
Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ER Morphology and Functions
2.1. Morphology of the ER
2.2. Functions of the ER
2.2.1. Protein Processing
2.2.2. Lipid Synthesis
2.2.3. Transfer of Molecules from the ER to Other Cellular Compartments
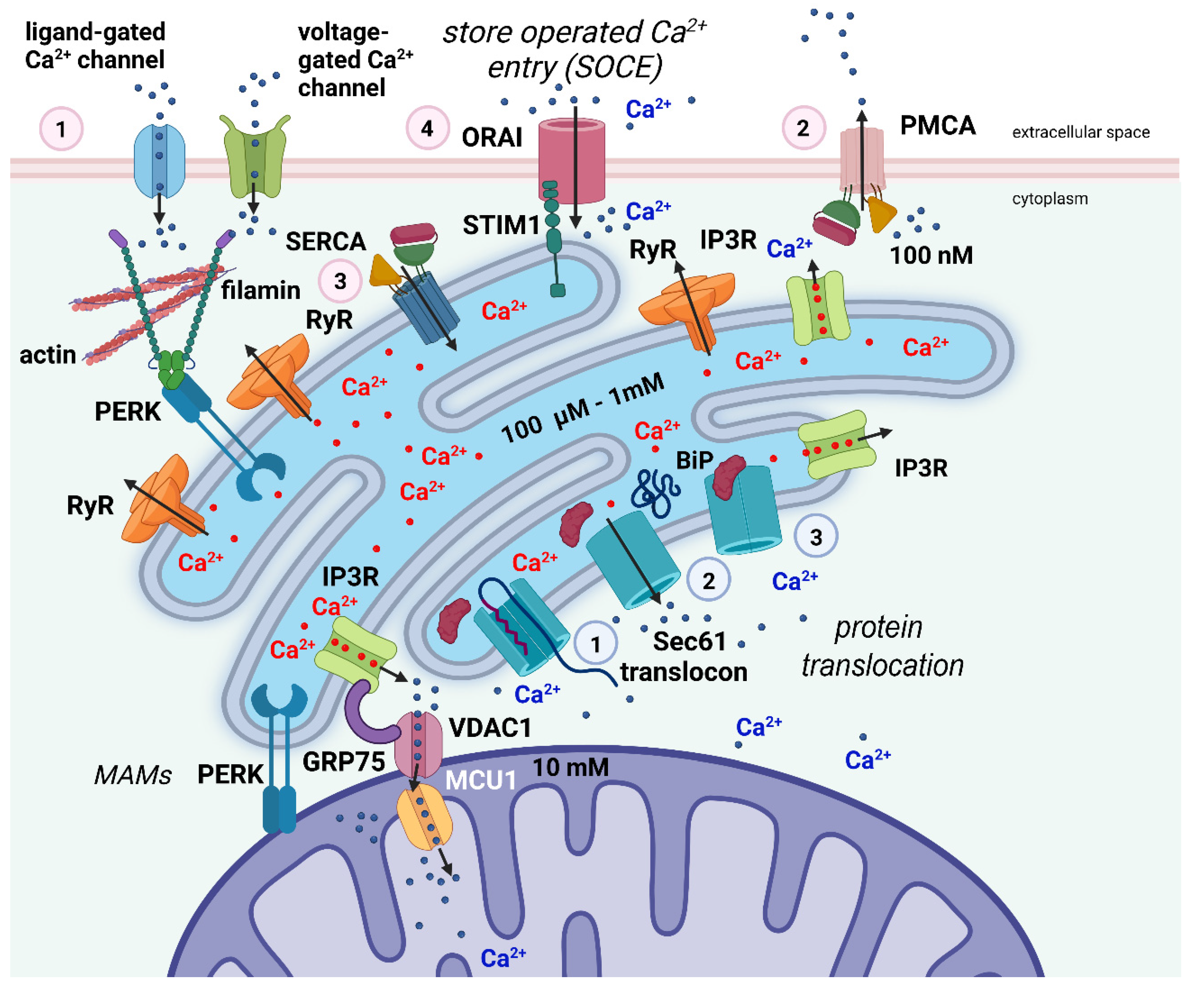
2.2.4. Regulation of Ca2+ Homeostasis
3. ER Stress
3.1. ER Stress and the Unfolded Protein Response
3.2. Effects of UPR Activation
3.3. ER Stress in Neurodegenerative Diseases
4. Death Modes Activated by ER Stress
4.1. Apoptosis
4.1.1. General Concepts
4.1.2. Caspases
4.1.3. BCL-2 Proteins
4.1.4. Apoptotic Pathways
Intrinsic Apoptosis
Extrinsic Apoptosis
Regulation of CASP8 Activity and DISC Formation
The Execution Phase of Apoptosis
4.2. Pathways Linking Apoptosis and ER Stress
4.3. Necrosis and Necroptosis
4.4. Pathways Linking Necroptosis and ER Stress
4.5. Autophagy
4.6. Pathways Linking Autophagy and ER Stress
4.7. Ferroptosis
4.8. Pathways Linking Ferroptosis and ER Stress
5. Adaptive Reticulum Stress and Its Relationship to Ca2+ Dynamics
6. The Cerebellar Granule Cells (CGCs) as a Model to Study the Link between ER Stress and Cell Death in Neurons
6.1. ER Stress and Apoptosis in CGCs

6.2. ER Stress and Autophagy in CGCs
7. Conclusions and Further Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-APB | 2-aminoethoxydiphenyl borate |
| 6-OHDA | 6-hydroxydopamine |
| AIF | apoptosis-inducing factor |
| AKT | protein kinase B (or PKB) |
| ALS | amyotrophic lateral sclerosis |
| Apaf1 | apoptosis protease-activated factor 1 |
| ATF4 | activating transcription factor 4 |
| ATF6 | activating transcription factor 6 |
| ATF6f | cytosolic activating transcription factor 6 |
| BH | BCL2-homology |
| BID | BH3 interacting domain death agonist |
| BiP | binding immunoglobulin protein (gene GRP78) |
| BSG | basigin |
| CAMK | Ca2+/calmodulin-dependent protein kinase |
| CASP1 | cysteinyl aspartate-specific protease 1 (or caspase 1, interleukin converting enzyme 1, ICE) |
| CASP10 | cysteinyl aspartate-specific protease 10 (or caspase 10) |
| CASP3 | cysteinyl aspartate-specific protease 3 (or caspase 3) |
| CASP7 | cysteinyl aspartate-specific protease 7 (or caspase 7) |
| CASP8 | cysteinyl aspartate-specific protease 8 (or caspase 8, FLICE, MASH, Mch5) |
| c-FLIP | cellular FLICE inhibitory protein |
| CGC | cerebellar granule cell |
| CHOP | CAAT/enhancer-binding protein (C/EBP) homologous protein |
| cIAP1 | cellular inhibitor of apoptosis protein 1 |
| cIAP2 | cellular inhibitor of apoptosis protein 2 |
| CICR | Ca2+-induced Ca2+ release |
| DISC | death-inducing signaling complex |
| EDAR | ectodysplasin A receptor |
| eIF2α | eukaryotic translation initiation factor 2α |
| ER | endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| FADD | FAS-associated protein with death domain |
| FAS | FS-7-associated surface antigen |
| FasL | FS-7-associated surface antigen ligand |
| FasR | FS-7-associated surface antigen receptor (or CD95 or APO-1) |
| GADD3 | growth arrest and DNA-damage-inducible 34 |
| GM1 | monosialoganglioside 1 |
| GRP78 | glucose-regulated protein 78 |
| GPX4 | glutathione peroxidase 4 |
| GSK3/3β | glycogen synthase kinase 3/3β |
| GSNO | S-nitrosoglutathione |
| GTP | guanosine triphosphate |
| IAPs | proteins that inhibit apoptosis |
| ICAD | caspase-activated DNase inhibitor |
| IP3R | inositol-3phosphate receptor |
