
C) and the MX
(M = Be, Mg, Zn and X = H, F, Cl, Br, Me) species. While the C⋯M contact has been shown to be certainly dominant in all of these dimers, some of them contain additional intermolecular interactions (either hydrogen bond or dihydrogen bond). Since the purpose of this article is to quantify the physical nature of the C⋯M bond, such dimers are not considered here. In other words, in this article, only those dimers in which the described interaction C⋯M is undoubtedly dominant will be considered. The structural characteristics of these dimers will be briefly discussed in the first subsection. Then, in the second and third subsections, the description of the C⋯M interaction obtained by the IQA and ETS-NOCV methods, respectively, will be presented. The correspondence of the results with the outcomes originating from the DLPNO-CCSD(T) method, and based on it, LED energy decomposition will be shown in the fourth subsection. Finally, in the fifth subsection, we will analyze the energy changes of dimers during the rotation of carbene with respect to the ZnBr
subunit.
2.1. Structural Characteristics of the Considered Dimers
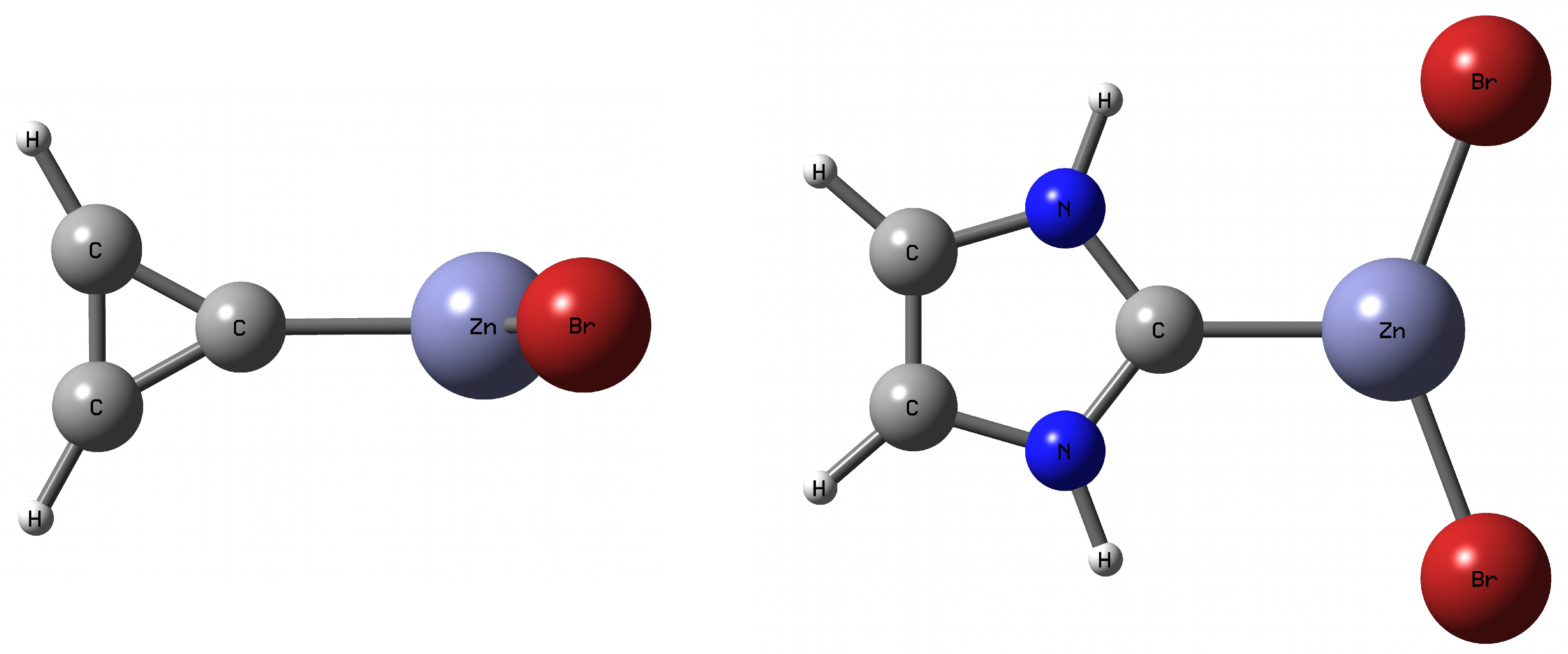
To meet the aforementioned condition, in this article only the cyclopropenylidene⋯MX
(M = Be, Mg, Zn; X = H, Br) and, additionally, imidazol-2-ylidene⋯MBr
dimers will be studied (see
Figures S1 and S2). It is worth noting that in the former case, the lack of additional intermolecular interactions (i.e., apart from the described C⋯M) is ensured by the perpendicular orientation of the cyclopropenylidene and MX
molecules (see the representative cyclopropenylidene⋯ZnBr
dimer in
Figure 1).
