
A Novel Huntington’s Disease Assessment Platform to Support Future Drug Discovery and Development

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. HD Pathogenesis
1.2. Current HD Therapy
1.3. Previously Used HD Mouse Models
1.4. zQ175 Mice and Novel HD Mice
2. Results
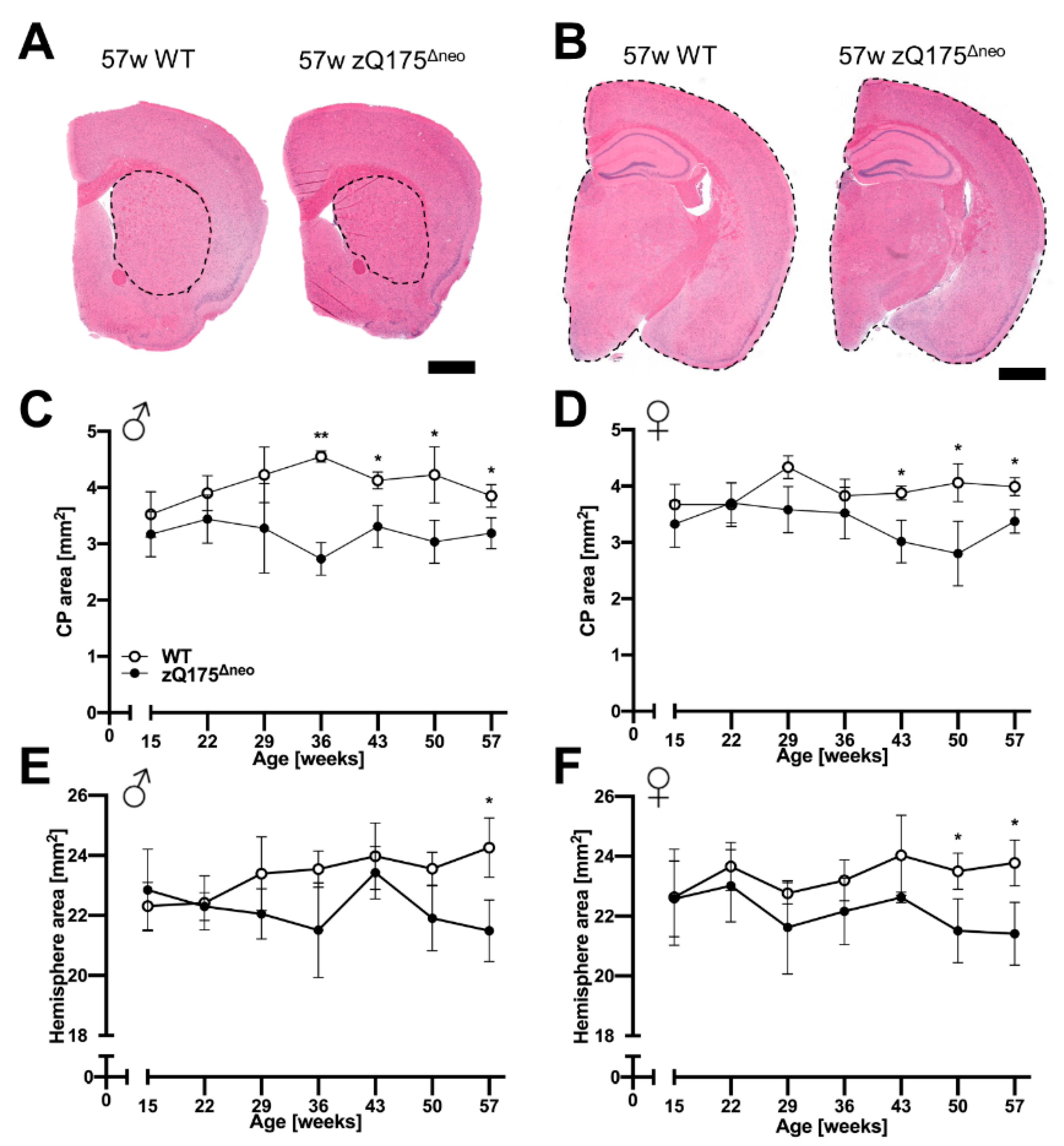
2.1. Development, Characterization, and Establishment of a Novel HD Mouse Model
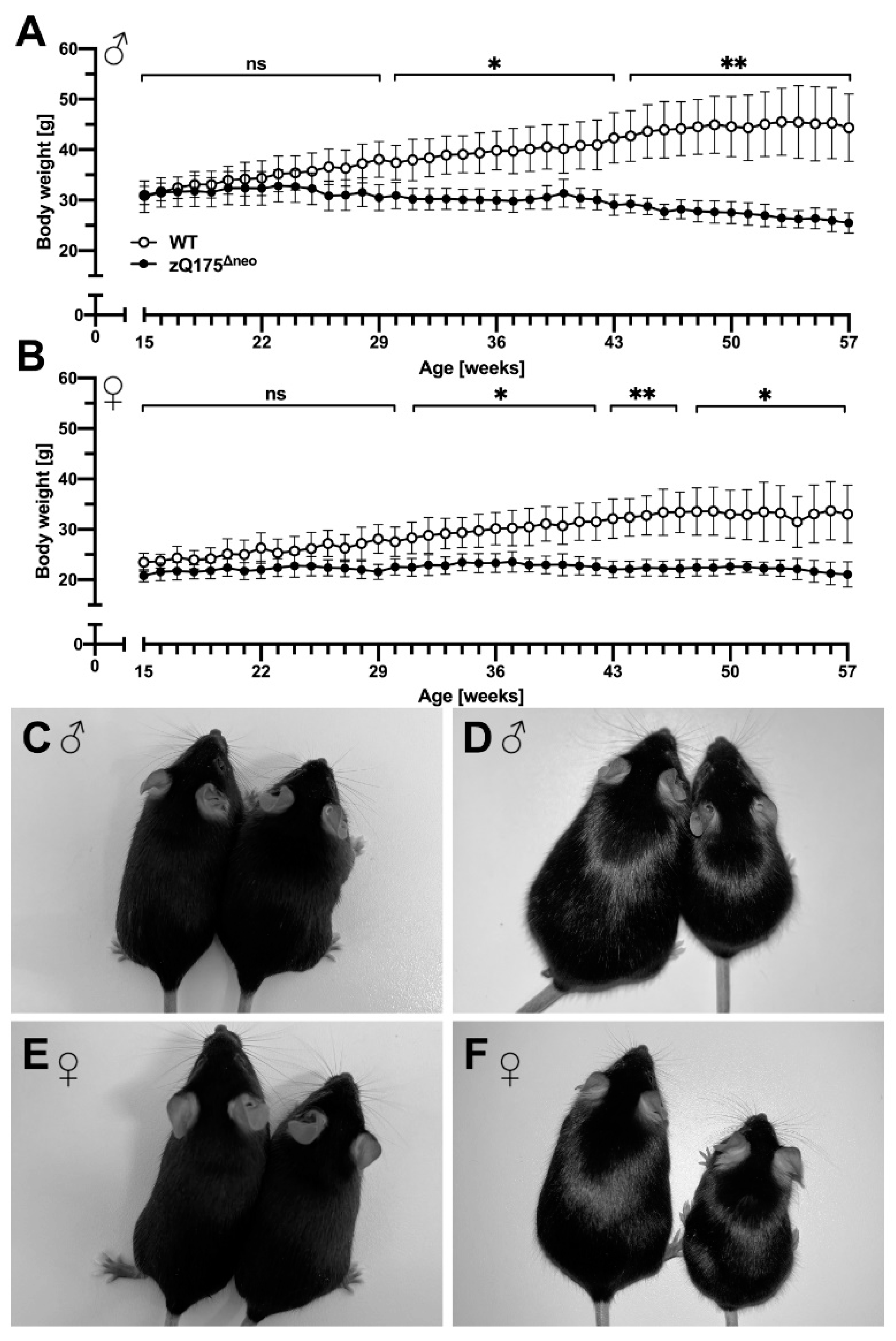
2.1.1. Body Weight and Food Consumption Analysis
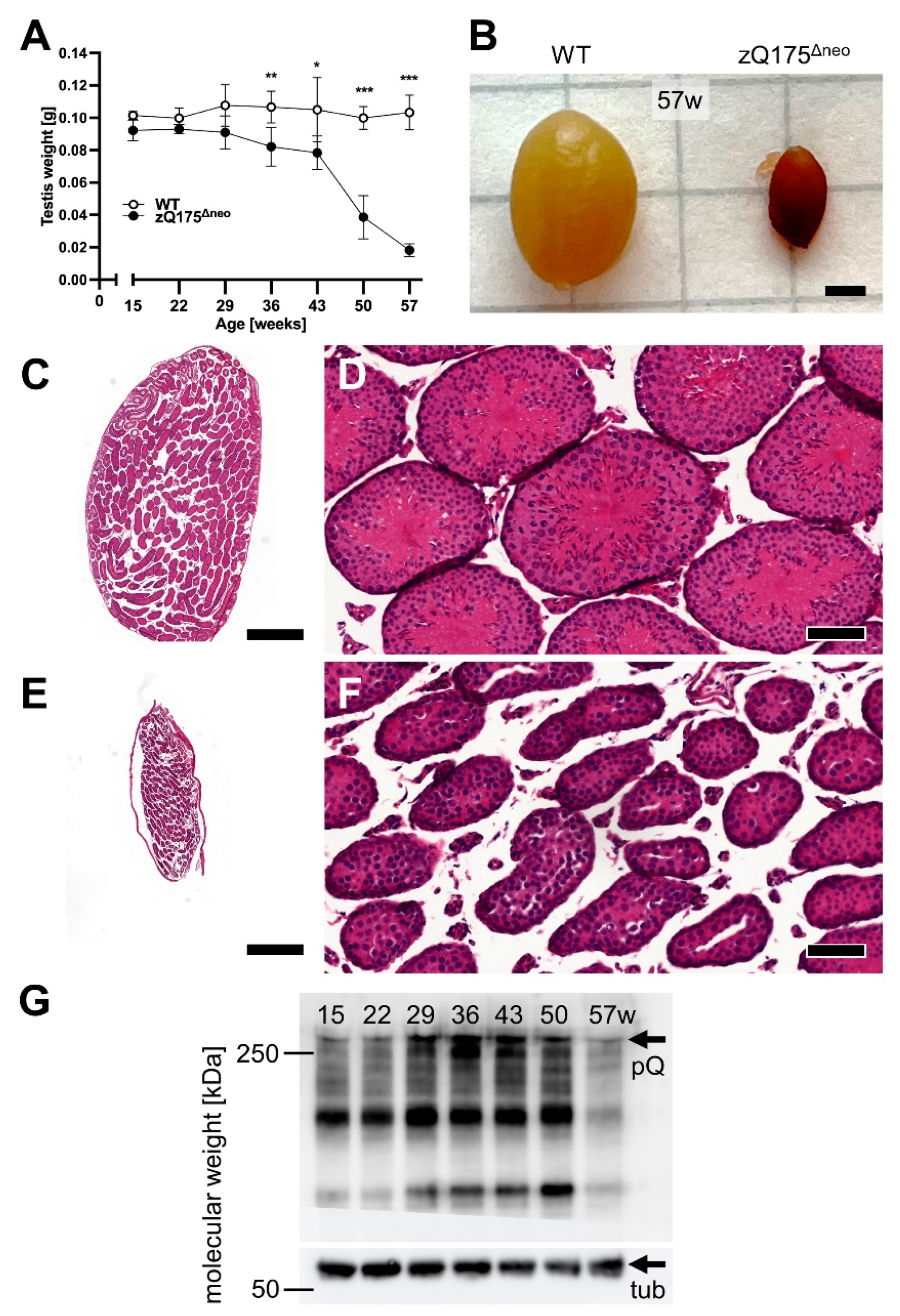
2.1.2. Testicular Atrophy
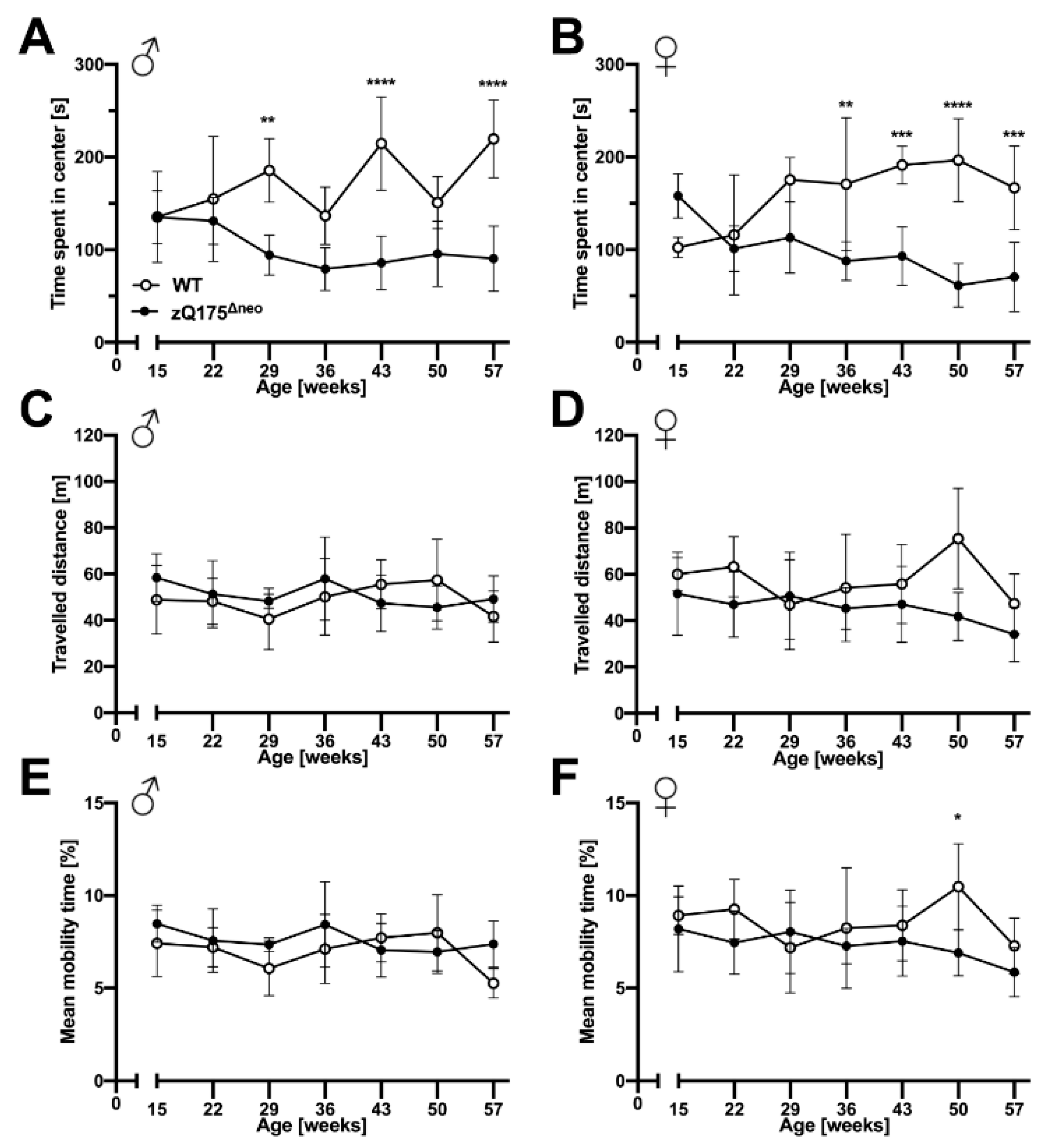
2.1.3. Motoric and Behavioral Changes
2.1.4. Atrophy of Brain Regions
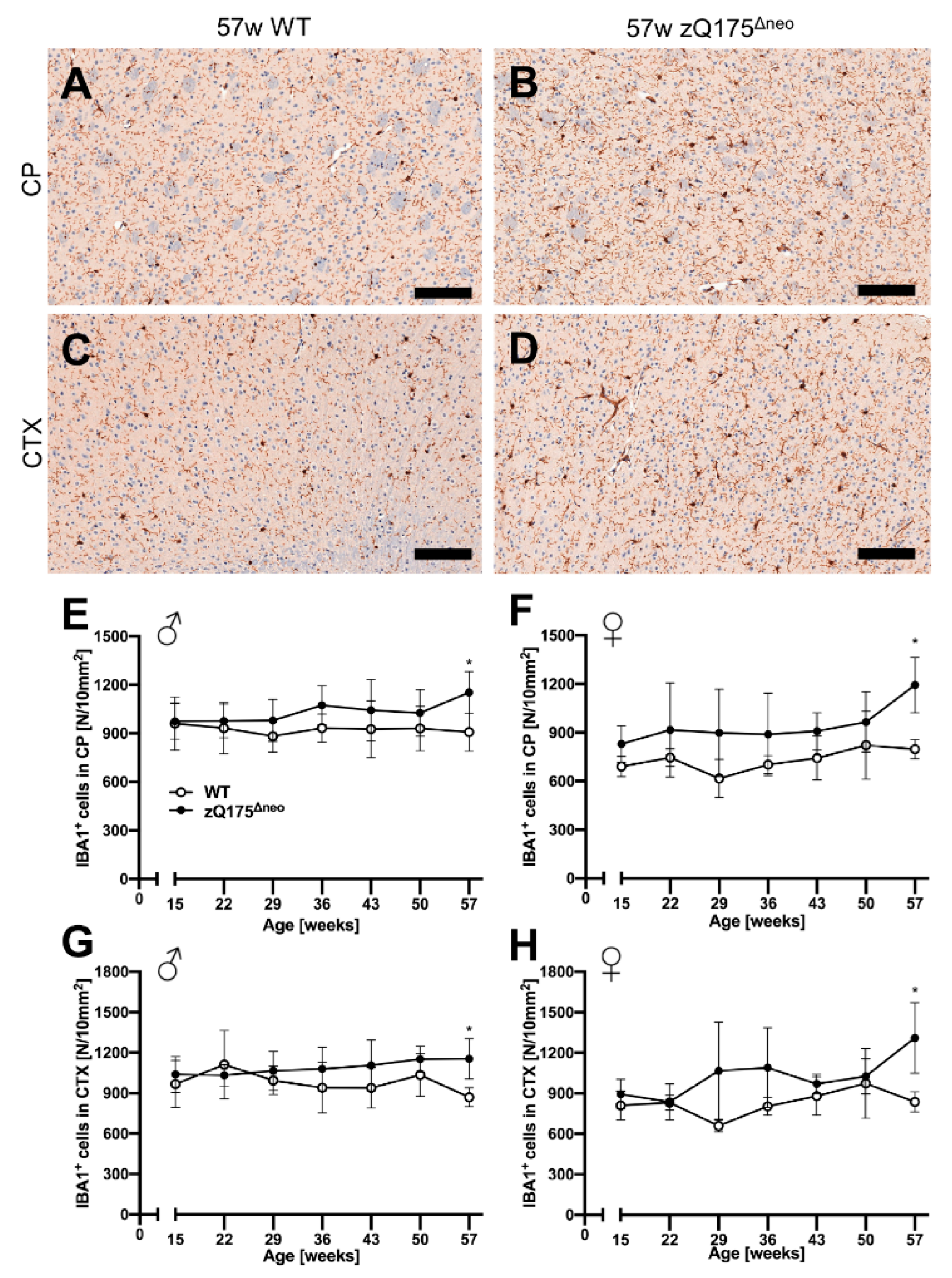
2.1.5. Neuroinflammatory/Glial Reaction
Astrocytes
Microglia

2.2. Charting the Bioactivity Network of Known HD-Targeting Agents
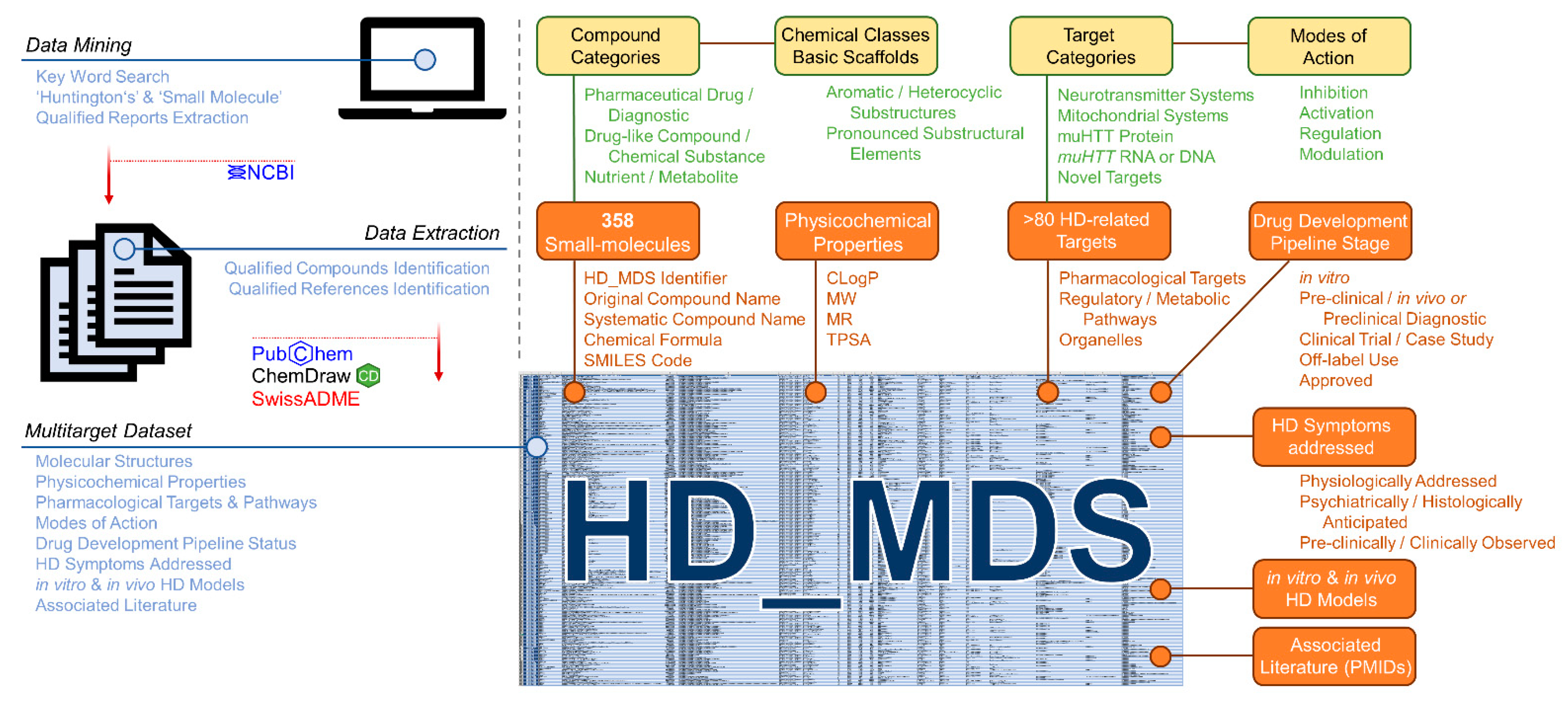
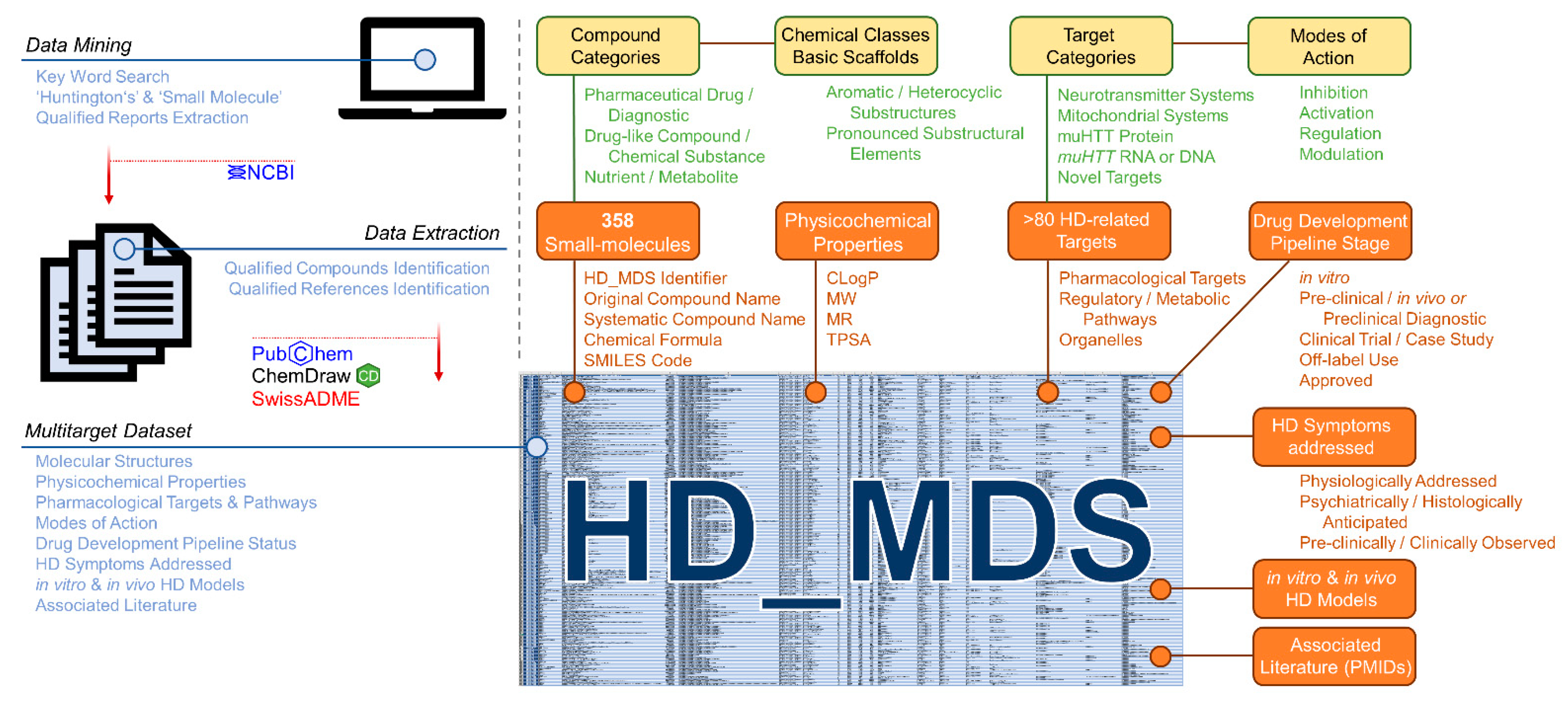
2.2.1. Compilation of the Huntingtin’s Disease Multitarget Dataset (HD_MDS)
2.2.2. HD_MDS Analysis and Validation—The HD Target Landscape
2.2.3. HD_MDS Analysis and Validation—The HD Polypharmacology Landscape
3. Discussion
3.1. Interpretation of the New In Vivo Assessment Platform
3.2. Interpretation of the HD_MDS
3.2.1. Recurring Targets: Solute Carriers and Other Transporters
3.2.2. Recurring Scaffolds: Perspective on Future HD Drug Discovery and Development
3.3. Conclusion, Outlook, and Hypotheses
4. Material and Methods
4.1. Animal Models and Breeding Scheme
4.2. Body Weight and Food Consumption
4.3. Experimental Design
4.4. Open Field Test
4.5. Accelerating Rotarod Performance Test
4.6. Pole Test
4.7. Wire Hang Test
4.8. Tissue Collection and Processing
4.9. Immunohistochemistry and Morphological Quantification
4.10. Histomorphologic Evaluation
4.11. Western Blot Analysis
4.12. Statistical Analysis
4.13. Compilation of the HD_MDS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A.; Hermans, J.; Vegter-van der Vlis, M.; van Ommen, G.J.; Bruyn, G.W. Duration of illness in Huntington’s disease is not related to age at onset. J. Neurol. Neurosurg. Psychiatry 1993, 56, 98–100. [Google Scholar] [CrossRef] [Green Version]
- Warby, S.C.; Montpetit, A.; Hayden, A.R.; Carroll, J.B.; Butland, S.L.; Visscher, H.; Collins, J.A.; Semaka, A.; Hudson, T.J.; Hayden, M.R. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 2009, 84, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2014, 29, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Langbehn, D.R.; Registry Investigators of the European Huntington Disease Network. Longer CAG repeat length is associated with shorter survival after disease onset in Huntington disease. Am. J. Hum. Genet. 2022, 109, 172–179. [Google Scholar] [CrossRef]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Joag, H.; Ghatpande, V.; Desai, M.; Sarkar, M.; Raina, A.; Shinde, M.; Chitale, R.; Deo, A.; Bose, T.; Majumdar, A. A role of cellular translation regulation associated with toxic Huntingtin protein. Cell Mol. Life Sci. 2020, 77, 3657–3670. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.W.; Bouzou, B.; Cha, J.H. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devadiga, S.J.; Bharate, S.S. Recent developments in the management of Huntington’s disease. Bioorg. Chem. 2022, 120, 105642. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and Possible Future Therapeutic Options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, G.; Wang, Z.; Wang, G.; Huang, J.; Liu, B. Repurposing small-molecule drugs for modulating toxic protein aggregates in neurodegenerative diseases. Drug Discov. Today 2022, 27, 1994–2007. [Google Scholar] [CrossRef]
- Dickey, A.S.; La Spada, A.R. Therapy development in Huntington disease: From current strategies to emerging opportunities. Am. J. Med. Genet. A 2018, 176, 842–861. [Google Scholar] [CrossRef]
- Coppen, E.M.; Roos, R.A. Current Pharmacological Approaches to Reduce Chorea in Huntington’s Disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Videnovic, A. Treatment of huntington disease. Curr. Treat. Options Neurol. 2013, 15, 424–438. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Jurcau, M.C. Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy. Biomedicines 2022, 10, 8. [Google Scholar] [CrossRef]
- Paul, R.; Nath, J.; Paul, S.; Mazumder, M.K.; Phukan, B.C.; Roy, R.; Bhattacharya, P.; Borah, A. Suggesting 7,8-dihydroxyflavone as a promising nutraceutical against CNS disorders. Neurochem. Int. 2021, 148, 105068. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Kumar, V.; Singh, S.K.; Dubey, A.K.; Kim, J.J. Flavonoids: Potential Candidates for the Treatment of Neurodegenerative Disorders. Biomedicines 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.L.; Breda, C.; Mason, R.P.; Kooner, G.; Luthi-Carter, R.; Gant, T.W.; Giorgini, F. Connectivity mapping uncovers small molecules that modulate neurodegeneration in Huntington’s disease models. J. Mol. Med. 2016, 94, 235–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyva, M.J.; Degiacomo, F.; Kaltenbach, L.S.; Holcomb, J.; Zhang, N.; Gafni, J.; Park, H.; Lo, D.C.; Salvesen, G.S.; Ellerby, L.M.; et al. Identification and evaluation of small molecule pan-caspase inhibitors in Huntington’s disease models. Chem. Biol. 2010, 17, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Varma, H.; Cheng, R.; Voisine, C.; Hart, A.C.; Stockwell, B.R. Inhibitors of metabolism rescue cell death in Huntington’s disease models. Proc. Natl. Acad. Sci. USA 2007, 104, 14525–14530. [Google Scholar] [CrossRef] [Green Version]
- Desai, U.A.; Pallos, J.; Ma, A.A.; Stockwell, B.R.; Thompson, L.M.; Marsh, J.L.; Diamond, M.I. Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum. Mol. Genet. 2006, 15, 2114–2124. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.K.; Jeong, S.; Lee, J. The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2022, 23, 11. [Google Scholar] [CrossRef]