| IRE1α | inositol-requiring enzyme 1 α (or RIDD) |
| JNK | jun amino-terminal kinase (or JNK-46 or JNK1or JNK1A2) |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAM | mitochondria-associated membrane |
| MANF | mesencephalic astrocyte-derived neurotrophic factor |
| MAPK | mitogen-activated protein kinase |
| MCU1 | mitochondrial Ca2+ uniporter 1 |
| MFN2 | mitofusin-2 |
| MKP-1 | mitogen-activated protein kinase phosphatase 1 |
| MLKL | mixed lineage kinase domain-like pseudokinase |
| MOMP | mitochondrial outer membrane permeabilization |
| MPT | mitochondrial permeability transition |
| mTOR | mammalian target of rapamycin |
| NF-Κb | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | nitric oxide |
| NOXA | phorbol-12-myristate-13-acetate-induced protein 1 |
| NPTN | neuroplastin protein |
| ORAI1 | calcium release-activated calcium channel protein 1 |
| p75NTR | p75 neurotrophin receptor |
| p-eIF2α | phospho-eIF2α |
| PERK | protein kinase RNA-like (PKR-like) endoplasmic reticulum kinase |
| PI3K | phosphoinositide 3-kinase |
| PCD | programmed cell death |
| PMCA | plasma membrane Ca2+ selective ATPases |
| PT | permeability transition |
| PUFA-PLs | polyunsaturated fatty acyl moieties in phospholipids |
| PUMA | p53 upregulated modulator of apoptosis (or BC3, JFY-1, BCL-2 binding component 3) |
| RER | rough endoplasmic reticulum |
| RIPIK1 | receptor-interacting serine/threonine kinase 1 |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| SER | smooth endoplasmic reticulum |
| SERCA | endoplasmic reticulum Ca2+-ATPase |
| SMAC/DIABLO | second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low PI |
| SOCE | store-operated Ca2+ entry |
| STARD3 | StAR (steroidogenic acute regulatory protein)-related lipid transfer domain-3 STARD3NL STARD3 N-terminal like |
| STIM1 | stromal interaction molecule 1 |
| TASK | TWIK-related acid-sensitive K+ |
| TNFR1 | tumor necrosis factor receptor 1 (or TNFRSF1A) |
| TNFRSF21 | tumor necrosis factor receptor superfamily21 (or DR6) |
| TNFRSF25 | tumor necrosis factor receptor superfamily 25 (or DR3) |
| TRAF2 | TNF receptor-associated factor 2 |
| TRAILR1 | TNF-related apoptosis-inducing ligand receptor 1 (or TNFRSF10A, DR4) |
| TRAILR2 | TNF-related apoptosis-inducing ligand receptor 2 (or TNFRSF10B, DR5) |
| UPR | unfolded protein response |
| VDAC1 | voltage-dependent anion channel 1 |
| XIAP | X-linked inhibitor of apoptosis protein |
| XIAP | X-linked inhibitor of apoptosis protein (or API3, BIRC4, IAP-3, ILP1, MIHA, XLP2) |
| XKR8 | XK-related protein 8 |
References
- Porter, K.R.; Claude, A.; Fullam, E.F. A study of tissue culture cells by electron microscopy: Methods and preliminary observations. J. Exp. Med. 1945, 81, 233–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palade, G.E.; Porter, K.R. Studies on the endoplasmic reticulum. I. Its identification in cells in situ. J. Exp. Med. 1954, 100, 641–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function, and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, P.; Demaurex, N. Redox regulation of store-operated Ca2+ entry. Antioxid. Redox Signal 2014, 21, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.; Khatiwada, S.; Costa-Mattioli, M.; Hetz, C. ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci. 2018, 41, 610–624. [Google Scholar] [CrossRef]