In the latter case, however, although the flatness of the entire dimer admits the possibility of the presence of N–H⋯Br hydrogen bonds, these interactions should be relatively weaker than C⋯M due to the large distance H⋯Br and the almost parallel orientation of the N–H and Zn–Br bonds relative to each other (see the representative imidazol-2-ylidene⋯ZnBr
dimer in
Figure 1). A thorough analysis of these intermolecular N-H⋯Br hydrogen bonds will also be performed. The values of some fundamental parameters characterizing the considered carbene⋯MX
dimers are presented in
Table 1.
As can be seen from
Figure 1 and
Table 1 (see the
angle), the characteristic feature of the considered dimers is the clearly bent structure of the MX
molecule. It is worth noting that this bend is similar in the case of MX
molecules with beryllium or zinc, while it is clearly smaller in the case of the magnesium atom, which forms more ionic and thus more resistant M–X bonds. Thus, the value of the angle
suggests the similarity between the Be and Zn atoms. It should be noted that the bending of the MX
molecule is a characteristic effect of the presence of a beryllium or magnesium bond in dimers with different Lewis bases [
85,
86,
88,
92]. Moreover, Martín-Sómer et al. [
86] have shown that in the case of H
N⋯BeH
X
(X = F, Cl, Br;
2) dimers, the nonlinearity of the BeH
X
molecule is due to the decrease in LUMO energy. It is also worth noting that for the bond length M–X the relation Be<Zn<Mg applies so that for a given X the magnesium atom forms the longest M–X bond. It should also be emphasized that the formation of the C⋯M bond leads to significant elongation of the M–X bond (see
in
Table 1) and a greater opening of the L–C–L angle in the carbene molecule (see
in
Table 1). For the same MX
molecule, both of these effects are clearly greater in imidazol-2-ylidene than in cyclopropenylidene, suggesting that the C⋯M interaction should be stronger in the dimers of imidazol-2-ylidene. This is confirmed by the computed dissociation energies [
35]. For imidazol-2-ylidene, these energies are in the 41.2–48.6 kcal/mol range, while for cyclopropenylidene, only 28.2–35.4 kcal/mol (for the same set of MX
molecules, i.e., MBr
), similar outcomes are valid when considering other XC and DLPNO-CCSD(T) method,
Table S1. The greatest dissociation energy value characterizes the dimers with Be, while the smallest
are noted in the dimers containing Zn, which can only partially be explained by the shortest distance C⋯M in the case when M = Be. This is because in the case of Mg and Zn, the distance C⋯Mg is longer than that of C⋯Zn, but the magnesium bond is stronger than the zinc bond. For example, for the cyclopropenylidene⋯MH
dimers, the C⋯M lengths for Be, Mg, and Zn, respectively, are 1.743, 2.268, and 2.121 Å, while the dissociation energies are 29.1, 20.9, and 15.2 kcal/mol. This result shows that the dissociation energy is not completely dependent on the distance C⋯M. Such a trend has already been observed in literature [
35].
It has already been mentioned in Introduction that the carbene⋯MX
dimers considered here are characterized by a very high charge transfer effect from the Lewis base, i.e., the carbene molecule, to the Lewis acid, i.e., the MX
molecule, and this effect is particularly large in the presence of highly polarizable bromine atoms. Indeed, in the case of the imidazol-2-ylidene⋯BeBr
dimer, the value of the charge transfer (based on chemically reliable [
128,
129,
130] Hirshfeld atomic charges [
126,
127]) is as high as −0.464 au. It is enough to mention that the charge transfer determined on the same level of theory in the case of the water dimer is only −0.098 au. This comparison suggests the presence of strong interorbital interactions between the carbene molecule and the MX
unit. Their significance, however, can be quantified by ETS-NOCV and LED methods.
2.2. IQA-Based Results
As already mentioned in the Methods section, the IQA approach is a unique method that enables the determination of the interaction energy of any two atoms and the decomposition of this energy into individual terms, according to Equations (
1) and (
2). The results of such decomposition carried out for the contact C⋯M are shown in
Table 2.