- Nelson, V.K.; Ali, A.; Dutta, N.; Ghosh, S.; Jana, M.; Ganguli, A.; Komarov, A.; Paul, S.; Dwivedi, V.; Chatterjee, S.; et al. Azadiradione ameliorates polyglutamine expansion disease in Drosophila by potentiating DNA binding activity of heat shock factor 1. Oncotarget 2016, 7, 78281–78296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Au, Q.; Yoon, I.S.; Tremblay, M.H.; Yip, G.; Zhou, Y.; Barber, J.R.; Ng, S.C. Identification of small-molecule HSF1 amplifiers by high content screening in protection of cells from stress induced injury. Biochem. Biophys. Res. Commun. 2009, 390, 925–930. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Trotta, C.R.; Narasimhan, J.; Wiedinger, K.J.; Li, W.; Effenberger, K.A.; Woll, M.G.; Jani, M.B.; Risher, N.; Yeh, S.; et al. Small molecule splicing modifiers with systemic HTT-lowering activity. Nat. Commun. 2021, 12, 7299. [Google Scholar] [CrossRef]
- Harding, R.J.; Tong, Y.F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharm. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.T.; Neubert, T.; Liu, M.; Sperry, S.; Zuccola, H.; Turnbull, A.; Fleck, B.; Kargo, W.; Woody, L.; Chiang, P.; et al. Identification of novel HSP90alpha/beta isoform selective inhibitors using structure-based drug design. demonstration of potential utility in treating CNS disorders such as Huntington’s disease. J. Med. Chem. 2014, 57, 3382–3400. [Google Scholar] [CrossRef] [PubMed]
- Herbst, M.; Wanker, E.E. Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation. A possible therapeutic strategy. Neuro-Degener. Dis. 2007, 4, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, D.; Mestre, T.A. Therapeutic Update on Huntington’s Disease: Symptomatic Treatments and Emerging Disease-Modifying Therapies. Neurotherapeutics 2020, 17, 1645–1659. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Bolton, M.M.; Shah, B.; Kanju, P.M.; Lewis, G.M.; Turmel, G.J.; Whaley, J.C.; Trask, O.J., Jr.; Lo, D.C. Composite primary neuronal high-content screening assay for Huntington’s disease incorporating non-cell-autonomous interactions. J. Biomol. Screen 2010, 15, 806–819. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Arbez, N.; Sahn, J.J.; Lu, Y.; Linkens, K.T.; Hodges, T.R.; Tang, A.; Wiseman, R.; Martin, S.F.; Ross, C.A. Neuroprotective Effects of sigma2R/TMEM97 Receptor Modulators in the Neuronal Model of Huntington’s Disease. ACS Chem. Neurosci. 2022, 13, 2852–2862. [Google Scholar] [CrossRef]
- Patel-Murray, N.L.; Adam, M.; Huynh, N.; Wassie, B.T.; Milani, P.; Fraenkel, E. A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules. Sci. Rep. 2020, 10, 954. [Google Scholar] [CrossRef] [Green Version]
- Pei, F.; Li, H.; Henderson, M.J.; Titus, S.A.; Jadhav, A.; Simeonov, A.; Cobanoglu, M.C.; Mousavi, S.H.; Shun, T.; McDermott, L.; et al. Connecting Neuronal Cell Protective Pathways and Drug Combinations in a Huntington’s Disease Model through the Application of Quantitative Systems Pharmacology. Sci. Rep. 2017, 7, 17803. [Google Scholar] [CrossRef]
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M.; et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 1993, 4, 387–392. [Google Scholar] [CrossRef]
- Ramaswamy, S.; McBride, J.L.; Kordower, J.H. Animal models of Huntington’s disease. ILAR J. 2007, 48, 356–373. [Google Scholar] [CrossRef] [Green Version]
- Ehrnhoefer, D.E.; Butland, S.L.; Pouladi, M.A.; Hayden, M.R. Mouse models of Huntington disease: Variations on a theme. Dis. Model. Mech. 2009, 2, 123–129. [Google Scholar] [CrossRef] [Green Version]
- De Paula Nascimento-Castro, C.; Winkelmann-Duarte, E.C.; Mancini, G.; Welter, P.G.; Placido, E.; Farina, M.; Gil-Mohapel, J.; Rodrigues, A.L.S.; de Bem, A.F.; Brocardo, P.S. Temporal Characterization of Behavioral and Hippocampal Dysfunction in the YAC128 Mouse Model of Huntington’s Disease. Biomedicines 2022, 10, 6. [Google Scholar] [CrossRef]
- Menalled, L.B.; Sison, J.D.; Dragatsis, I.; Zeitlin, S.; Chesselet, M.F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 2003, 465, 11–26. [Google Scholar] [CrossRef]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Wu, B.; Jiang, M.; Jin, J.; Hou, Z.; Zheng, J.; Zhang, J.; Duan, W. Characterization of Behavioral, Neuropathological, Brain Metabolic and Key Molecular Changes in zQ175 Knock-In Mouse Model of Huntington’s Disease. PLoS ONE 2016, 11, e0148839. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.T.; MacIvor, D.M.; Hug, B.A.; Heusel, J.W.; Ley, T.J. Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. USA 1996, 93, 13090–13095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southwell, A.L.; Smith-Dijak, A.; Kay, C.; Sepers, M.; Villanueva, E.B.; Parsons, M.P.; Xie, Y.; Anderson, L.; Felczak, B.; Waltl, S.; et al. An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum. Mol. Genet. 2016, 25, 3654–3675. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, F.; Baron, U.; Rajewsky, K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995, 23, 5080–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef]
- Lo, D.C.; Hughes, R.E. Neurobiology of Huntington’s Disease: Applications to Drug Discovery; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suarez-Farinas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; Andre, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoglio, D.; Bard, J.; Hessmann, M.; Liu, L.; Gartner, A.; de Lombaerde, S.; Huscher, B.; Zajicek, F.; Miranda, A.; Peters, F.; et al. Development of a ligand for in vivo imaging of mutant huntingtin in Huntington’s disease. Sci. Transl. Med. 2022, 14, eabm3682. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.; Hessmann, M.; Schaertl, S.; Berg-Rosseburg, K.; Brown, C.J.; Bursow, G.; Chiki, A.; Ebneth, A.; Gehrmann, M.; Hoeschen, N.; et al. Pharmacological characterization of mutant huntingtin aggregate-directed PET imaging tracer candidates. Sci. Rep. 2021, 11, 17977. [Google Scholar] [CrossRef]
- Moretti, D.; Tambone, S.; Cerretani, M.; Fezzardi, P.; Missineo, A.; Sherman, L.T.; Munoz-Sajuan, I.; Harper, S.; Dominquez, C.; Pacifici, R.; et al. NRF2 activation by reversible KEAP1 binding induces the antioxidant response in primary neurons and astrocytes of a Huntington’s disease mouse model. Free Radic. Biol. Med. 2021, 162, 243–254. [Google Scholar] [CrossRef]
