
Calciprotein Particles Cause Physiologically Significant Pro-Inflammatory Response in Endothelial Cells and Systemic Circulation
 , , ,
, , ,
Abstract
1. Introduction
2. Results
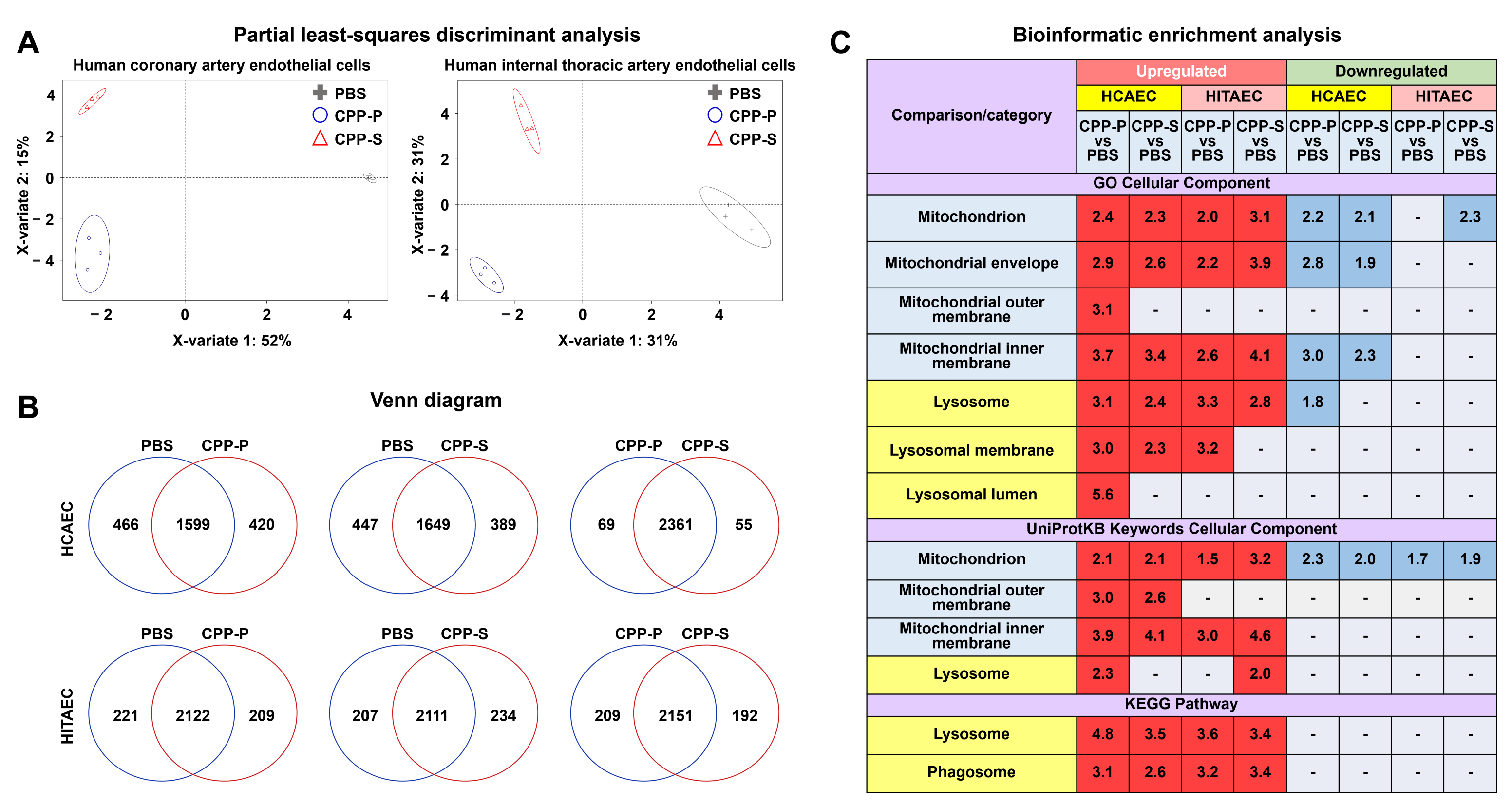
2.1. Proteomic Profiling Reveals a Significant Response of ECs to CPP-P or CPP-S Treatment
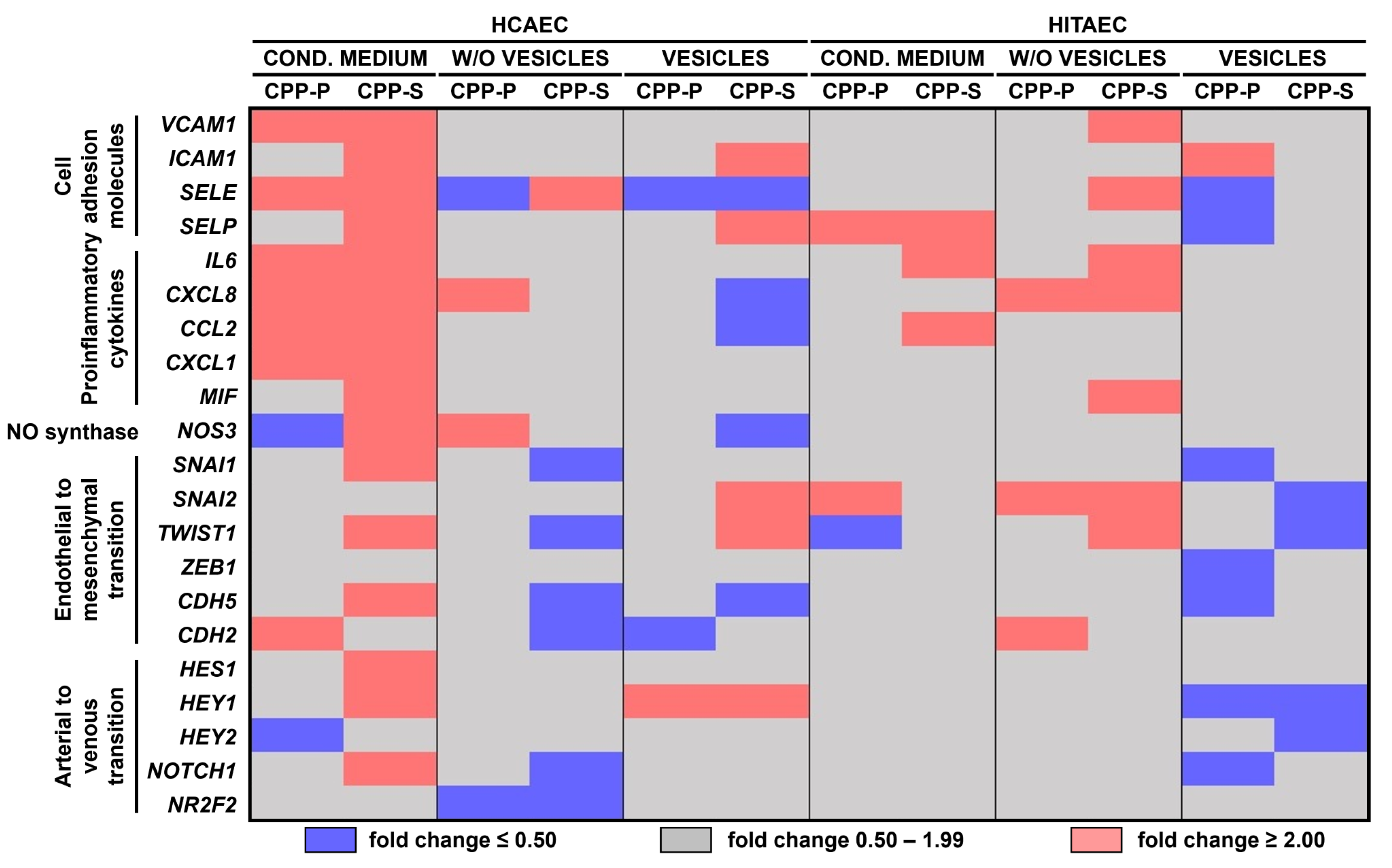
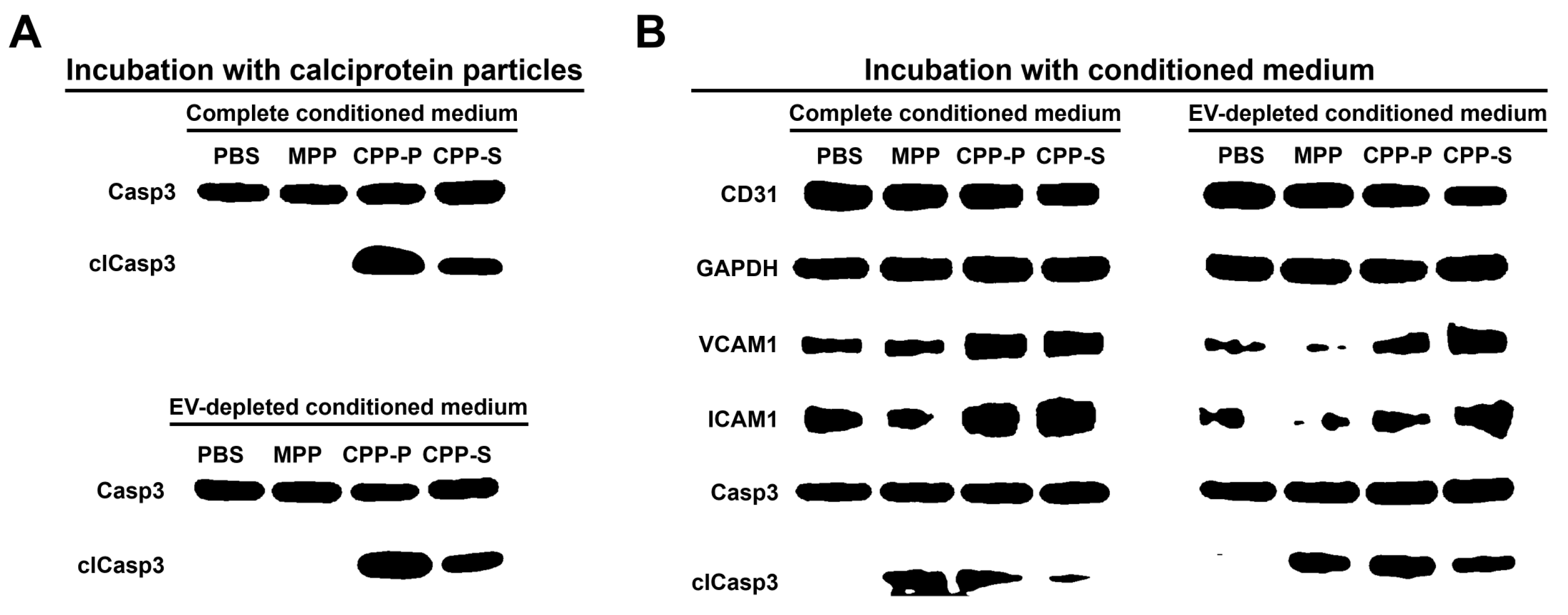
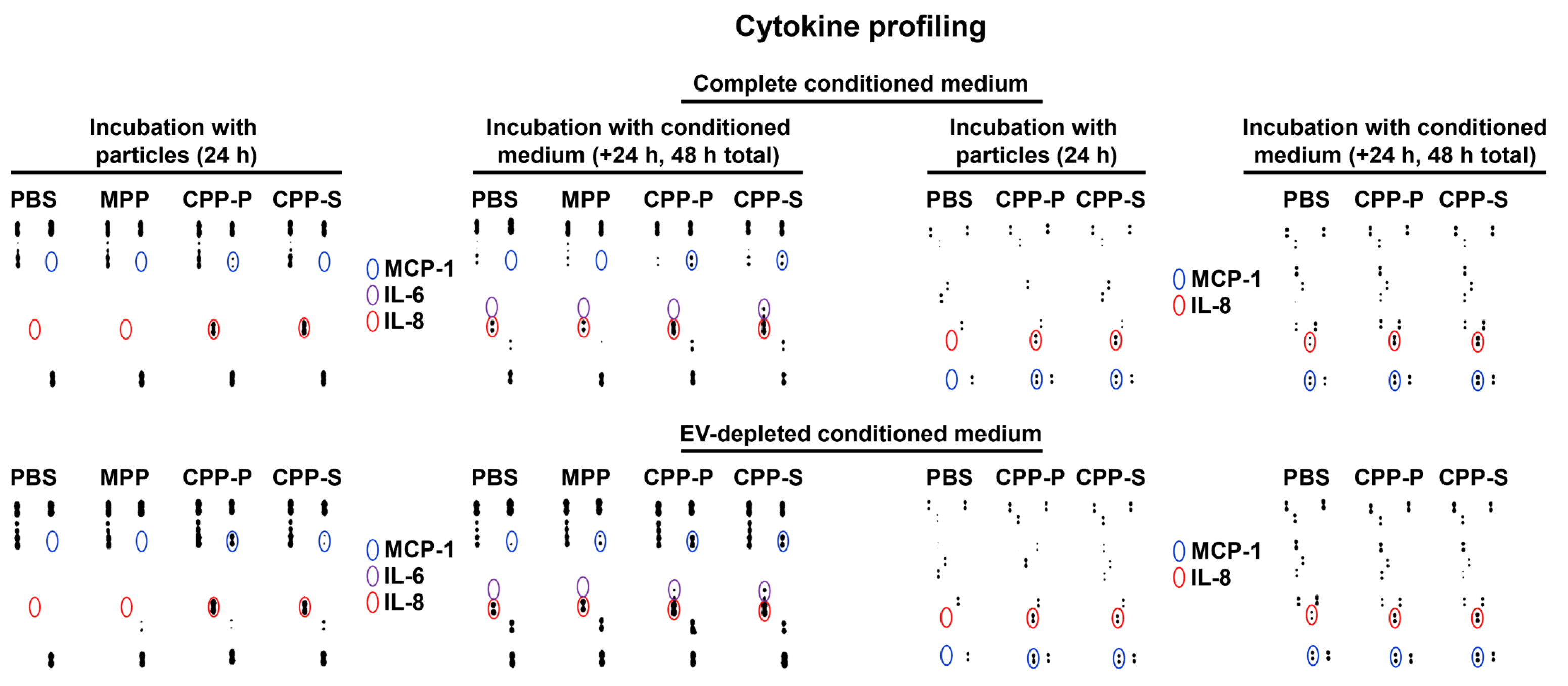
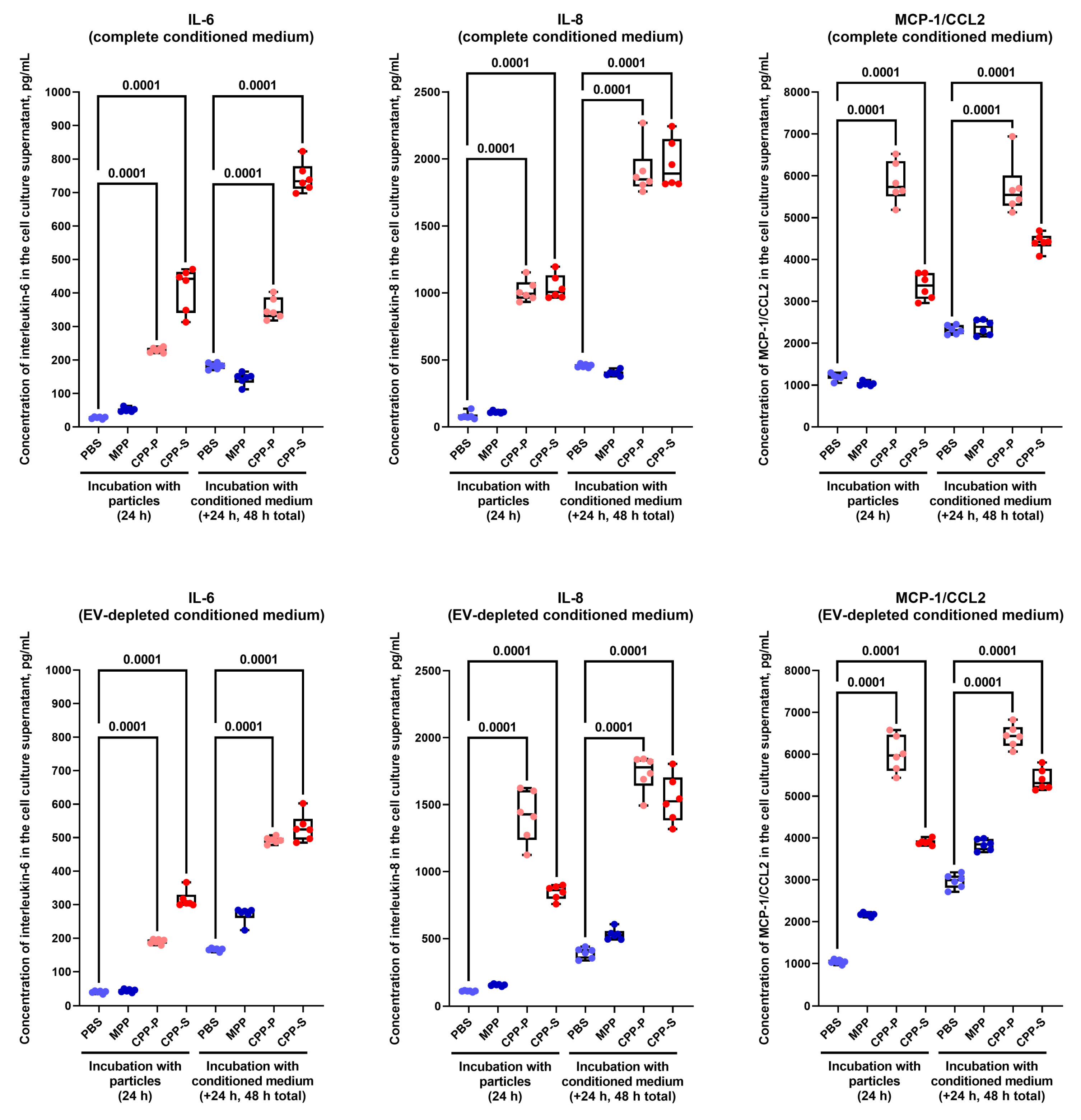
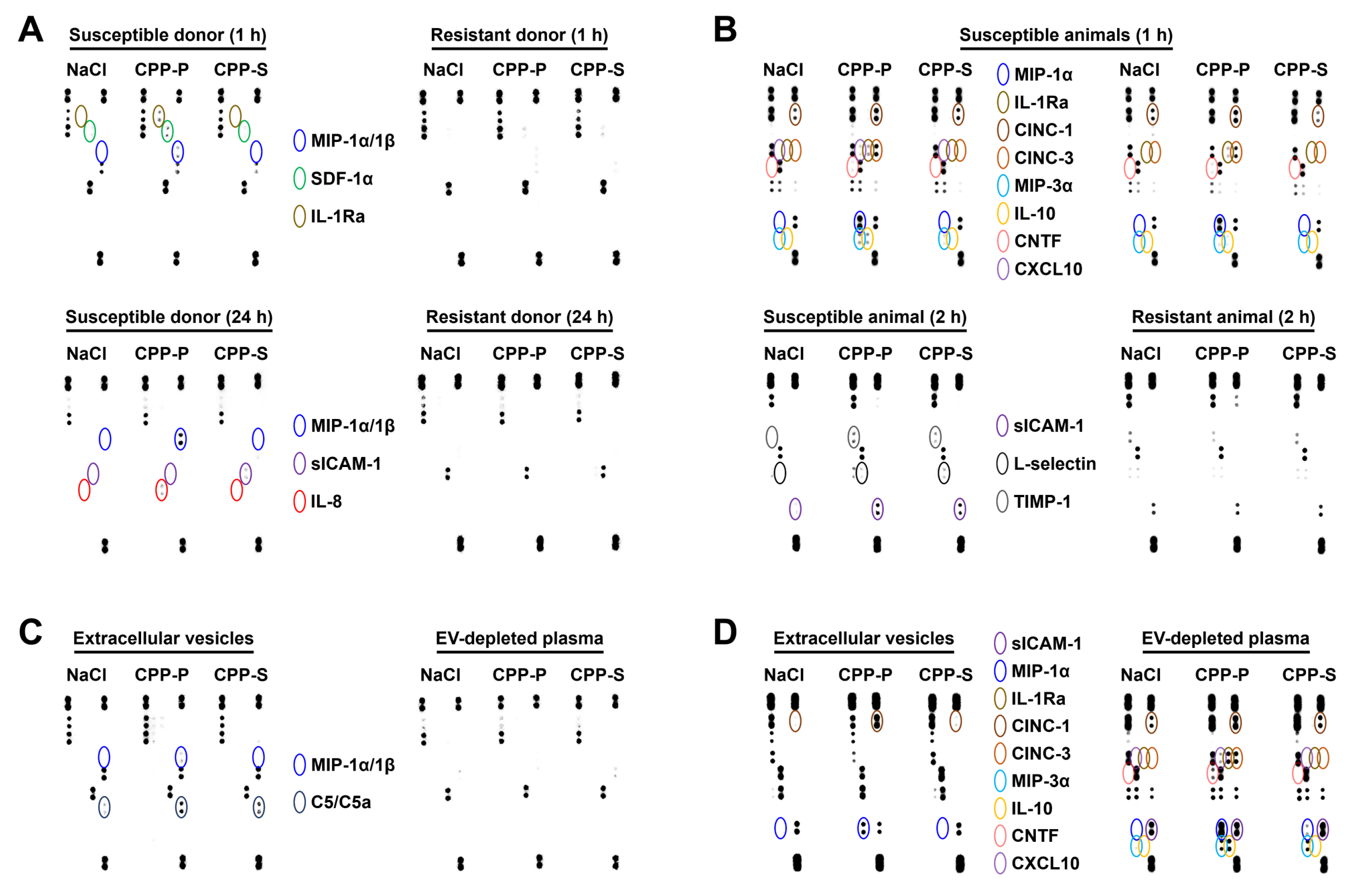
2.2. Pro-Inflammatory Activation of ECs by CPP-P and CPP-S Causes Pathological Paracrine Effects on Intact ECs
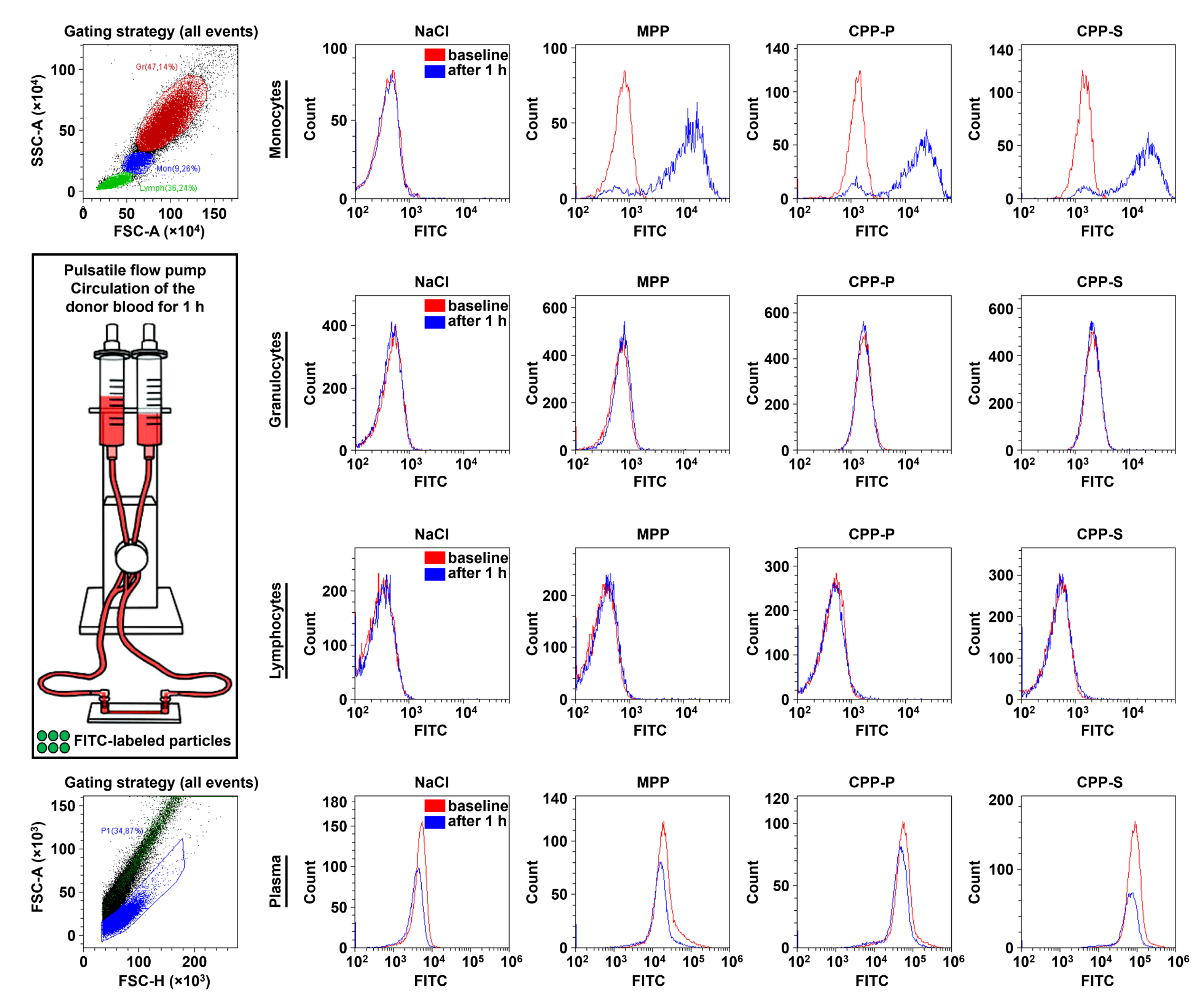
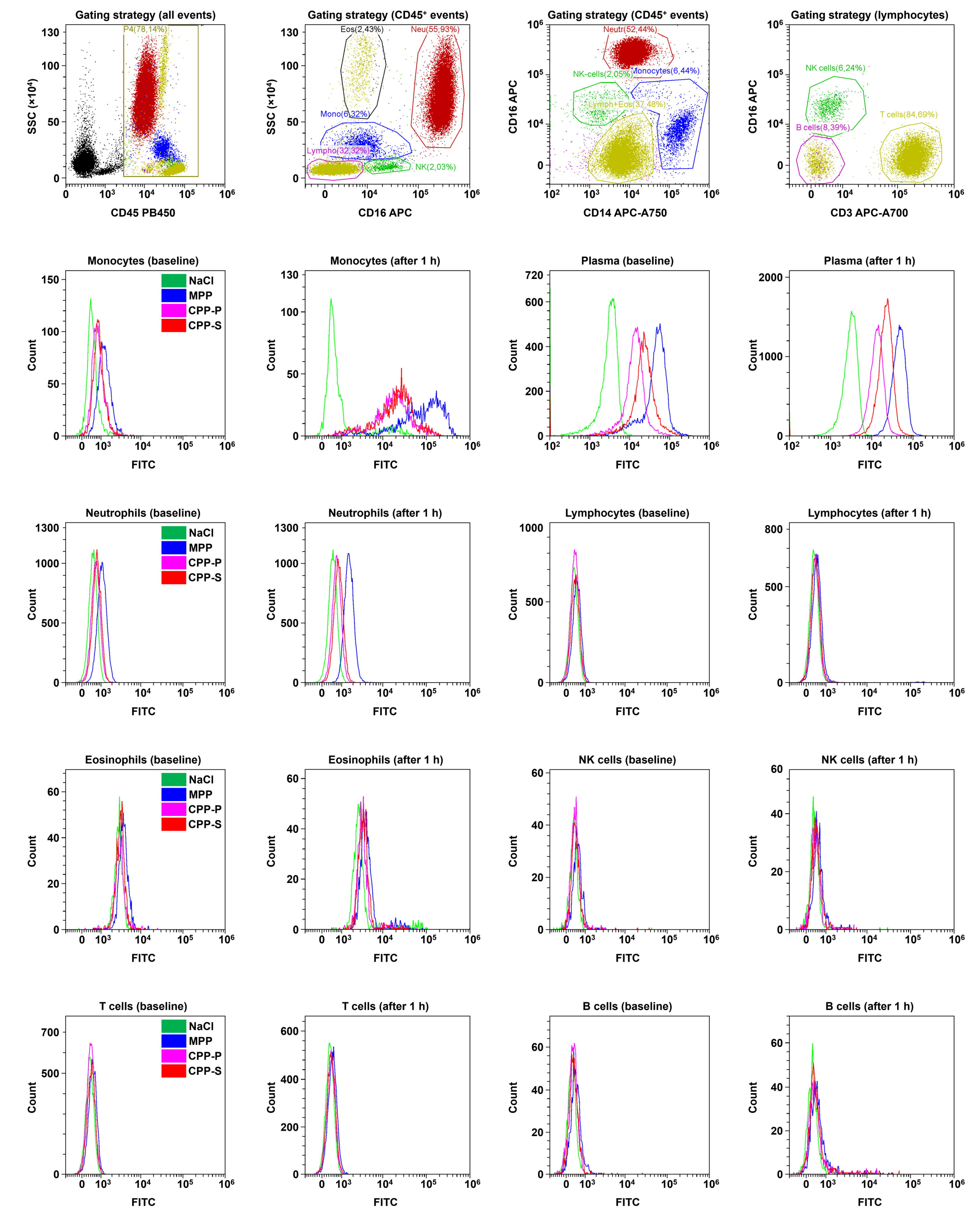
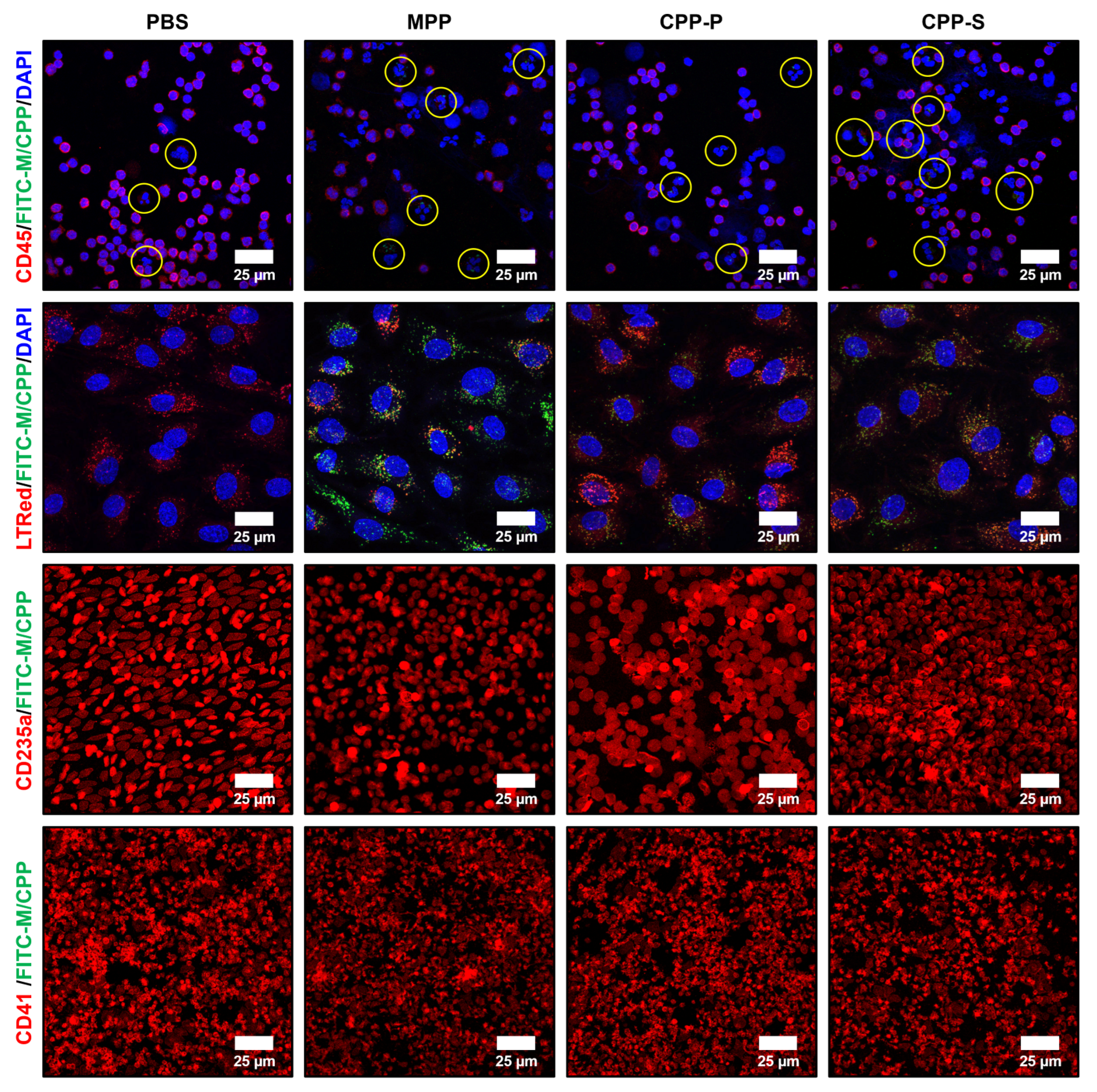
2.3. Circulating CPPs Are Internalised by Monocytes and Trigger a Systemic Inflammatory Response
3. Discussion
4. Materials and Methods
4.1. Artificial Synthesis and Quantification of CPPs
4.2. Cell Culture
4.3. Proteomic Profiling
4.4. Modelling of Endothelial Cell Paracrine Effects
4.5. Molecular Profiling
4.6. Internalisation Assays
4.7. Cytotoxicity and Inflammation Assays
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Jahnen-Dechent, W.; Endo, H.; Schwahn, D. Structural dynamics of a colloidal protein-mineral complex bestowing on calcium phosphate a high solubility in biological fluids. Biointerphases 2007, 2, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Eckert, T.; Aretz, A.; Richtering, W.; van Dorp, W.; Schäfer, C.; Jahnen-Dechent, W. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J. Biol. Chem. 2008, 283, 14815–14825. [Google Scholar] [CrossRef] [PubMed]
- Rochette, C.N.; Rosenfeldt, S.; Heiss, A.; Narayanan, T.; Ballauff, M.; Jahnen-Dechent, W. A shielding topology stabilizes the early stage protein-mineral complexes of fetuin-A and calcium phosphate: A time-resolved small-angle X-ray study. Chembiochem 2009, 10, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Pipich, V.; Jahnen-Dechent, W.; Schwahn, D. Fetuin-A is a mineral carrier protein: Small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys. J. 2010, 99, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Schäfer, C.; Heiss, A.; Gräber, S.; Kinkeldey, A.; Büscher, A.; Schmitt, M.M.; Bornemann, J.; Nimmerjahn, F.; Herrmann, M.; et al. Clearance of fetuin-A—Containing calciprotein particles is mediated by scavenger receptor-A. Circ. Res. 2012, 111, 575–584. [Google Scholar] [CrossRef]
- Köppert, S.; Büscher, A.; Babler, A.; Ghallab, A.; Buhl, E.M.; Latz, E.; Hengstler, J.G.; Smith, E.R.; Jahnen-Dechent, W. Cellular Clearance and Biological Activity of Calciprotein Particles Depend on Their Maturation State and Crystallinity. Front. Immunol. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Koeppert, S.; Ghallab, A.; Peglow, S.; Winkler, C.F.; Graeber, S.; Büscher, A.; Hengstler, J.G.; Jahnen-Dechent, W. Live Imaging of Calciprotein Particle Clearance and Receptor Mediated Uptake: Role of Calciprotein Monomers. Front. Cell Dev. Biol. 2021, 9, 633925. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Velikanova, E.A.; Mukhamadiyarov, R.A.; Glushkova, T.V.; Borisov, V.V.; Matveeva, V.G.; Antonova, L.V.; Filip’ev, D.E.; Golovkin, A.S.; Shishkova, D.K.; et al. Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci. Rep. 2016, 6, 27255. [Google Scholar] [CrossRef]