A certain inconvenience of IQA (which is also a characteristic feature of energy decomposition methods) is that the components of the interaction energy are generally much larger than the interaction energy itself. This is obviously due to the close cancellation of the negative and largest and the positive sum of and . It is obvious that the rapid growth of the relevant components with the change Be→Mg→Zn results from both the increase in the atomic number of M, i.e., the charge of the atomic nucleus, and the increase in the number of electrons. However, it is much more important that according to IQA, the C⋯M (M = Be, Mg, Zn) interaction in all the dimers with the participation of cyclopropenylidene is stabilizing, which is indicated by negative values of the obtained interaction energies. For the same X, the binding effect increases in the Zn→Mg→Be direction and is slightly stronger when X = Br, so that the strongest C⋯M bond is for BeBr (−0.3644 a.u.).
It is interesting to look at the values of the exchange-correlation energy (
) as well as the percentage share of this component in the total interaction energy (%
). Of course, independently of X,
increases (i.e., becomes more negative) in the direction Mg→Be→Zn, with the corresponding values being slightly greater for Br than for H. Importantly, the values for Zn are significantly greater than those for Be or Mg (e.g.,
is −0.1091 a.u. in cyclopropenylidene⋯ZnH
and only −0.0458 a.u. and −0.0307 a.u. in cyclopropenylidene⋯BeH
and cyclopropenylidene⋯MgH
, respectively). The contribution of negative electrostatic energy (
) in the case of the cyclopropenylidene dimers quickly decreases in the Be→Mg→Zn series, as a consequence of which the percentage of the exchange-correlation energy quickly increases from ca. 12–13% for the beryllium bond, to ca. 16% for the magnesium bond and up to ca. 63–66% for the zinc bond. This result shows that the IQA method suggests that the zinc bond is highly covalent, whereas the beryllium and magnesium bonds are electrostatic in nature. Of course, such a result was to be expected, because, as already mentioned in the Introduction section, the same trend has already been obtained earlier for the delocalization index
(C,M) [
35], which, as is known [
107,
108], correlates very well with the exchange-correlation energy.
Interestingly, IQA has given quite small interaction energies for the imidazol-2-ylidene⋯MBr
dimers, which do not match the significant dissociation energies for these complexes;
Table 1. Even more surprisingly, a positive interaction energy value (0.0379 a.u.) has been obtained for the imidazol-2-ylidene⋯MgBr
dimer. This destabilizing effect of C⋯Mg (0.0379 a.u.) is due to a significant destabilizing electrostatic contribution (0.0777 a.u.), which exceeds the negative exchange-correlation energy (−0.0397, i.e., ca. 51% only). In the case of imidazol-2-ylidene⋯ZnBr
,
is also positive (0.1190 a.u.; and therefore the purely Coulombic contribution gives no bonding), but unlike for imidazol-2-ylidene⋯MgBr
, this contribution is excessively overshadowed (over 110%) by
(−0.1317 a.u.), which gives stabilizing C⋯Zn bond, which could be considered as anti-electrostatic and covalent.
These diatomic IQA data do not explain the stability of the imidazol-2-ylidene⋯MgBr
species. To this end, we have also summed the interaction energies corresponding to the pairs of atoms in the groups representing the {carbene}⋯{MX
} fragmentation (i.e., all the combinations where one of the atoms is in the {carbene} moiety and the other one in {MX
}). In such treatment, which in fact ignores many-body effects, all the
values are negative, as expected. Furthermore, the IQA fragment-based data (see
Table 3) indicate domination of
over
for the Be- and Mg-based systems ( ca. 51–58% of E
), with the Zn-based species as the only exceptions and the share of electrostatics in their total stabilization reaching ca. 16–21% only.
For the imidazol-2-ylidene complexes, among all the {carbene}⋯{MX
} atomic pairs, the most stabilizing according to IQA are N⋯M interactions, providing from ca. −150 kcal/mol in the case of Zn via. ca. −220 kcal/mol (Mg) to ca. −250 kcal/mol (Be), followed by C⋯Br (from ca. −43 kcal/mol to ca. −39 kcal/mol) and H⋯Br (from ca. −48 kcal/mol to ca. −32 kcal/mol),
Table S2. Surprisingly, the C⋯M interactions are not among the most stabilizing ones, ranging from +20 kcal/mol (Mg) to −40 kcal/mol (Be). In the case of cyclopropenylidene complexes, the C⋯M interactions are definitely the most important, ranging from ca. −100 to over −200 kcal/mol,
Tables S3 and S4.