- Liu, L.; Prime, M.E.; Lee, M.R.; Khetarpal, V.; Brown, C.J.; Johnson, P.D.; Miranda-Azpiazu, P.; Chen, X.; Clark-Frew, D.; Coe, S.; et al. Imaging Mutant Huntingtin Aggregates: Development of a Potential PET Ligand. J. Med. Chem. 2020, 63, 8608–8633. [Google Scholar] [CrossRef]
- Abd-Elrahman, K.S.; Hamilton, A.; Hutchinson, S.R.; Liu, F.; Russell, R.C.; Ferguson, S.S.G. mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington’s disease. Sci. Signal. 2017, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Back, M.K.; Kurzawa, J.; Ruggieri, S.; von Engelhardt, J. C57BL/6 Background Attenuates mHTT Toxicity in the Striatum of YAC128 Mice. Int. J. Mol. Sci. 2021, 22, 23. [Google Scholar] [CrossRef]
- Kaartinen, V.; Nagy, A. Removal of the floxed neo gene from a conditional knockout allele by the adenoviral Cre recombinase in vivo. Genesis 2001, 31, 126–129. [Google Scholar] [CrossRef]
- Sjogren, M.; Soylu-Kucharz, R.; Dandunna, U.; Stan, T.L.; Cavalera, M.; Sandelius, A.; Zetterberg, H.; Bjorkqvist, M. Leptin deficiency reverses high metabolic state and weight loss without affecting central pathology in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2019, 132, 104560. [Google Scholar] [CrossRef]
- Djousse, L.; Knowlton, B.; Cupples, L.A.; Marder, K.; Shoulson, I.; Myers, R.H. Weight loss in early stage of Huntington’s disease. Neurology 2002, 59, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Block, R.C.; Dorsey, E.R.; Beck, C.A.; Brenna, J.T.; Shoulson, I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010, 4, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Burg, J.M.M.; Weydt, P.; Landwehrmeyer, G.B.; Aziz, N.A. Effect of Body Weight on Age at Onset in Huntington Disease: A Mendelian Randomization Study. Neurol. Genet. 2021, 7, e603. [Google Scholar] [CrossRef]
- Petersen, A.; Bjorkqvist, M. Hypothalamic-endocrine aspects in Huntington’s disease. Eur. J. Neurosci. 2006, 24, 961–967. [Google Scholar] [CrossRef]
- Ransome, M.I. Androgen function in the pathophysiology and treatment of male Huntington’s disease patients. J. Neuroendocr. 2012, 24, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Murphy, Z.; Selva, D.M.; Hamidizadeh, R.; Pearson, J.; Petersen, A.; Bjorkqvist, M.; Muir, C.; Mackenzie, I.R.; Hammond, G.L.; et al. Testicular degeneration in Huntington disease. Neurobiol. Dis. 2007, 26, 512–520. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Bissada, N.; Vogl, A.W.; Hayden, M.R.; Leavitt, B.R. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2005, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, B.; Publicover, S.J.; Castro, L.F.C.; Esteves, P.J.; Fardilha, M. Brain and testis: More alike than previously thought? Open Biol. 2021, 11, 200322. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Metzler, M.; Slow, E.; Pearson, J.; Schwab, C.; Carroll, J.; Graham, R.K.; Leavitt, B.R.; Hayden, M.R. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol. Dis. 2007, 26, 189–200. [Google Scholar] [CrossRef]
- Goh, A.M.; Wibawa, P.; Loi, S.M.; Walterfang, M.; Velakoulis, D.; Looi, J.C. Huntington’s disease: Neuropsychiatric manifestations of Huntington’s disease. Australas Psychiatry 2018, 26, 366–375. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Q.; Liu, H.; Cheng, L.; Hou, Z.; Mori, S.; Hua, J.; Ross, C.A.; Zhang, J.; Nopoulos, P.C.; et al. Abnormal Brain Development in Huntington’ Disease Is Recapitulated in the zQ175 Knock-In Mouse Model. Cereb. Cortex Commun. 2020, 1, tgaa044. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Moller, T. Neuroinflammation in Huntington’s disease. J. Neural. Transm. 2010, 117, 1001–1008. [Google Scholar] [CrossRef]
- Krohn, M.; Bracke, A.; Avchalumov, Y.; Schumacher, T.; Hofrichter, J.; Paarmann, K.; Frohlich, C.; Lange, C.; Bruning, T.; von Bohlen Und Halbach, O.; et al. Accumulation of murine amyloid-beta mimics early Alzheimer’s disease. Brain 2015, 138 Pt 8, 2370–2382. [Google Scholar] [CrossRef]
- Mohle, L.; Bascunana, P.; Brackhan, M.; Pahnke, J. Development of deep learning models for microglia analyses in brain tissue using DeePathology STUDIO. J. Neurosci. Methods 2021, 364, 109371. [Google Scholar] [CrossRef]
- Bascunana, P.; Brackhan, M.; Pahnke, J. Machine Learning-Supported Analyses Improve Quantitative Histological Assessments of Amyloid-beta Deposits and Activated Microglia. J. Alzheimers Dis. 2021, 79, 597–605. [Google Scholar] [CrossRef]
- Wilton, D.K.; Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 2020, 143, 104963. [Google Scholar] [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 16. [Google Scholar] [CrossRef] [PubMed]
- Stefan, S.M.; Jansson, P.J.; Pahnke, J.; Namasivayam, V. A curated binary pattern multitarget dataset of focused ATP-binding cassette transporter inhibitors. Sci. Data 2022, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Johnson, P.D.; Prime, M.E.; Khetarpal, V.; Lee, M.R.; Brown, C.J.; Chen, X.; Clark-Frew, D.; Coe, S.; Conlon, M.; et al. [(11)C]CHDI-626, a PET Tracer Candidate for Imaging Mutant Huntingtin Aggregates with Reduced Binding to AD Pathological Proteins. J. Med. Chem. 2021, 64, 12003–12021. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; Mills, B.D.; Butler Iii, R.R.; Kuan, J.; McHugh, T.L.M.; Akers, C.; Zhou, J.; Syriani, W.; Grouban, M.; Zeineh, M.; et al. Neuroimaging, Urinary, and Plasma Biomarkers of Treatment Response in Huntington’s Disease: Preclinical Evidence with the p75(NTR) Ligand LM11A-31. Neurotherapeutics 2021, 18, 1039–1063. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; James, M.L.; Belichenko, N.P.; Semaan, S.; Condon, C.; Kuan, J.; Shuhendler, A.J.; Miao, Z.; Chin, F.T.; Longo, F.M. TSPO-PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: Preclinical evidence with the p75NTR ligand LM11A-31. Hum. Mol. Genet. 2018, 27, 2893–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namasivayam, V.; Stefan, K.; Gorecki, L.; Korabecny, J.; Soukup, O.; Jansson, P.J.; Pahnke, J.; Stefan, S.M. Physicochemistry shapes bioactivity landscape of pan-ABC transporter modulators: Anchor point for innovative Alzheimer’s disease therapeutics. Int. J. Biol. Macromol. 2022, 217, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease-Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Mestre, T.; Ferreira, J.; Coelho, M.M.; Rosa, M.; Sampaio, C. Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst. Rev. 2009, 3, CD006456. [Google Scholar] [CrossRef]