- Shishkova, D.; Markova, V.; Sinitsky, M.; Tsepokina, A.; Velikanova, E.; Bogdanov, L.; Glushkova, T.; Kutikhin, A. Calciprotein Particles Cause Endothelial Dysfunction under Flow. Int. J. Mol. Sci. 2020, 21, 8802. [Google Scholar] [CrossRef]
- Shishkova, D.K.; Velikanova, E.A.; Bogdanov, L.A.; Sinitsky, M.Y.; Kostyunin, A.E.; Tsepokina, A.V.; Gruzdeva, O.V.; Mironov, A.V.; Mukhamadiyarov, R.A.; Glushkova, T.V.; et al. Calciprotein Particles Link Disturbed Mineral Homeostasis with Cardiovascular Disease by Causing Endothelial Dysfunction and Vascular Inflammation. Int. J. Mol. Sci. 2021, 22, 12458. [Google Scholar] [CrossRef] [PubMed]
- Shishkova, D.; Velikanova, E.; Sinitsky, M.; Tsepokina, A.; Gruzdeva, O.; Bogdanov, L.; Kutikhin, A. Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. Int. J. Mol. Sci. 2019, 20, 5728. [Google Scholar] [CrossRef] [PubMed]
- Jäger, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 2020, 11, 4243. [Google Scholar] [CrossRef] [PubMed]
- Anzai, F.; Karasawa, T.; Komada, T.; Yamada, N.; Miura, Y.; Sampilvanjil, A.; Baatarjav, C.; Fujimura, K.; Matsumura, T.; Tago, K.; et al. Calciprotein Particles Induce IL-1β/α-Mediated Inflammation through NLRP3 Inflammasome-Dependent and -Independent Mechanisms. Immunohorizons 2021, 5, 602–614. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Peng, H.H.; Wu, C.Y.; Young, D.; Martel, J.; Young, A.; Ojcius, D.M.; Lee, Y.H.; Young, J.D. Physicochemical and biological properties of biomimetic mineralo-protein nanoparticles formed spontaneously in biological fluids. Small 2013, 9, 2297–2307. [Google Scholar] [CrossRef]
- Bundy, J.D.; Cai, X.; Mehta, R.C.; Scialla, J.J.; de Boer, I.H.; Hsu, C.Y.; Go, A.S.; Dobre, M.A.; Chen, J.; Rao, P.S.; et al. Serum Calcification Propensity and Clinical Events in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 1562–1571. [Google Scholar] [CrossRef]
- Pruijm, M.; Lu, Y.; Megdiche, F.; Piskunowicz, M.; Milani, B.; Stuber, M.; Bachtler, M.; Vogt, B.; Burnier, M.; Pasch, A. Serum calcification propensity is associated with renal tissue oxygenation and resistive index in patients with arterial hypertension or chronic kidney disease. J. Hypertens. 2017, 35, 2044–2052. [Google Scholar] [CrossRef]
- Mencke, R.; van der Vaart, A.; Pasch, A.; Harms, G.; Waanders, F.; Bilo, H.J.G.; van Goor, H.; Hillebrands, J.L.; van Dijk, P.R. Serum calcification propensity is associated with HbA1c in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2021, 9, e002016. [Google Scholar] [CrossRef]
- Dahdal, S.; Devetzis, V.; Chalikias, G.; Tziakas, D.; Chizzolini, C.; Ribi, C.; Trendelenburg, M.; Eisenberger, U.; Hauser, T.; Pasch, A.; et al. Serum calcification propensity is independently associated with disease activity in systemic lupus erythematosus. PLoS ONE 2018, 13, e0188695. [Google Scholar] [CrossRef]