In the fragment-based treatment, a quite different picture of the C⋯M bond nature has been obtained. Namely, for cyclopropenylidene⋯MX
, the exchange-correlation energy component became more prominent (ca. 45% for Be and Mg, and up to 80% in the case of Zn;
Table 3). Concurrently, in the fragment-based approach, all the MX
complexes with imidazol-2-ylidene have negative
values, as well as their electrostatic and exchange-correlation components;
Table 3. Moreover, the character of these interactions changed in the case of Be and Mg as the electrostatic energy became dominant.
Clearly, the fragment-based approach seems to be much more credible for the description of the carbene⋯metal bonds in both cyclopropenylidene and imidazol-2-ylidene complexes. However, the lack of the C⋯M interaction among the most important contributions is alarming, as much as the counterintuitive positive
value for the imidazol-2-ylidene⋯MgBr
dimer (
Table 2). The most probable explanation is a quite significant positive QTAIM charge on the metal atoms (e.g., the QTAIM-based atomic charge of Mg is as much as 1.687 a.u. compared to 0.616 a.u. only obtained from the Hirshfeld method) [
35]. This, coupled with the incorrect sign of charges on the carbene carbon atom (already stressed in the first work concerning these systems [
35]), puts the reliability of IQA electrostatics in question. Since the IQA method clearly depends on the charge distribution between QTAIM atomic basins and therefore provides variable outcomes for different charge density partitionings, we will switch to ETS-NOCV, which estimates
in a more reliable way through the quasi-classical approach based on self-consistently converged fragment electron densities; see Methods.
2.3. ETS-NOCV-Based Results
In this section, we will present the results obtained by means of the molecular orbital-based ETS-NOCV method. We have used singlet {carbene} and {MX
} fragments which have appeared to be more suitable than triplets since they deliver the smallest orbital interaction contribution. The obtained values of the ETS-NOCV energy terms (see Equation (
5)) are listed in
Table 4.
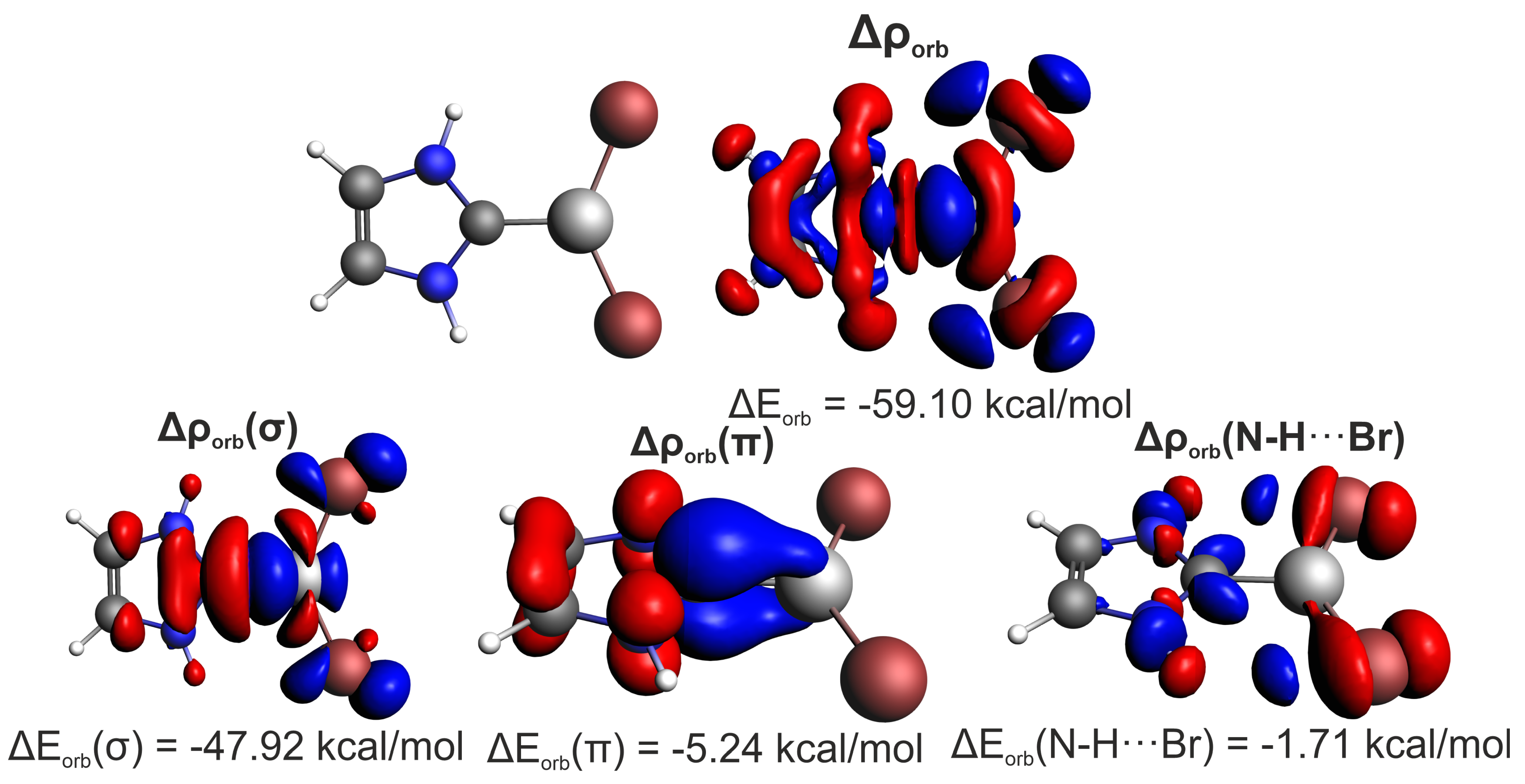
Contrary to the atomic resolution-based IQA results, all ETS-NOCV interaction energies (
) are negative and correspond better to the bond dissociation energies (
values in
Table 1). Bonds formed by beryllium are the strongest, followed by energetically similar magnesium and zinc bonds. Systems with Br atoms are bonded noticeably stronger than with H atoms, and complexes with imidazol-2-ylidene are stabilized more than their cyclopropenylidene counterparts. The carbene⋯metal bonds are dominated by electrostatics (
,
Table 4), with its percentage contribution to the total stabilization being 67–71% in the case of Mg and Zn dimers and 56–61% in systems with Be. This is similar to other reports of bonding in transition metal complexes of imidazol-2-ylidene-based NHCs [