- Cankurtaran, E.S.; Ozalp, E.; Soygur, H.; Cakir, A. Clinical experience with risperidone and memantine in the treatment of Huntington’s disease. J. Natl. Med. Assoc. 2006, 98, 1353–1355. [Google Scholar]
- Rozpedek-Kaminska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Sun, X.; Magpusao, A.; Fedorov, Y.; Thompson, M.; Wang, B.; Lundberg, K.; Adams, D.J.; Qi, X. Small-molecule suppression of calpastatin degradation reduces neuropathology in models of Huntington’s disease. Nat. Commun. 2021, 12, 5305. [Google Scholar] [CrossRef]
- HORIZON Investigators of the Huntington Study Group; European Huntington’s Disease Network. A randomized, double-blind, placebo-controlled study of latrepirdine in patients with mild to moderate Huntington disease. JAMA Neurol. 2013, 70, 25–33. [Google Scholar]
- Sabbagh, M.N.; Shill, H.A. Latrepirdine, a potential novel treatment for Alzheimer’s disease and Huntington’s chorea. Curr. Opin. Investig. Drugs 2010, 11, 80–91. [Google Scholar] [PubMed]
- Keller, C.G.; Shin, Y.; Monteys, A.M.; Renaud, N.; Beibel, M.; Teider, N.; Peters, T.; Faller, T.; St-Cyr, S.; Knehr, J.; et al. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat. Commun. 2022, 13, 1150. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Barrero, C.A.; Vasquez-Del Carpio, R.; Reddy, E.P.; Fecchio, C.; Merali, S.; Deglincerti, A.; Fang, C.; Rogers, J.; Maccecchini, M.L. Posiphen Reduces the Levels of Huntingtin Protein through Translation Suppression. Pharmaceutics 2021, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Matthes, F.; Massari, S.; Bochicchio, A.; Schorpp, K.; Schilling, J.; Weber, S.; Offermann, N.; Desantis, J.; Wanker, E.; Carloni, P.; et al. Reducing Mutant Huntingtin Protein Expression in Living Cells by a Newly Identified RNA CAG Binder. ACS Chem. Neurosci. 2018, 9, 1399–1408. [Google Scholar] [CrossRef] [Green Version]
- Khan, E.; Tawani, A.; Mishra, S.K.; Verma, A.K.; Upadhyay, A.; Kumar, M.; Sandhir, R.; Mishra, A.; Kumar, A. Myricetin Reduces Toxic Level of CAG Repeats RNA in Huntington’s Disease (HD) and Spino Cerebellar Ataxia (SCAs). ACS Chem. Biol. 2018, 13, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Khan, E.; Mishra, S.K.; Mishra, R.; Mishra, A.; Kumar, A. Discovery of a potent small molecule inhibiting Huntington’s disease (HD) pathogenesis via targeting CAG repeats RNA and Poly Q protein. Sci. Rep. 2019, 9, 16872. [Google Scholar] [CrossRef] [Green Version]
- Khan, E.; Biswas, S.; Mishra, S.K.; Mishra, R.; Samanta, S.; Mishra, A.; Tawani, A.; Kumar, A. Rationally designed small molecules targeting toxic CAG repeat RNA that causes Huntington’s disease (HD) and spinocerebellar ataxia (SCAs). Biochimie 2019, 163, 21–32. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, S.; Hu, Y.; Wong, C.H.; Kwan, K.M.; Chan, H.Y.E.; Zuo, Z. Efficient brain uptake and distribution of an expanded CAG RNA inhibitor DB213 via intranasal administration. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2019, 127, 240–251. [Google Scholar] [CrossRef]
- Nakamori, M.; Panigrahi, G.B.; Lanni, S.; Gall-Duncan, T.; Hayakawa, H.; Tanaka, H.; Luo, J.; Otabe, T.; Li, J.; Sakata, A.; et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet. 2020, 52, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Degradation of huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorg. Med. Chem. Lett. 2018, 28, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules that Induce the Degradation of Huntingtin. Angew Chem. Int. Ed. Engl. 2017, 56, 11530–11533. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.; Shin, D. Chemical-Mediated Targeted Protein Degradation in Neurodegenerative Diseases. Life 2021, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Heiser, V.; Scherzinger, E.; Boeddrich, A.; Nordhoff, E.; Lurz, R.; Schugardt, N.; Lehrach, H.; Wanker, E.E. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: Implications for Huntington’s disease therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 6739–6744. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.C.; Chi, C.S.; Cheng, S.N.; Lee, H.F.; Tsai, J.D.; Lin, S.Z.; Harn, H.J. Targeting New Candidate Genes by Small Molecules Approaching Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 17, 26. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.J.; Hyeon, S.J.; Kim, Y.; Lim, S.; Lee, M.Y.; Kim, J.; Londhe, A.M.; Gotina, L.; Kim, Y.; Pae, A.N.; et al. Modulation of SETDB1 activity by APQ ameliorates heterochromatin condensation, motor function, and neuropathology in a Huntington’s disease mouse model. J. Enzym. Inhib. Med. Chem. 2021, 36, 856–868. [Google Scholar] [CrossRef]
- Park, I.; Hwang, Y.J.; Kim, T.; Viswanath, A.N.I.; Londhe, A.M.; Jung, S.Y.; Sim, K.M.; Min, S.J.; Lee, J.E.; Seong, J.; et al. In silico probing and biological evaluation of SETDB1/ESET-targeted novel compounds that reduce tri-methylated histone H3K9 (H3K9me3) level. J. Comput. Aided Mol. Des. 2017, 31, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [Green Version]