- Tiong, M.K.; Smith, E.R.; Toussaint, N.D.; Al-Khayyat, H.F.; Holt, S.G. Reduction of Calciprotein Particles in Adults Receiving Infliximab for Chronic Inflammatory Disease. JBMR Plus 2021, 5, e10497. [Google Scholar] [CrossRef] [PubMed]
- Bressendorff, I.; Hansen, D.; Schou, M.; Pasch, A.; Brandi, L. The Effect of Increasing Dialysate Magnesium on Serum Calcification Propensity in Subjects with End Stage Kidney Disease: A Randomized, Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2018, 13, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Eelderink, C.; Zeper, L.W.; Pasch, A.; Bakker, S.J.L.; de Borst, M.H.; Hoenderop, J.G.J.; de Baaij, J.H.F. Calciprotein particle inhibition explains magnesium-mediated protection against vascular calcification. Nephrol. Dial. Transplant. 2020, 35, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Keyzer, C.A.; de Borst, M.H.; van den Berg, E.; Jahnen-Dechent, W.; Arampatzis, S.; Farese, S.; Bergmann, I.P.; Floege, J.; Navis, G.; Bakker, S.J.; et al. Calcification Propensity and Survival among Renal Transplant Recipients. J. Am. Soc. Nephrol. 2016, 27, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Block, G.A.; Bachtler, M.; Smith, E.R.; Jahnen-Dechent, W.; Arampatzis, S.; Chertow, G.M.; Parfrey, P.; Ma, X.; Floege, J. Blood Calcification Propensity, Cardiovascular Events, and Survival in Patients Receiving Hemodialysis in the EVOLVE Trial. Clin. J. Am. Soc. Nephrol. 2017, 12, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Bodenham, E.; McMahon, L.P.; Farese, S.; Rajkumar, C.; Holt, S.G.; Pasch, A. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J. Am. Soc. Nephrol. 2014, 25, 339–348. [Google Scholar] [CrossRef]
- Smith, E.R.; Hewitson, T.D.; Cai, M.M.X.; Aghagolzadeh, P.; Bachtler, M.; Pasch, A.; Holt, S.G. A novel fluorescent probe-based flow cytometric assay for mineral-containing nanoparticles in serum. Sci. Rep. 2017, 7, 5686. [Google Scholar] [CrossRef]
- Kraler, S.; Libby, P.; Evans, P.C.; Akhmedov, A.; Schmiady, M.O.; Reinehr, M.; Camici, G.G.; Lüscher, T.F. Resilience of the Internal Mammary Artery to Atherogenesis: Shifting From Risk to Resistance to Address Unmet Needs. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2237–2251. [Google Scholar] [CrossRef]
- Otsuka, F.; Yahagi, K.; Sakakura, K.; Virmani, R. Why is the mammary artery so special and what protects it from atherosclerosis? Ann. Cardiothorac. Surg. 2013, 2, 519–526. [Google Scholar] [CrossRef]
- Lind, L.; Skarfors, E.; Berglund, L.; Lithell, H.; Ljunghall, S. Serum calcium: A new, independent, prospective risk factor for myocardial infarction in middle-aged men followed for 18 years. J. Clin. Epidemiol. 1997, 50, 967–973. [Google Scholar] [CrossRef]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G.; Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef]