12,
56,
57,
58,
59,
61] and other model carbenes [
55,
58,
61]. For example, it turned out that the electrostatic contribution to C⋯M bonds in imidazol-2-ylidene complexes with Cu, Ag and Au halides accounts for more than 65% of the binding energy [
56]. This contribution was also very similar (ca. 2/3 of the interaction energy) in the various NHC–TM complexes studied by Tonner et al. [
58]. It is worth mentioning at this point the excellent review on the NHC–M bond by Jacobsen et al. [
12], in which it was found that NHC–M bonds are mainly electrostatic in origin with a percentage contribution from 60% to 80%.
Orbital interaction is the second strongest energy term, reaching up to 40% of total stabilization in beryllium systems and 25–30% in the case of Mg and Zn systems,
Table 4. The carbene→metal donation (the LP(C)
(MX) transfer in MO language) dominates by far (72–87%) over the metal→carbene back-donation in
symmetry, with 16–20% contribution in the case of systems with X = H and 7–12% for X = Br. The clearly lower back-donation effect is visible in the latter case, resulting from the electron-withdrawing properties of Br atoms. It should be pointed out that in many transition metal complexes, the
-back-bonding may be far more pronounced and can reach even 40% of the overall orbital interaction energy [
60]. Finally, it should be commented that the geometry distortion term appeared to be the most pronounced for Be-species,
Table S5. Also noteworthy, weak (up to −1.7 kcal/mol, i.e., ca. 3% of the orbital interaction energy) side N–H⋯Br hydrogen bonds have been identified in the imidazol-2-ylidene⋯MBr
complexes,
Figure 2.
Such a negligible share of N–H⋯Br hydrogen bonds really confirms the correct choice of systems with the dominant C⋯M interaction. As ETS-NOCV is only capable of separating the
contribution, it is necessary to support it with complementary IQA results, which indicate a strong electrostatic component (
represents 95% of
amounting to −45.5 kcal/mol). Such pronounced interaction energies are common for a two-atomic approach. For example, for the hydrogen bond in water, the
E
value amounts to ca. −100 kcal/mol [
111].
2.4. LED-Based Results
Local Energy Decomposition [
124,
125] allows for partitioning of the binding energy at the DLPNO-CCSD(T) [
120,
121,
122] level of theory, often considered as the `gold standard’ for calculating interaction energies. The same fragments as before, i.e., {carbene} and {MX
}, have been chosen. The binding energies obtained at the DLPNO-CCSD(T) level of theory are consistent with the DFT-based energies, that is, the beryllium bonds are the strongest, followed by magnesium and zinc (spodium) bonds; compare
Table 1 and
Table 5.