- Quinti, L.; Dayalan Naidu, S.; Trager, U.; Chen, X.; Kegel-Gleason, K.; Lleres, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Cho, I.H. Sulforaphane Ameliorates 3-Nitropropionic Acid-Induced Striatal Toxicity by Activating the Keap1-Nrf2-ARE Pathway and Inhibiting the MAPKs and NF-kappaB Pathways. Mol. Neurobiol. 2016, 53, 2619–2635. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, G.; Kaplan, A.; Gaschler, M.M.; Zhang, X.; Hou, Z.; Jiang, M.; Zott, R.; Cremers, S.; Stockwell, B.R.; et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1545–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.; Stockwell, B.R. Structural Elucidation of a Small Molecule Inhibitor of Protein Disulfide Isomerase. ACS Med. Chem. Lett. 2015, 6, 966–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, P.O.; Wong, H.K.; Oyama, F.; Goswami, A.; Okuno, M.; Kino, Y.; Miyazaki, H.; Nukina, N. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J. Biol. Chem. 2009, 284, 13153–13164. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zheng, J.; Peng, Q.; Hou, Z.; Zhang, J.; Mori, S.; Ellis, J.L.; Vlasuk, G.P.; Fries, H.; Suri, V.; et al. Sirtuin 1 activator SRT2104 protects Huntington’s disease mice. Ann. Clin. Transl. Neurol. 2014, 1, 1047–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, T. Bi-phasic dose response in the preclinical and clinical developments of sigma-1 receptor ligands for the treatment of neurodegenerative disorders. Expert Opin. Drug Discov. 2021, 16, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, P.E.; Auguet, M. Pharmacological properties of BN82451: A novel multitargeting neuroprotective agent. CNS Drug Rev. 2007, 13, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Sepp, K.J.; Wu, C.; Hong, P.; Littleton, J.T. High-content chemical and RNAi screens for suppressors of neurotoxicity in a Huntington’s disease model. PLoS ONE 2011, 6, e23841. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Wang, C.; Zhu, C.; Wang, Z.; Yang, H.; Wu, P.; Cui, X.; Botas, J.; Dang, Y.; Ding, Y.; et al. Suppression of toxicity of the mutant huntingtin protein by its interacting compound, desonide. Proc. Natl. Acad. Sci. USA 2022, 119, e2114303119. [Google Scholar] [CrossRef]
- Weber, J.J.; Ortiz Rios, M.M.; Riess, O.; Clemens, L.E.; Nguyen, H.P. The calpain-suppressing effects of olesoxime in Huntington’s disease. Rare Dis. 2016, 4, e1153778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, T.; Kumar, V.; Kaznacheyeva, E.V.; Jana, N.R. Withaferin A Induces Heat Shock Response and Ameliorates Disease Progression in a Mouse Model of Huntington’s Disease. Mol. Neurobiol. 2021, 58, 3992–4006. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Popovic, N.; Brundin, P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx 2005, 2, 447–464. [Google Scholar] [CrossRef] [Green Version]
- Hickey, M.A.; Kosmalska, A.; Enayati, J.; Cohen, R.; Zeitlin, S.; Levine, M.S.; Chesselet, M.F. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience 2008, 157, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Kudwa, A.E.; Menalled, L.B.; Oakeshott, S.; Murphy, C.; Mushlin, R.; Fitzpatrick, J.; Miller, S.F.; McConnell, K.; Port, R.; Torello, J.; et al. Increased Body Weight of the BAC HD Transgenic Mouse Model of Huntington’s Disease Accounts for Some but Not All of the Observed HD-like Motor Deficits. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Van der Burg, J.M.M.; Gardiner, S.L.; Ludolph, A.C.; Landwehrmeyer, G.B.; Roos, R.A.C.; Aziz, N.A. Body weight is a robust predictor of clinical progression in Huntington disease. Ann. Neurol. 2017, 82, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.R.; Dubeau, L.; de Young, M.; Wexler, N.S.; Arnheim, N. Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc. Natl. Acad. Sci. USA 2003, 100, 8834–8838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, L.; Chen, J.; Berggren, K.; Fox, J. Characterization of Striatal Neuronal Loss and Atrophy in the R6/2 Mouse Model of Huntington’s Disease. PLoS Curr. 2014, 6, ecurrents.hd.48727b68b39b82d5fe350f753984bcf9. [Google Scholar] [CrossRef]
- DiFiglia, M. Clinical Genetics, II. Huntington’s disease: From the gene to pathophysiology. Am. J. Psychiatry 1997, 154, 1046. [Google Scholar] [PubMed] [Green Version]
- Zhang, J.; Peng, Q.; Li, Q.; Jahanshad, N.; Hou, Z.; Jiang, M.; Masuda, N.; Langbehn, D.R.; Miller, M.I.; Mori, S.; et al. Longitudinal characterization of brain atrophy of a Huntington’s disease mouse model by automated morphological analyses of magnetic resonance images. Neuroimage 2010, 49, 2340–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikkinen, T.; Lehtimaki, K.; Vartiainen, N.; Puolivali, J.; Hendricks, S.J.; Glaser, J.R.; Bradaia, A.; Wadel, K.; Touller, C.; Kontkanen, O.; et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PLoS ONE 2012, 7, e50717. [Google Scholar] [CrossRef] [Green Version]
- Etxeberria-Rekalde, E.; Alzola-Aldamizetxebarria, S.; Flunkert, S.; Hable, I.; Daurer, M.; Neddens, J.; Hutter-Paier, B. Quantification of Huntington’s Disease Related Markers in the R6/2 Mouse Model. Front. Mol. Neurosci. 2020, 13, 617229. [Google Scholar] [CrossRef]
- Yu, X.; Nagai, J.; Marti-Solano, M.; Soto, J.S.; Coppola, G.; Babu, M.M.; Khakh, B.S. Context-Specific Striatal Astrocyte Molecular Responses Are Phenotypically Exploitable. Neuron 2020, 108, 1146–1162.e10. [Google Scholar] [CrossRef]
- Benraiss, A.; Mariani, J.N.; Osipovitch, M.; Cornwell, A.; Windrem, M.S.; Villanueva, C.B.; Chandler-Militello, D.; Goldman, S.A. Cell-intrinsic glial pathology is conserved across human and murine models of Huntington’s disease. Cell Rep. 2021, 36, 109308. [Google Scholar] [CrossRef]
- Carty, N.; Berson, N.; Tillack, K.; Thiede, C.; Scholz, D.; Kottig, K.; Sedaghat, Y.; Gabrysiak, C.; Yohrling, G.; von der Kammer, H.; et al. Characterization of HTT inclusion size, location, and timing in the zQ175 mouse model of Huntington’s disease: An in vivo high-content imaging study. PLoS ONE 2015, 10, e0123527. [Google Scholar] [CrossRef]
- Savage, J.C.; St-Pierre, M.K.; Carrier, M.; El Hajj, H.; Novak, S.W.; Sanchez, M.G.; Cicchetti, F.; Tremblay, M.E. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflamm. 2020, 17, 98. [Google Scholar] [CrossRef]