- Foley, R.N.; Collins, A.J.; Ishani, A.; Kalra, P.A. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. Heart J. 2008, 156, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Larsson, T.E.; Olauson, H.; Hagstrom, E.; Ingelsson, E.; Arnlov, J.; Lind, L.; Sundstrom, J. Conjoint effects of serum calcium and phosphate on risk of total, cardiovascular, and noncardiovascular mortality in the community. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 333–339. [Google Scholar] [CrossRef] [PubMed]
- McGovern, A.P.; de Lusignan, S.; van Vlymen, J.; Liyanage, H.; Tomson, C.R.; Gallagher, H.; Rafiq, M.; Jones, S. Serum phosphate as a risk factor for cardiovascular events in people with and without chronic kidney disease: A large community based cohort study. PLoS ONE 2013, 8, e74996. [Google Scholar] [CrossRef]
- Lutsey, P.L.; Alonso, A.; Michos, E.D.; Loehr, L.R.; Astor, B.C.; Coresh, J.; Folsom, A.R. Serum magnesium, phosphorus, and calcium are associated with risk of incident heart failure: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Clin. Nutr. 2014, 100, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, S.; Garmo, H.; Malmstrom, H.; Hammar, N.; Jungner, I.; Walldius, G.; Van Hemelrijck, M. Association between serum calcium concentration and risk of incident and fatal cardiovascular disease in the prospective AMORIS study. Atherosclerosis 2016, 251, 85–93. [Google Scholar] [CrossRef]
- Reid, I.R.; Gamble, G.D.; Bolland, M.J. Circulating calcium concentrations, vascular disease and mortality: A systematic review. J. Intern. Med. 2016, 279, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Campos-Obando, N.; Lahousse, L.; Brusselle, G.; Stricker, B.H.; Hofman, A.; Franco, O.H.; Uitterlinden, A.G.; Zillikens, M.C. Serum phosphate levels are related to all-cause, cardiovascular and COPD mortality in men. Eur. J. Epidemiol. 2018, 33, 859–871. [Google Scholar] [CrossRef]
- Kobylecki, C.J.; Nordestgaard, B.G.; Afzal, S. Plasma Ionized Calcium and Risk of Cardiovascular Disease: 106 774 Individuals from the Copenhagen General Population Study. Clin. Chem. 2021, 67, 265–275. [Google Scholar] [CrossRef]
- Ginsberg, C.; Houben, A.J.H.M.; Malhotra, R.; Berendschot, T.T.J.M.; Dagnelie, P.C.; Kooman, J.P.; Webers, C.A.; Stehouwer, C.D.A.; Ix, J.H. Serum Phosphate and Microvascular Function in a Population-Based Cohort. Clin. J. Am. Soc. Nephrol. 2019, 14, 1626–1633. [Google Scholar] [CrossRef]
- Anderson, T.J.; Charbonneau, F.; Title, L.M.; Buithieu, J.; Rose, M.S.; Conradson, H.; Hildebrand, K.; Fung, M.; Verma, S.; Lonn, E.M. Microvascular function predicts cardiovascular events in primary prevention: Long-term results from the Firefighters and Their Endothelium (FATE) study. Circulation 2011, 123, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Folsom, A.R.; Brancati, F.L. Is low magnesium concentration a risk factor for coronary heart disease? The Atherosclerosis Risk in Communities (ARIC) Study. Am. Heart J. 1998, 136, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Reffelmann, T.; Ittermann, T.; Dorr, M.; Volzke, H.; Reinthaler, M.; Petersmann, A.; Felix, S.B. Low serum magnesium concentrations predict cardiovascular and all-cause mortality. Atherosclerosis 2011, 219, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Joosten, M.M.; Gansevoort, R.T.; Mukamal, K.J.; van der Harst, P.; Geleijnse, J.M.; Feskens, E.J.; Navis, G.; Bakker, S.J.; PREVEND Study Group. Urinary and plasma magnesium and risk of ischemic heart disease. Am. J. Clin. Nutr. 2013, 97, 1299–1306. [Google Scholar] [CrossRef]
- Qu, X.; Jin, F.; Hao, Y.; Li, H.; Tang, T.; Wang, H.; Yan, W.; Dai, K. Magnesium and the risk of cardiovascular events: A meta-analysis of prospective cohort studies. PLoS ONE 2013, 8, e57720. [Google Scholar] [CrossRef]
- Adebamowo, S.N.; Jimenez, M.C.; Chiuve, S.E.; Spiegelman, D.; Willett, W.C.; Rexrode, K.M. Plasma magnesium and risk of ischemic stroke among women. Stroke 2014, 45, 2881–2886. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Khan, H.; Laukkanen, J.A. Serum magnesium and risk of new onset heart failure in men: The Kuopio Ischemic Heart Disease Study. Eur. J. Epidemiol. 2016, 31, 1035–1043. [Google Scholar] [CrossRef]
- Kieboom, B.C.; Niemeijer, M.N.; Leening, M.J.; van den Berg, M.E.; Franco, O.H.; Deckers, J.W.; Hofman, A.; Zietse, R.; Stricker, B.H.; Hoorn, E.J. Serum Magnesium and the Risk of Death From Coronary Heart Disease and Sudden Cardiac Death. J. Am. Heart Assoc. 2016, 5, e002707. [Google Scholar] [CrossRef]
- Wannamethee, S.G.; Papacosta, O.; Lennon, L.; Whincup, P.H. Serum magnesium and risk of incident heart failure in older men: The British Regional Heart Study. Eur. J. Epidemiol. 2018, 33, 873–882. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, M.E.; Gomez-Delgado, F.; Arenas de Larriva, A.P.; Canalejo, A.; Gomez-Luna, P.; Herencia, C.; Lopez-Moreno, J.; Rodriguez, M.; Lopez-Miranda, J.; Almaden, Y. Serum Magnesium is associated with Carotid Atherosclerosis in patients with high cardiovascular risk (CORDIOPREV Study). Sci. Rep. 2019, 9, 8013. [Google Scholar] [CrossRef]
- Rooney, M.R.; Alonso, A.; Folsom, A.R.; Michos, E.D.; Rebholz, C.M.; Misialek, J.R.; Chen, L.Y.; Dudley, S.; Lutsey, P.L. Serum magnesium and the incidence of coronary artery disease over a median 27 years of follow-up in the Atherosclerosis Risk in Communities (ARIC) Study and a meta-analysis. Am. J. Clin. Nutr. 2020, 111, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, Q.; Zhang, H.; Xu, Z.; Wang, X.; Pang, J.; Ma, J.; Ling, W.; Li, D. Associations of serum magnesium levels and calcium-magnesium ratios with mortality in patients with coronary artery disease. Diabetes Metab. 2020, 46, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Y.; Chen, Q.; Li, Q.; Li, Y.; Ling, W. Lower Plasma Fetuin-A Levels Are Associated With a Higher Mortality Risk in Patients With Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2213–2219. [Google Scholar] [CrossRef]