- Laguesse, S.; Creppe, C.; Nedialkova, D.D.; Prévot, P.-P.; Borgs, L.; Huysseune, S.; Franco, B.; Duysens, G.; Krusy, N.; Lee, G.; et al. A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev. Cell 2015, 35, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Mimeault, M. New advances on structural and biological functions of ceramide in apoptotic/necrotic cell death and cancer. FEBS Lett. 2002, 530, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Segal, M. Dendritic spines and long-term plasticity. Nat. Rev. Neurosci. 2005, 6, 277–284. [Google Scholar] [CrossRef]
- Ramírez, O.A.; Couve, A. The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol. 2011, 21, 219–227. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Belforte, J.E.; Lu, Y.; Yabe, Y.; Pickel, J.; Smith, C.B.; Je, H.S.; Lu, B.; Nakazawa, K. eIF2α phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J. Neurosci. 2010, 30, 2582–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, K.Y.; Danilova, T.; Domanskyi, A.; Saarma, M.; Lindahl, M.; Airavaara, M. MANF is essential for neurite extension and neuronal migration in the developing cortex. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Sims, S.G.; Cisney, R.N.; Lipscomb, M.M.; Meares, G.P. The role of endoplasmic reticulum stress in astrocytes. Glia 2022, 70, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, M.; Yin, H. Signaling pathways involved in endoplasmic reticulum stress-induced neuronal apoptosis. Int. J. Neurosci. 2013, 123, 155–162. [Google Scholar] [CrossRef]
- Mattson, M.P.; LaFerla, F.M.; Chan, S.L.; Leissring, M.A.; Shepel, P.N.; Geiger, J.D. Calcium signaling in the ER: Its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000, 23, 222–229. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Borgese, N.; Francolini, M.; Snapp, E. Endoplasmic reticulum architecture: Structures in flux. Curr. Opin. Cell Biol. 2006, 18, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulmay, A.; Prinz, W.A. Lipid transfer and signaling at organelle contact sites: The tip of the iceberg. Curr. Opin. Cell Biol. 2011, 23, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Zhu, L.; Balogi, Z.; Stefan, C.; Pleiss, J.A.; Emr, S.D. Mdm1/Snx13 is a novel ER-endolysosomal interorganelle tethering protein. J. Cell Biol. 2015, 210, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy 2010, 6, 301–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, T.; Yamamoto, M.; Kametaka, A.; Sou, Y.S.; Yabashi, A.; Yamada, A.; Annoh, H.; Kametaka, S.; Komatsu, M.; Waguri, S. A cluster of thin tubular structures mediate the transformation of the endoplasmic reticulum to autophagic isolation membrane. Mol. Cell. Biol. 2014, 34, 1695–1706. [Google Scholar] [CrossRef] [Green Version]
- Benyair, R.; Ron, E.; Lederkremer, G.Z. Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell Mol. Biol. 2011, 292, 197–280. [Google Scholar] [PubMed]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Wallis, A.K.; Freedman, R.B. Assisting oxidative protein folding: How do protein disulphide-isomerases couple conformational and chemical processes in protein folding? Top. Curr. Chem. 2013, 328, 1–34. [Google Scholar]
- Aebi, M.; Bernasconi, R.; Clerc, S.; Molinari, M. N-glycan structures: Recognition and processing in the ER. Trends Biochem. Sci. 2010, 35, 74–82. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef] [Green Version]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef]