- Franciosi, S.; Ryu, J.K.; Shim, Y.; Hill, A.; Connolly, C.; Hayden, M.R.; McLarnon, J.G.; Leavitt, B.R. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2012, 45, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Pido-Lopez, J.; Andre, R.; Benjamin, A.C.; Ali, N.; Farag, S.; Tabrizi, S.J.; Bates, G.P. In vivo neutralization of the protagonist role of macrophages during the chronic inflammatory stage of Huntington’s disease. Sci. Rep. 2018, 8, 11447. [Google Scholar] [CrossRef] [Green Version]
- Kwan, W.; Magnusson, A.; Chou, A.; Adame, A.; Carson, M.J.; Kohsaka, S.; Masliah, E.; Moller, T.; Ransohoff, R.; Tabrizi, S.J.; et al. Bone marrow transplantation confers modest benefits in mouse models of Huntington’s disease. J. Neurosci. 2012, 32, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garland, M.; Kirkpatrick, P. Atomoxetine hydrochloride. Nat. Rev. Drug Discov. 2004, 3, 385–386. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, N.; Daniele, F.; Ragone, M.A. Fluoxetine in the treatment of Huntington’s disease. Psychopharmacology 2001, 153, 264–266. [Google Scholar] [CrossRef]
- Carbone, M.; Duty, S.; Rattray, M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem. Int. 2012, 60, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Pico, S.; Parras, A.; Santos-Galindo, M.; Pose-Utrilla, J.; Castro, M.; Fraga, E.; Hernandez, I.H.; Elorza, A.; Anta, H.; Wang, N.; et al. CPEB alteration and aberrant transcriptome-polyadenylation lead to a treatable SLC19A3 deficiency in Huntington’s disease. Sci. Transl. Med. 2021, 13, eabe7104. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010, 30, 4552–4561. [Google Scholar] [CrossRef] [Green Version]
- Lievens, J.C.; Woodman, B.; Mahal, A.; Spasic-Boscovic, O.; Samuel, D.; Kerkerian-Le Goff, L.; Bates, G.P. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 2001, 8, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Petr, G.T.; Bakradze, E.; Frederick, N.M.; Wang, J.; Armsen, W.; Aizenman, E.; Rosenberg, P.A. Glutamate transporter expression and function in a striatal neuronal model of Huntington’s disease. Neurochem. Int. 2013, 62, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Desmond, T.J.; Albin, R.L.; Frey, K.A. Vesicular neurotransmitter transporters in Huntington’s disease: Initial observations and comparison with traditional synaptic markers. Synapse 2001, 41, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Chung, H.; Rundquist, S.; Maat-Schieman, M.L.; Colgan, L.; Englund, E.; Liu, Y.J.; Roos, R.A.; Faull, R.L.; Brundin, P.; et al. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum. Mol. Genet. 2006, 15, 3119–3131. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.P.; Reiner, A. Cholinergic interneurons in the Q140 knockin mouse model of Huntington’s disease: Reductions in dendritic branching and thalamostriatal input. J. Comp. Neurol. 2016, 524, 3518–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinius, B.; Blunder, M.; Brett, F.M.; Eriksson, A.; Patra, K.; Jonsson, J.; Jazin, E.; Kullander, K. Conditional targeting of medium spiny neurons in the striatal matrix. Front. Behav. Neurosci. 2015, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dargaei, Z.; Bang, J.Y.; Mahadevan, V.; Khademullah, C.S.; Bedard, S.; Parfitt, G.M.; Kim, J.C.; Woodin, M.A. Restoring GABAergic inhibition rescues memory deficits in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E1618–E1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.T.; Chang, Y.G.; Liu, Y.C.; Wang, K.Y.; Chen, H.M.; Lee, D.J.; Yang, S.S.; Tsai, C.H.; Lien, C.C.; Chern, Y. Enhanced Na(+) -K(+) -2Cl(−) cotransporter 1 underlies motor dysfunction in huntington’s disease. Mov. Disord. 2019, 34, 845–857. [Google Scholar] [CrossRef]
- Gamberino, W.C.; Brennan, W.A., Jr. Glucose transporter isoform expression in Huntington’s disease brain. J. Neurochem. 1994, 63, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Krzyszton-Russjan, J.; Zielonka, D.; Jackiewicz, J.; Kusmirek, S.; Bubko, I.; Klimberg, A.; Marcinkowski, J.T.; Anuszewska, E.L. A study of molecular changes relating to energy metabolism and cellular stress in people with Huntington’s disease: Looking for biomarkers. J. Bioenerg. Biomembr. 2013, 45, 71–85. [Google Scholar] [CrossRef]
- Szeliga, M.; Rozycka, A.; Jedrak, P.; Baranska, S.; Janik, P.; Jamrozik, Z.; Albrecht, J. Expression of RNAs Coding for Metal Transporters in Blood of Patients with Huntington’s Disease. Neurochem. Res. 2016, 41, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Guitart, X.; Bonaventura, J.; Rea, W.; Orru, M.; Cellai, L.; Dettori, I.; Pedata, F.; Brugarolas, M.; Cortes, A.; Casado, V.; et al. Equilibrative nucleoside transporter ENT1 as a biomarker of Huntington disease. Neurobiol. Dis. 2016, 96, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becanovic, K.; Pouladi, M.A.; Lim, R.S.; Kuhn, A.; Pavlidis, P.; Luthi-Carter, R.; Hayden, M.R.; Leavitt, B.R. Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Hum. Mol. Genet. 2010, 19, 1438–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Chen, M.; Song, W.; Lin, G.N. Label propagation-based semi-supervised feature selection on decoding clinical phenotypes with RNA-seq data. BMC Med. Genom. 2021, 14 (Suppl. S1), 141. [Google Scholar] [CrossRef]
- Wessels, D.; Lusche, D.F.; Scherer, A.; Kuhl, S.; Myre, M.A.; Soll, D.R. Huntingtin regulates Ca(2+) chemotaxis and K(+)-facilitated cAMP chemotaxis, in conjunction with the monovalent cation/H(+) exchanger Nhe1, in a model developmental system: Insights into its possible role in Huntingtons disease. Dev. Biol. 2014, 394, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cristo, F.; Calarco, A.; Digilio, F.A.; Sinicropi, M.S.; Rosano, C.; Galderisi, U.; Melone, M.A.B.; Saturnino, C.; Peluso, G. The Discovery of Highly Potent THP Derivatives as OCTN2 Inhibitors: From Structure-Based Virtual Screening to In Vivo Biological Activity. Int. J. Mol. Sci. 2020, 21, 19. [Google Scholar] [CrossRef]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Acuna, A.I.; Esparza, M.; Kramm, C.; Beltran, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef] [Green Version]