- Peng, W.; Zhang, C.; Wang, Z.; Yang, W. Prediction of all-cause mortality with hypoalbuminemia in patients with heart failure: A meta-analysis. Biomarkers 2019, 24, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.M.; Ran, L.S.; Jiang, J.; Chen, Y.S.; Ji, H.Y.; Quan, X.Q. Association between fetuin-A and prognosis of CAD: A systematic review and meta-analysis. Eur. J. Clin. Investig. 2019, 49, e13091. [Google Scholar] [CrossRef]
- Ronit, A.; Kirkegaard-Klitbo, D.M.; Dohlmann, T.L.; Lundgren, J.; Sabin, C.A.; Phillips, A.N.; Nordestgaard, B.G.; Afzal, S. Plasma Albumin and Incident Cardiovascular Disease: Results From the CGPS and an Updated Meta-Analysis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 473–482. [Google Scholar] [CrossRef]
- Seidu, S.; Kunutsor, S.K.; Khunti, K. Serum albumin, cardiometabolic and other adverse outcomes: Systematic review and meta-analyses of 48 published observational cohort studies involving 1,492,237 participants. Scand. Cardiovasc. J. 2020, 54, 280–293. [Google Scholar] [CrossRef]
- Pignatelli, P.; Farcomeni, A.; Menichelli, D.; Pastori, D.; Violi, F. Serum albumin and risk of cardiovascular events in primary and secondary prevention: A systematic review of observational studies and Bayesian meta-regression analysis. Intern. Emerg. Med. 2020, 15, 135–143. [Google Scholar] [CrossRef]
- Moe, S.M.; Chertow, G.M.; Parfrey, P.S.; Kubo, Y.; Block, G.A.; Correa-Rotter, R.; Drueke, T.B.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis: The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Circulation 2015, 132, 27–39. [Google Scholar] [CrossRef]
- Chung, M.; Tang, A.M.; Fu, Z.; Wang, D.D.; Newberry, S.J. Calcium Intake and Cardiovascular Disease Risk: An Updated Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 165, 856–866. [Google Scholar] [CrossRef]
- Kahwati, L.C.; Weber, R.P.; Pan, H.; Gourlay, M.; LeBlanc, E.; Coker-Schwimmer, M.; Viswanathan, M. Vitamin D, Calcium, or Combined Supplementation for the Primary Prevention of Fractures in Community-Dwelling Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2018, 319, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Barbarawi, M.; Kheiri, B.; Zayed, Y.; Barbarawi, O.; Dhillon, H.; Swaid, B.; Yelangi, A.; Sundus, S.; Bachuwa, G.; Alkotob, M.L.; et al. Vitamin D Supplementation and Cardiovascular Disease Risks in More Than 83 000 Individuals in 21 Randomized Clinical Trials: A Meta-analysis. JAMA Cardiol. 2019, 4, 765–776. [Google Scholar] [CrossRef]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceicao, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Collin-Osdoby, P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ. Res. 2004, 95, 1046–1057. [Google Scholar] [CrossRef]
- Yasuda, H. Discovery of the RANKL/RANK/OPG system. J. Bone Miner. Metab. 2021, 39, 2–11. [Google Scholar] [CrossRef]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; McKee, M.D.; Liaw, L.; Almeida, M.; Yang, H.; Giachelli, C.M. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am. J. Pathol. 2002, 161, 2035–2046. [Google Scholar] [CrossRef]