Local Energy Decomposition partitioning confirms the importance of electrostatics in metal–carbene bonding, with E
90% of total stabilization for all compounds,
Table 5. Dispersion contribution of ca. 2% of the total interaction energy proved to be very small. The charge-transfer contributions, bottom of the
Table 5, confirm qualitatively the picture obtained by ETS-NOCV, i.e., the carbene → MX
donation being far more important than MX
carbene back–donation.
2.5. Flat vs. Perpendicular Structure of the Carben⋯MX Dimer
Looking at
Figure 1, showing the cyclopropenylidene ⋯ZnBr
and imidazol-2-ylidene⋯ZnBr
dimers, the fundamental difference between them in the orientation of the ZnBr
molecule relative to the carbene plane is clearly visible. Namely, the ZnBr
and carbene units are perpendicular to each other in cyclopropenylidene⋯ZnBr
, while imidazol-2-ylidene⋯ZnBr
is planar. Taking into account the fact that the cause of the twisting of the metal-unit in relation to the carbene plane was described in the literature only very sporadically [
78], and taking advantage of the relatively high structural simplicity of the studied systems, it was tempting to investigate the cause of the structural differences regarding the twisting of the MX
and carbene planes in relation to each other. For this purpose, we have performed additional calculations for the twisted structures of both dimers, i.e., cyclopropenylidene⋯ZnBr
and imidazol-2-ylidene⋯ZnBr
, in which the twist angle (denoted by
) L-C-Zn-Br (L is either C or N) was changed every 10
from 0
to 90
.
Figure 3 (left) shows the energy profile related to the change in
from 0
to 90
.
It is clearly visible that although the equilibrium geometry of the cyclopropenylidene ⋯ZnBr
dimer corresponds to the perpendicular structure (i.e., with
= 90
), the planar structure (
= 0
) is energetically less stable by only 1 kcal/mol so that the rotation of the ZnBr
unit around the C⋯Zn bond axis is practically free,
Figure 3. Dimer imidazol-2-ylidene⋯ZnBr
presents a completely different situation, namely the flat structure of this dimer is more stable than the perpendicular structure by about 6.5 kcal/mol which proves hindered rotation of ZnBr
due to the presence of adjacent N–H bonds,
Figure 1. Planarization clearly favors the formation of residual hydrogen bonds N–H⋯Br giving rise to Zn⋯C bond contraction by ca. 0.023 Å;
Figure 3, right.
According to the Walsh rule [
131], a molecule adopts the conformation that most ensures stabilization of HOMO. A Walsh diagram showing the dependence of the orbital energies of the three highest occupied orbitals, i.e., HOMO, HOMO-1 and HOMO-2, on the twist angle
is shown in
Figure 4.
It is clearly visible that in the case of imi-ZnBr
, i.e., the imidazol-2-yliden⋯ZnBr
complex, the 90
0
rotation leads to a greater stabilization of all the orbitals considered, and thus also of HOMO. Thus, based on Walsh’s rule, the planar structure should be more stable for this complex than the perpendicular structure, which is indeed true (see
Figure 1 and
Figure 3). By the way, it is worth noting that both structures are characterized by reversed positions of HOMO and HOMO-1. In the case of the cyclopropenylidene⋯ZnBr
dimer, the 0
90
rotation leads to the stabilization of HOMO, but this stabilization is so minimal that it is doubtful to see this stabilization as the cause of the perpendicular structure of this dimer (
Figure 1). Nevertheless, small changes in orbital energies may perhaps explain a small change in total energy (
Figure 3) and almost unhindered rotation around the C⋯Zn axis.
However, in order to make a more reliable assessment of structural changes, ETS-NOCV contributions along rotation angle have been determined and depicted in
Figure 5.