- Barlowe, C.; Orci, L.; Yeung, T.; Hosobuchi, M.; Hamamoto, S.; Salama, N.; Rexach, M.F.; Ravazzola, M.; Amherdt, M.; Schekman, R. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Meldolesi, J.; Pozzan, T. The endoplasmic reticulum Ca2+ store: A view from the lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Prischi, F.; Ali, M.M. UPR Signal Activation by Luminal Sensor Domains. Int. J. Mol. Sci. 2013, 14, 6454–6466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewska, S.; Kochan, K.; Madanecki, P.; Piotrowski, A.; Ochocka, R.; Collawn, J.F.; Bartoszewski, R. Regulation of the unfolded protein response by microRNAs. Cell. Mol. Biol. Lett. 2013, 18, 555–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Healy, S.J.; Jäger, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Jäättelä, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef]
- Pablo Muñoz, J.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Isabel Hernández-Alvarez, M.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2014, 33, 171. [Google Scholar] [CrossRef] [Green Version]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P.; et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2511. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.M.; Ries, V.; Oo, T.F.; Yarygina, O.; Jackson-Lewis, V.; Ryu, E.J.; Lu, P.D.; Marciniak, S.J.; Ron, D.; Przedborski, S.; et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 2005, 95, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sado, M.; Yamasaki, Y.; Iwanaga, T.; Onaka, Y.; Ibuki, T.; Nishihara, S.; Mizuguchi, H.; Momota, H.; Kishibuchi, R.; Hashimoto, T.; et al. Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 2009, 1257, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Popko, B.; Roos, R.P. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011, 20, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Bruch, J.; Xu, H.; Rösler, T.W.; De Andrade, A.; Kuhn, P.H.; Lichtenthaler, S.F.; Arzberger, T.; Winklhofer, K.F.; Müller, U.; Höglinger, G.U. PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol. Med. 2017, 9, 371–384. [Google Scholar] [CrossRef]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [Green Version]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Kang, J. A small molecule targeting protein translation does not rescue spatial learning and memory deficits in the hAPP-J20 mouse model of Alzheimer’s disease. PeerJ 2016, 4, e2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [Green Version]
- Loewen, C.A.; Feany, M.B. The unfolded protein response protects from tau neurotoxicity in vivo. PLoS ONE 2010, 5, e13084. [Google Scholar] [CrossRef]
- Cissé, M.; Duplan, E.; Lorivel, T.; Dunys, J.; Bauer, C.; Meckler, X.; Gerakis, Y.; Lauritzen, I.; Checler, F. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in the Alzheimer model. Mol. Psychiatry 2017, 22, 1562–1575. [Google Scholar] [CrossRef]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef]
- Zuleta, A.; Vidal, R.L.; Armentano, D.; Parsons, G.; Hetz, C. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2012, 420, 558–563. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.G.; Posada, A.; Primi, M.P.; Castagne, V. Neuronal death in the central nervous system during development. Biomed. Pharmacother. 1998, 52, 356–362. [Google Scholar] [CrossRef]