- Speer, M.Y.; McKee, M.D.; Guldberg, R.E.; Liaw, L.; Yang, H.Y.; Tung, E.; Karsenty, G.; Giachelli, C.M. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: Evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J. Exp. Med. 2002, 196, 1047–1055. [Google Scholar] [CrossRef]
- Paloian, N.J.; Leaf, E.M.; Giachelli, C.M. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int. 2016, 89, 1027–1036. [Google Scholar] [CrossRef]
- Busch, E.; Hohenester, E.; Timpl, R.; Paulsson, M.; Maurer, P. Calcium affinity, cooperativity, and domain interactions of extracellular EF-hands present in BM-40. J. Biol. Chem. 2000, 275, 25508–25515. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Viegas, C.S.; Rafael, M.S.; Enriquez, J.L.; Teixeira, A.; Vitorino, R.; Luis, I.M.; Costa, R.M.; Santos, S.; Cavaco, S.; Neves, J.; et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Rafael, M.S.; Cavaco, S.; Viegas, C.S.; Santos, S.; Ramos, A.; Willems, B.A.; Herfs, M.; Theuwissen, E.; Vermeer, C.; Simes, D.C. Insights into the association of Gla-rich protein and osteoarthritis, novel splice variants and γ-carboxylation status. Mol. Nutr. Food Res. 2014, 58, 1636–1646. [Google Scholar] [CrossRef]
- Tesfamariam, B. Involvement of Vitamin K-Dependent Proteins in Vascular Calcification. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 323–333. [Google Scholar] [CrossRef]
- Shioi, A.; Morioka, T.; Shoji, T.; Emoto, M. The Inhibitory Roles of Vitamin K in Progression of Vascular Calcification. Nutrients 2020, 12, 583. [Google Scholar] [CrossRef]
- Xiao, H.; Chen, J.; Duan, L.; Li, S. Role of emerging vitamin K-dependent proteins: Growth arrest-specific protein 6, Gla-rich protein and periostin (Review). Int. J. Mol. Med. 2021, 47, 2. [Google Scholar] [CrossRef]
- Jahnen-Dechent, W.; Heiss, A.; Schäfer, C.; Ketteler, M. Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Feenstra, L.; Kostyunin, A.E.; Yuzhalin, A.E.; Hillebrands, J.L.; Krenning, G. Calciprotein Particles: Balancing Mineral Homeostasis and Vascular Pathology. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1607–1624. [Google Scholar] [CrossRef]
- Liang, J.J.; Fraser, I.D.C.; Bryant, C.E. Lipid regulation of NLRP3 inflammasome activity through organelle stress. Trends Immunol. 2021, 42, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.E.; Iruela-Arispe, M.L. The many flavors of monocyte/macrophage--endothelial cell interactions. Curr. Opin. Hematol. 2020, 27, 181–189. [Google Scholar] [CrossRef]
- Atehortúa, L.; Rojas, M.; Vásquez, G.M.; Castaño, D. Endothelial Alterations in Systemic Lupus Erythematosus and Rheumatoid Arthritis: Potential Effect of Monocyte Interaction. Mediators Inflamm. 2017, 2017, 9680729. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 2011, 12, 35. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G. Impute: Imputation for Microarray Data, R package version 1.70.0; R package: Vienna, Austria, 2022.
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling, R package version 1.14.0; R package: Vienna, Austria, 2022.