It can be seen that the significant shortening (0.024 Å) of the C⋯Zn distance upon planarization of the imidazol-2-ylidene⋯ZnBr
dimer (see
Figure 3, right) is due to an increase in the orbital interaction term and, to a larger extent, the electrostatic contributions, which causes a drop of
by ca. 4.7 kcal/mol. The former, i.e., the orbital interactions, results mainly from the amplification of the
(Zn–C) component and, to a lesser extent, the
(Zn–C) component,
Figure 5. Interestingly, the effect of twisting (0
90
) the cyclopropenylidene⋯ZnBr
dimer also causes a similar shortening (0.025 Å) of the distance C⋯Zn (
Figure 3, right). Nevertheless, the overall C⋯Zn interaction energy (
) changes only slightly by ca. 1.3 kcal/mol, which is consistent with a facile rotation. Such small variations in C⋯Zn bond interaction energies are caused by modifications of both orbital interaction and electrostatic contributions,
Figure 5. Finally, it is worth mentioning that rotation around the C⋯Zn axis in the cyclopropenylidene⋯ZnBr
dimer has practically no effect on the Zn–Br bond length, while in the imidazol-2-ylidene⋯ZnBr
dimer the transition from perpendicular to planar structure slightly lengthens the bond by 0.006 Å. This effect can be attributed to a larger LP(C)
(Zn–Br) donation.
3. Methods and Materials
Geometries of monomers and dimers were fully optimized on the
B97X-D/6-311++G (2df,2p) level of theory, that is utilizing the
B97X-D range-separated hybrid GGA exchange-correlation functional [
132] of Density Functional Theory (DFT) [
133,
134,
135,
136] and the 6-311++G(2df,2p) basis set [
137,
138,
139,
140,
141,
142,
143], which includes both polarization and diffuse functions. Mardirossian and Head-Gordon have shown [
136] that
B97X-D is one of the best exchange-correlation functionals among 200 for general purposes including intermolecular interactions. To increase the accuracy of the optimization procedure and numerical integration, cutoffs on forces and step size that are used to determine convergence were additionally tightened (0.000015 and 0.000010 for maximum force and its root mean square, respectively, and 0.000060 and 0.000040 for maximum displacement and its root mean square, respectively) and integration grid was increased to the (99, 590) one (UltraFine) having 99 radial shells and 590 angular points per shell. Vibration analysis showed that all the considered systems correspond to real minima on potential energy surfaces. Both geometry optimization and vibration analysis were performed by means of Gaussian 09 [
144].
The Interacting Quantum Atoms (IQA) approach [
111,
112], which is based on Bader’s QTAIM (i.e., Quantum Theory of Atoms in Molecules) [
101], allows the total energy of a system to be divided into mono- and polyatomic contributions. Of the many useful terms defined by the IQA method, this article will utilize the interatomic interaction energy:
where
is the repulsion energy between nuclei of atoms E
and E
,
is the attraction energy between the nucleus of the atom E
and the electrons of the atom E
,
is the attraction energy between electrons of the atom E
and the nucleus of the atom E
and
is the interatomic two-electron repulsion energy. The sum of the middle two terms gives the energy of the interatomic nucleus–electron attraction (
). Importantly, the interelectron repulsion energy can be further divided into a sum of the purely classical (Coulombic) contribution and the exchange-correlation (i.e., the non-classical term) energy:
Moreover, the sum of the first three terms in Equation (
1) and the classical interelectron energy contribution gives the electrostatic energy, such that:
As one can see, all the terms in the IQA equations have strictly defined physical meanings. It is also worth emphasizing that an extremely valuable feature of the IQA decomposition method is the fact that it does not require any reference system and that the interatomic interaction energy can be determined for any pair of E
and E
atoms, e.g., not necessarily connected with each other by a bond path. In this article, the interatomic interaction energy and its components will be determined for the C⋯M (M = Be, Mg, Zn) interaction. Moreover, for an estimation of the total interaction energy between carbene and metal moieties, pair atomic energies between all atoms in both fragments were summed, such as for moieties
and
:
The AIMAll program [
145] was utilized for this purpose. MP2/6-311+G(d,p)-based [
146,
147] wave functions generated in Gaussian09 were used based on the aforementioned geometries.