- Christodoulou, C.C.; Zachariou, M.; Tomazou, M.; Karatzas, E.; Demetriou, C.A.; Zamba-Papanicolaou, E.; Spyrou, G.M. Investigating the Transition of Pre-Symptomatic to Symptomatic Huntington’s Disease Status Based on Omics Data. Int. J. Mol. Sci. 2020, 21, 19. [Google Scholar] [CrossRef]
- Stefan, K.; Wen Leck, L.Y.; Namasivayam, V.; Bascuñana, P.; Huang, M.L.H.; Riss, P.J.; Pahnke, J.; Jansson, P.J.; Stefan, S.M. Vesicular ATP-binding cassette transporters in human disease: Relevant aspects of their organization for future drug development. Future Drug Discov. 2020, 2, FDD51. [Google Scholar] [CrossRef]
- Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Pahnke, J.; Bascunana, P.; Brackhan, M.; Stefan, K.; Namasivayam, V.; Koldamova, R.; Wu, J.; Mohle, L.; Stefan, S.M. Strategies to gain novel Alzheimer’s disease diagnostics and therapeutics using modulators of ABCA transporters. Free Neuropathol. 2021, 2, 33. [Google Scholar] [PubMed]
- Schumacher, T.; Krohn, M.; Hofrichter, J.; Lange, C.; Stenzel, J.; Steffen, J.; Dunkelmann, T.; Paarmann, K.; Frohlich, C.; Uecker, A.; et al. ABC transporters B1, C1 and G2 differentially regulate neuroregeneration in mice. PLoS ONE 2012, 7, e35613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Alfen, F.; et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Pahnke, J.; Walker, L.C.; Scheffler, K.; Krohn, M. Alzheimer’s disease and blood-brain barrier function-Why have anti-beta-amyloid therapies failed to prevent dementia progression? Neurosci. Biobehav. Rev. 2009, 33, 1099–1108. [Google Scholar] [CrossRef] [Green Version]
- Pahnke, J.; Wolkenhauer, O.; Krohn, M.; Walker, L.C. Clinico-pathologic function of cerebral ABC transporters—Implications for the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 396–405. [Google Scholar] [CrossRef]
- Bartels, A.L.; Kortekaas, R.; Bart, J.; Willemsen, A.T.; de Klerk, O.L.; de Vries, J.J.; van Oostrom, J.C.; Leenders, K.L. Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: A possible role in progressive neurodegeneration. Neurobiol. Aging 2009, 30, 1818–1824. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural. Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, H.G.; Hildebrandt, J.; Dobrowolny, H.; Steiner, J.; Bogerts, B.; Pahnke, J. Morphometric analysis of the cerebral expression of ATP-binding cassette transporter protein ABCB1 in chronic schizophrenia: Circumscribed deficits in the habenula. Schizophr. Res. 2016, 177, 52–58, Erratum in Schizophr. Res. 2018, 197, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Holzl, G.; Dobrowolny, H.; Hildebrandt, J.; Trubner, K.; Krohn, M.; Bogerts, B.; Pahnke, J. Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech. Ageing Dev. 2014, 141–142, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, S.; Warzok, R.W.; Cascorbi, I.; Kunert-Keil, C.; Schroeder, E.; Kroemer, H.K.; Siegmund, W.; Walker, L.C.; Pahnke, J. The Role of P-glycoprotein in Cerebral Amyloid Angiopathy; Implications for the Early Pathogenesis of Alzheimer’s Disease. Curr. Alzheimer Res. 2004, 1, 121–125. [Google Scholar] [CrossRef]
- Vogelgesang, S.; Cascorbi, I.; Kroemer, H.K.; Schroeder, E.; Pahnke, J.; Siegmund, W.; Keil, C.; Warzok, R.W.; Walker, L.C. Deposition of amyloid is inversely correlated with the expression of p-glycoprotein—Implications on the possibility of prevention of Alzheimer’s disease. Acta Neuropathol. 2001, 12, 545. [Google Scholar]
- Picataggi, A.; Rodrigues, A.; Cromley, D.A.; Wang, H.; Wiener, J.P.; Garliyev, V.; Billheimer, J.T.; Grabiner, B.C.; Hurt, J.A.; Chen, A.C.; et al. Specificity of ABCA7-mediated cell lipid efflux. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159157. [Google Scholar] [CrossRef]
- Katzov, H.; Chalmers, K.; Palmgren, J.; Andreasen, N.; Johansson, B.; Cairns, N.J.; Gatz, M.; Wilcock, G.K.; Love, S.; Pedersen, N.L.; et al. Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to beta-amyloid metabolism. Hum. Mutat. 2004, 23, 358–367. [Google Scholar] [CrossRef]
- Wollmer, M.A.; Streffer, J.R.; Lutjohann, D.; Tsolaki, M.; Iakovidou, V.; Hegi, T.; Pasch, T.; Jung, H.H.; Bergmann, K.; Nitsch, R.M.; et al. ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 421–426. [Google Scholar] [CrossRef]
- Futter, M.; Diekmann, H.; Schoenmakers, E.; Sadiq, O.; Chatterjee, K.; Rubinsztein, D.C. Wild-type but not mutant huntingtin modulates the transcriptional activity of liver X receptors. J. Med. Genet. 2009, 46, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Guevara, E.; Cardenas, G.; Perez-Severiano, F.; Martinez-Lazcano, J.C. Dysregulated Brain Cholesterol Metabolism Is Linked to Neuroinflammation in Huntington’s Disease. Mov. Disord. 2020, 35, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Marullo, M.; Di Paolo, E.; Cesana, E.; Zuccato, C.; Biella, G.; Cattaneo, E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015, 22, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Obeidat, M.; Fishbane, N.; Nie, Y.; Chen, V.; Hollander, Z.; Tebbutt, S.J.; Bosse, Y.; Ng, R.T.; Miller, B.E.; McManus, B.; et al. The Effect of Statins on Blood Gene Expression in COPD. PLoS ONE 2015, 10, e0140022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Caccia, C. The impairment of cholesterol metabolism in Huntington disease. Biochim. Biophys. Acta 2015, 1851, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Zoufal, V.; Mairinger, S.; Krohn, M.; Wanek, T.; Filip, T.; Sauberer, M.; Stanek, J.; Kuntner, C.; Pahnke, J.; Langer, O. Measurement of cerebral ABCC1 transport activity in wild-type and APP/PS1-21 mice with positron emission tomography. J. Cereb. Blood Flow Metab. 2020, 40, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, F.; Liu, C.; Jin, B.; Jiang, Y.; Tang, M.; Qi, X.; Guo, X. Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalathur, R.K.; Giner-Lamia, J.; Machado, S.; Barata, T.; Ayasolla, K.R.; Futschik, M.E. The unfolded protein response and its potential role in Huntington’s disease elucidated by a systems biology approach. F1000Res 2015, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Galea, E.; Launay, N.; Portero-Otin, M.; Ruiz, M.; Pamplona, R.; Aubourg, P.; Ferrer, I.; Pujol, A. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: A paradigm for multifactorial neurodegenerative diseases? Biochim. Biophys. Acta 2012, 1822, 1475–1488. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.; Rosenstock, T.R.; Cunha-Oliveira, T.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. Glutathione redox cycle dysregulation in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2012, 53, 1857–1867. [Google Scholar] [CrossRef]