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA—Efficient Multi-Omics Comparative Pathway Analysis. Mol. Cell. Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison/Category (Fold Change and Number of Observed and Expected Differentially Expressed Proteins) | Upregulated | Downregulated | ||||||
|---|---|---|---|---|---|---|---|---|
| HCAEC | HITAEC | HCAEC | HITAEC | |||||
| CPP-P vs. PBS | CPP-S vs. PBS | CPP-P vs. PBS | CPP-S vs. PBS | CPP-P vs. PBS | CPP-S vs. PBS | CPP-P vs. PBS | CPP-S vs. PBS | |
| GO Biological Process | ||||||||
| Nitrogen compound metabolic process | 1.49 191 vs. 129.39 | 1.57 188 vs. 119.51 | - | - | 1.55 222 vs. 143.29 | 1.67 230 vs. 137.73 | 1.47 100 vs. 67.94 | 1.64 105 vs. 63.92 |
| Cellular nitrogen compound metabolic process | - | - | - | - | 2.27 163 vs. 71.94 | 2.40 166 vs. 69.15 | 2.08 71 vs. 34.11 | 2.31 74 vs. 32.09 |
| Cellular nitrogen compound catabolic process | - | - | - | - | 2.63 19 vs. 7.23 | 2.59 18 vs. 6.95 | - | - |
| Cellular nitrogen compound biosynthetic process | - | 1.78 43 vs. 24.22 | - | - | 2.34 68 vs. 29.04 | 2.04 57 vs. 27.91 | 2.18 30 vs. 13.77 | 2.62 34 vs. 12.96 |
| Regulation of nitrogen compound metabolic process | - | - | - | - | 1.31 164 vs. 124.75 | 1.32 158 vs. 119.91 | - | - |
| Negative regulation of nitrogen compound metabolic process | - | - | - | - | 1.51 81 vs. 53.62 | 1.59 82 vs. 51.54 | 1.77 45 vs. 25.42 | - |
| Organonitrogen compound metabolic process | 1.71 167 vs. 97.39 | 1.78 160 vs. 89.95 | - | - | 1.33 143 vs. 107.85 | - | - | 1.62 78 vs. 48.11 |
| Organonitrogen compound catabolic process | 2.01 42 vs. 20.93 | 1.97 38 vs. 19.34 | - | - | - | - | - | - |
| Organonitrogen compound biosynthetic process | 2.44 65 vs. 22.43 | 2.68 63 vs. 23.49 | - | - | 2.38 67 vs. 28.16 | 1.99 54 vs. 27.07 | 2.25 30 vs. 13.35 | 2.39 30 vs. 12.56 |
| Nitrogen compound transport | 2.18 70 vs. 32.10 | 2.19 65 vs. 29.65 | - | - | 1.97 70 vs. 35.55 | 1.84 63 vs. 34.17 | - | 2.14 34 vs. 15.86 |
| Cellular response to nitrogen compound | 2.04 26 vs. 12.74 | - | - | - | - | - | - | - |
| Response to nitrogen compound | 2.02 44 vs. 21.79 | 1.99 40 vs. 20.13 | - | - | - | - | - | - |
| Response to organonitrogen compound | 2.00 40 vs. 20.02 | 1.95 36 vs. 18.49 | - | - | - | - | - | - |
| Response to hydrogen peroxide | - | 3.96 9 vs. 2.27 | - | - | - | - | - | - |
| Response to reactive oxygen species | - | 4.01 14 vs. 3.50 | - | - | - | - | - | - |
| Response to oxygen-containing compound | 1.69 54 vs. 31.88 | 1.66 49 vs. 29.45 | 2.08 33 vs. 15.90 | - | - | - | - | - |
| Response to oxidative stress | 2.44 19 vs. 7.77 | 3.62 27 vs. 7.18 | - | - | - | - | - | - |
| Cellular response to reactive oxygen species | - | 3.89 9 vs. 2.31 | - | - | - | - | - | - |
| Cellular response to oxidative stress | - | 3.56 15 vs. 4.21 | - | - | - | - | - | - |
| Cellular respiration | 4.10 15 vs. 3.66 | 3.84 13 vs. 3.38 | - | 5.40 11 vs. 2.04 | - | - | - | - |
| Energy derivation by oxidation of organic compounds | 3.01 15 vs. 4.98 | 3.26 15 vs. 4.60 | - | 4.33 12 vs. 2.77 | - | - | - | - |
| Macroautophagy | - | - | - | - | 4.55 16 vs. 3.51 | - | - | - |
| Regulation of macroautophagy | 4.53 13 vs. 2.87 | 4.53 12 vs. 2.65 | - | - | - | - | - | - |
| Response to wounding | 3.22 28 vs. 8.71 | 3.48 28 vs. 8.04 | 3.68 16 vs. 4.34 | 3.30 16 vs. 4.84 | - | - | - | - |
| Response to stress | 1.62 110 vs. 67.87 | 1.72 108 vs. 62.69 | 1.77 60 vs. 33.85 | - | - | - | - | - |
| Response to endoplasmic reticulum stress | 3.17 14 vs. 4.41 | 3.19 13 vs. 4.08 | - | - | - | - | - | - |
| Response to hypoxia | 3.03 18 vs. 5.94 | 3.46 19 vs. 5.49 | - | 3.63 12 vs. 3.30 | - | - | - | - |
| Vacuolar acidification | - | - | 16.42 4 vs. 0.24 | - | - | - | - | - |
| Regulation of pH | - | - | 6.43 6 vs. 0.93 | - | - | - | - | - |
| Regulation of release of cytochrome c from mitochondria | - | - | - | 9.21 5 vs. 0.54 | - | - | - | - |
| Positive regulation of epithelial to mesenchymal transition | - | - | 10.06 5 vs. 0.50 | 9.02 5 vs. 0.55 | - | - | - | - |
| Regulation of peptidase activity | 2.28 21 vs. 9.20 | 2.71 23 vs. 8.49 | - | 3.91 20 vs. 5.11 | - | - | - | - |
| Negative regulation of peptidase activity | 3.28 17 vs. 5.19 | 3.76 18 vs. 4.79 | 3.86 10 vs. 2.59 | 4.16 12 vs. 2.88 | - | - | - | - |
| Regulation of endopeptidase activity | 2.32 20 vs. 8.63 | 2.76 22 vs. 7.97 | - | 3.75 18 vs. 4.80 | - | - | - | - |
| Negative regulation of endopeptidase activity | 3.38 17 vs. 5.03 | 3.88 18 vs. 4.64 | 3.86 10 vs. 2.59 | 3.94 11 vs. 2.79 | - | - | - | - |
| Regulation of hydrolase activity | 1.81 38 vs. 21.02 | 1.91 37 vs. 19.41 | - | 2.99 35 vs. 11.69 | - | - | - | - |
| Negative regulation of hydrolase activity | 2.80 21 vs. 7.51 | 3.17 22 vs. 6.93 | - | 3.59 15 vs. 4.17 | - | - | - | - |
| Positive regulation of hydrolase activity | - | - | - | 2.86 19 vs. 6.65 | - | - | - | - |
| Regulation of proteolysis | 2.40 36 vs. 15.01 | 2.67 37 vs. 13.87 | 2.67 20 vs. 7.49 | 3.23 27 vs. 8.35 | - | - | - | - |
| Negative regulation of proteolysis | 3.01 21 vs. 6.98 | 3.41 22 vs. 6.45 | 3.45 12 vs. 3.48 | 3.87 15 vs. 3.88 | - | - | - | - |
| GO Molecular Function | ||||||||
| Exopeptidase activity | 4.73 10 vs. 2.12 | 5.12 10 vs. 1.95 | 6.63 7 vs. 1.06 | - | - | - | - | - |
| Hydrolase activity | 1.53 74 vs. 48.28 | 1.64 73 vs. 44.59 | - | - | - | - | - | - |
| Metallopeptidase activity | 3.31 13 vs. 3.93 | 3.31 12 vs. 3.63 | - | - | - | - | - | - |
| Metalloexopeptidase activity | - | - | 8.10 6 vs. 0.74 | - | - | - | - | - |
| Calcium ion binding | 2.29 34 vs. 14.85 | - | 2.70 20 vs. 7.41 | 2.91 24 vs. 8.26 | - | - | - | - |
| Reactome pathways | ||||||||
| Vpr-mediated induction of apoptosis by mitochondrial outer membrane permeabilisation | 49.15 3 vs. 0.06 | 53.22 3 vs. 0.06 | - | - | - | - | - | - |
| Response to elevated cytosolic Ca2+ | 8.94 24 vs. 2.69 | 8.87 22 vs. 2.48 | 9.70 13 vs. 1.34 | 8.71 13 vs. 1.49 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishkova, D.; Lobov, A.; Zainullina, B.; Matveeva, V.; Markova, V.; Sinitskaya, A.; Velikanova, E.; Sinitsky, M.; Kanonykina, A.; Dyleva, Y.; et al. Calciprotein Particles Cause Physiologically Significant Pro-Inflammatory Response in Endothelial Cells and Systemic Circulation. Int. J. Mol. Sci. 2022, 23, 14941. https://doi.org/10.3390/ijms232314941
Shishkova D, Lobov A, Zainullina B, Matveeva V, Markova V, Sinitskaya A, Velikanova E, Sinitsky M, Kanonykina A, Dyleva Y, et al. Calciprotein Particles Cause Physiologically Significant Pro-Inflammatory Response in Endothelial Cells and Systemic Circulation. International Journal of Molecular Sciences. 2022; 23(23):14941. https://doi.org/10.3390/ijms232314941
Chicago/Turabian StyleShishkova, Daria, Arseniy Lobov, Bozhana Zainullina, Vera Matveeva, Victoria Markova, Anna Sinitskaya, Elena Velikanova, Maxim Sinitsky, Anastasia Kanonykina, Yulia Dyleva, and et al. 2022. "Calciprotein Particles Cause Physiologically Significant Pro-Inflammatory Response in Endothelial Cells and Systemic Circulation" International Journal of Molecular Sciences 23, no. 23: 14941. https://doi.org/10.3390/ijms232314941
APA StyleShishkova, D., Lobov, A., Zainullina, B., Matveeva, V., Markova, V., Sinitskaya, A., Velikanova, E., Sinitsky, M., Kanonykina, A., Dyleva, Y., & Kutikhin, A. (2022). Calciprotein Particles Cause Physiologically Significant Pro-Inflammatory Response in Endothelial Cells and Systemic Circulation. International Journal of Molecular Sciences, 23(23), 14941. https://doi.org/10.3390/ijms232314941