The ETS-NOCV method [
118] is a combination of the ETS Ziegler–Rauk method [
113] with the Natural Orbitals for Chemical Valence (NOCV) method [
114,
115,
116,
117]. In the ETS method, the total interaction energy between fragments A and B (atoms, fragments and molecules) is decomposed according to the following equation:
where
is the distortion energy that is needed to transform the structures of the isolated fragments A and B into the geometries they have in the created A–B system,
is the energy of the electrostatic interaction of fragments A and B (in their final positions in the molecule, but frozen electron densities),
is the Pauli repulsion energy resulting from the repulsion between electrons with same spins of fragments A and B,
is the dispersion energy, and
is the orbital interaction energy associated with the formation of the A–B system (e.g., an A–B bond) and results from the interaction between the occupied orbitals of the A subsystem with the virtual orbitals of the B subsystem and vice versa. This term also contains the polarization energy related to the electron density changes in each of the A and B subsystems and the interfragment charge transfer. The last four terms of Equation (
5) form the interaction energy,
. On the other hand, the NOCV orbitals are eigenvectors of the differential charge and bond orders matrix,
, where
is the density matrix of a molecule (e.g., A–B), and
is the sum of the density matrices of the orthogonalized fragments A and B,
. The overriding feature of the NOCV orbitals representation is the ability to decompose the differential electron density,
, into chemically meaningful
diagonal contributions:
where
are the eigenvalues obtained by diagonalizing the matrix
, and
M is the number of basis set functions. Visualization of contributions
enables the identification and characterization of the individual components (
,
,
, etc.) of a bond. Importantly, under the ETS-NOCV method, it is possible to quantify the energies (
) corresponding to the individual
components:
Calculations under the ETS-NOCV method were performed on the BLYP-D3(BJ) [
148,
149,
150]/ TZP-ZORA [
151,
152] level of theory using the ADF program [
153]. Such a computational protocol proved to be adequate for bonding analysis both in our and other previous studies [
154,
155]. In this work, we will focus on the instantaneous interaction energy
, since total energy has already been studied in Ref. [
35].
The Domain-based Localized Pair-Natural Orbital Singles and Doubles Coupled Cluster with perturbative Triples [
120,
121,
122,
123] (DLPNO-CCSD(T)) approach is a relatively new approximation of the Coupled Clusters method, which in turn is often referred to as a `golden standard’ for quantum-chemical calculations [
142]. DLPNO-CCSD(T) itself recovers >95% of triples contribution and 99.8% of correlation energy, and thanks to its scaling being close to DFT, it is the first CCSD(T) approximation capable of calculating the entire protein [
122]. It is performed by localizing the orbitals and generating the Projected Atomic Orbitals from occupied localized orbitals, followed by creation of Pair Natural Orbitals from selected PAOs [
119,
120].
Local Energy Decomposition [
124,
125] was created for the DLPNO-CCSD(T) approach in order to harness its accuracy in determining interaction energies and provide an energy decomposition scheme on the CCSD(T) level of theory. It decomposes the binding energy into the following quantities:
The first component is the energy needed to distort fragments A and B from their optimal geometries to their final state in the AB adduct. On the other hand,
is the interaction energy of said fragments, which can be further decomposed into reference (HF) energy,
, and a correlation contribution (
):
The Hartree–Fock part can in turn be expanded into:
where
is the energy needed to distort the electronic structure of fragments into the state optimal for fragment interaction. Electrostatic interaction between monomers is denoted by
, and the HF exchange by
. Correlation energy is decomposed into:
Terms
and
are correlation energy contributions associated with dispersion and non-dispersive corrections, respectively. The term denoted by
is the triples correction arising from CCSD(T).
4. Conclusions
This article presents a comparative study of a series of metal–carbene bonds between main- and transition group metals: beryllium, magnesium and zinc, and model carbenes: cyclopropenylidene (a regular carbene), and imidazol-2-ylidene (N-heterocyclic carbene, NHC). The physical nature of these carbene⋯MX (X = H, Br) interactions has been described for the first time by the joint use of topological QTAIM-based IQA decomposition scheme, molecular orbital-based ETS-NOCV charge and energy decomposition method, as well as LED energy decomposition basing on the state-of-the-art DLPNO-CCSD(T) method.
All methods agree on the increasing bond strength in a series: Zn < Mg Be. For beryllium and magnesium bonds, electrostatics proved to be the most important bond component (55–90% of total stabilization), followed by stabilization due to electron sharing (10–45% of total stabilization), while dispersion interactions proved to be marginal (2–5%). QTAIM/IQA underestimates electrostatic contribution for zinc bonds with respect to both ETS-NOCV and LED schemes which, similarly to Be and Mg species, identify the following importance of Zn–carbene bond constituents: > > . Both ETS-NOCV and LED also provide a consistent picture of these carbene bonds on an orbital level, with the carbene→MX donation strongly dominating over the MX carbene back-donation of symmetry. Substitution of hydrogen atoms by bromines (X in MX) strengthens the metal–carbene bond in all cases due to the amplification of electrostatic forces as well as donation and back-donation effects. The former is likely due to making metal atoms more electrophilic. Finally, the conformational preferences (planar vs twisted) are controlled by the nature of C–Zn bonding as unveiled by ETS-NOCV analyses performed along rotation coordinate pathways.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}