- Namasivayam, V.; Stefan, K.; Silbermann, K.; Pahnke, J.; Wiese, M.; Stefan, S.M. Structural feature-driven pattern analysis for multitarget modulator landscapes. Bioinformatics 2021, 38, 1385–1392. [Google Scholar] [CrossRef]
- Namasivayam, V.; Silbermann, K.; Pahnke, J.; Wiese, M.; Stefan, S.M. Scaffold fragmentation and substructure hopping reveal potential, robustness, and limits of computer-aided pattern analysis (C@PA). Comput. Struct. Biotechnol. J. 2021, 19, 3269–3283. [Google Scholar] [CrossRef]
- Namasivayam, V.; Silbermann, K.; Wiese, M.; Pahnke, J.; Stefan, S.M. C@PA: Computer-Aided Pattern Analysis to Predict Multitarget ABC Transporter Inhibitors. J. Med. Chem. 2021, 64, 3350–3366. [Google Scholar] [CrossRef] [PubMed]
- Lamartiniere, Y.; Boucau, M.C.; Dehouck, L.; Krohn, M.; Pahnke, J.; Candela, P.; Gosselet, F.; Fenart, L. ABCA7 Downregulation Modifies Cellular Cholesterol Homeostasis and Decreases Amyloid-beta Peptide Efflux in an in vitro Model of the Blood-Brain Barrier. J. Alzheimers Dis. 2018, 64, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Bossaerts, L.; Cacace, R.; van Broeckhoven, C. The role of ATP-binding cassette subfamily A in the etiology of Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Huang, Y.; Orth, M.; Gillis, T.; Siciliano, J.; Hong, E.; Mysore, J.S.; Lucente, D.; Wheeler, V.C.; Seong, I.S.; et al. Genetic modifiers of Huntington disease differentially influence motor and cognitive domains. Am. J. Hum. Genet. 2022, 109, 885–899. [Google Scholar] [CrossRef]
- Moss, D.J.H.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Correia, K.; Harold, D.; Kim, K.H.; Holmans, P.; Jones, L.; Orth, M.; Myers, R.H.; Kwak, S.; Wheeler, V.C.; MacDonald, M.E.; et al. The Genetic Modifiers of Motor OnsetAge (GeM MOA) Website: Genome-wide Association Analysis for Genetic Modifiers of Huntington’s Disease. J. Huntingt. Dis. 2015, 4, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Lupton, M.K.; Proitsi, P.; Lin, K.; Hamilton, G.; Daniilidou, M.; Tsolaki, M.; Powell, J.F. The role of ABCA1 gene sequence variants on risk of Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 897–906. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Lefterov, I.; Fitz, N.F.; Cronican, A.; Lefterov, P.; Staufenbiel, M.; Koldamova, R. Memory deficits in APP23/Abca1+/− mice correlate with the level of Abeta oligomers. ASN Neuro 2009, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.; Staufenbiel, M.; Lefterov, I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 2005, 280, 43224–43235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brackhan, M.; Calza, G.; Lundgren, K.; Bascunana, P.; Bruning, T.; Soliymani, R.; Kumar, R.; Abelein, A.; Baumann, M.; Lalowski, M.; et al. Isotope-labeled amyloid-beta does not transmit to the brain in a prion-like manner after peripheral administration. EMBO Rep. 2022, 23, e54405. [Google Scholar] [CrossRef]
- Mohle, L.; Brackhan, M.; Bascunana, P.; Pahnke, J. Dimethyl fumarate does not mitigate cognitive decline and beta-amyloidosis in female APPPS1 mice. Brain Res. 2021, 1768, 147579. [Google Scholar] [CrossRef]
- Upite, J.; Bruning, T.; Mohle, L.; Brackhan, M.; Bascunana, P.; Jansone, B.; Pahnke, J. A New Tool for the Analysis of the Effect of Intracerebrally Injected Anti-Amyloid-beta Compounds. J. Alzheimers Dis. 2021, 84, 1677–1690. [Google Scholar] [CrossRef]
- Rai, S.P.; Krohn, M.; Pahnke, J. Early Cognitive Training Rescues Remote Spatial Memory but Reduces Cognitive Flexibility in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2020, 75, 1301–1317. [Google Scholar] [CrossRef]
- Rai, S.P.; Bascunana, P.; Brackhan, M.; Krohn, M.; Mohle, L.; Paarmann, K.; Pahnke, J. Detection and Prediction of Mild Cognitive Impairment in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2020, 77, 1209–1221. [Google Scholar] [CrossRef]
- Steffen, J.; Krohn, M.; Schwitlick, C.; Bruning, T.; Paarmann, K.; Pietrzik, C.U.; Biverstal, H.; Jansone, B.; Langer, O.; Pahnke, J. Expression of endogenous mouse APP modulates beta-amyloid deposition in hAPP-transgenic mice. Acta Neuropathol. Commun. 2017, 5, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, J.; Krohn, M.; Paarmann, K.; Schwitlick, C.; Bruning, T.; Marreiros, R.; Muller-Schiffmann, A.; Korth, C.; Braun, K.; Pahnke, J. Revisiting rodent models: Octodon degus as Alzheimer’s disease model? Acta Neuropathol. Commun. 2016, 4, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, K.B.; Paxinos, G. Mouse Brain in Stereotaxic Coordinates; Academic Press: Waltham, MA, USA, 1997. [Google Scholar]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Möhle, L.; Brüning, T.; Eiriz, I.; Rafehi, M.; Stefan, K.; Stefan, S.M.; Pahnke, J. A Novel Huntington’s Disease Assessment Platform to Support Future Drug Discovery and Development. Int. J. Mol. Sci. 2022, 23, 14763. https://doi.org/10.3390/ijms232314763
Wu J, Möhle L, Brüning T, Eiriz I, Rafehi M, Stefan K, Stefan SM, Pahnke J. A Novel Huntington’s Disease Assessment Platform to Support Future Drug Discovery and Development. International Journal of Molecular Sciences. 2022; 23(23):14763. https://doi.org/10.3390/ijms232314763
Chicago/Turabian StyleWu, Jingyun, Luisa Möhle, Thomas Brüning, Iván Eiriz, Muhammad Rafehi, Katja Stefan, Sven Marcel Stefan, and Jens Pahnke. 2022. "A Novel Huntington’s Disease Assessment Platform to Support Future Drug Discovery and Development" International Journal of Molecular Sciences 23, no. 23: 14763. https://doi.org/10.3390/ijms232314763