- Green, D.R.; Oguin, T.H.; Martinez, J. The clearance of dying cells: Table for two. Cell Death Differ. 2016, 23, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis, and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J. Molecular control of life and death. Curr. Opin. Cell Biol. 1995, 7, 211–214. [Google Scholar] [CrossRef]
- Meier, P.; Evan, G. Dying like flies. Cell 1998, 95, 295–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.A.; Hengartner, M.O. The molecular mechanism of programmed cell death in C. elegans. Ann. N. Y. Acad. Sci. 1999, 887, 92–104. [Google Scholar] [CrossRef]
- Hengartner, M.O. Apoptosis: Corralling the corpses. Cell 2001, 104, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Hengartner, M.O. Cell death. In C. elegans II; Riddle, D., Blumenthal, T., Meyer, B., Priess, J., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; pp. 383–415. [Google Scholar]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Yuan, J.; Shaham, S.; Ledoux, S.; Ellis, H.M.; Horvitz, H.R. The C.elegans cell death gene ced-9 encodes a protein similar to mammalian interleukin-1beta converting enzyme. Cell 1993, 75, 641–652. [Google Scholar] [CrossRef]
- Schwartz, L.M.; Milligan, C.E. Cold thoughts of death: The role of ICE proteases in neuronal cell death. Trends Neurosci. 1996, 19, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Salvesen, G.S. Catalytic properties of the caspases. Cell Death Differ. 1999, 6, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.C.; Bonzon, C.; Green, D.R. The machinery of programmed cell death. Pharmacol. Ther. 2001, 92, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell death surface receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. Apoptosomes: Engines for caspase activation. Curr. Opin. Cell Biol. 2002, 14, 715–720. [Google Scholar] [CrossRef]
- Hu, Y.; Benedict, M.A.; Wu, D.; Inohara, N.; Núñez, G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA 1998, 95, 4386–4391. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; O’Rourke, K.; Dixit, V.M. Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J. Biol. Chem. 1998, 273, 5841–5845. [Google Scholar] [CrossRef]
- Moriishi, K.; Huang, D.C.; Cory, S.; Adams, J.M. Bcl-2 family members do not inhibit apoptosis by binding the caspase activator Apaf-1. Proc. Natl. Acad. Sci. USA 1999, 96, 9683–9688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conus, S.; Rossé, T.; Borner, C. Failure of Bcl-2 family members to interact with Apaf-1 in normal and apoptotic cells. Cell Death Differ. 2000, 7, 947–954. [Google Scholar] [CrossRef]
- Newmeyer, D.D.; Bossy-Wetzel, E.; Kluck, R.M.; Wolf, B.B.; Beere, H.M.; Green, D.R. Bcl-xL does not inhibit the function of Apaf-1. Cell Death Differ. 2000, 7, 402–407. [Google Scholar] [CrossRef]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 family: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Strasser, A. The Bcl-2 family and cell death regulation. Curr. Opin. Genet. Dev. 1998, 8, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, R. Bcl-2 family members in the development and degenerative pathologies of the nervous system. Cell Death Differ. 1998, 5, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, I.; Yuan, J. A convoluted way to die. Neuron 2001, 29, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengartner, M.O.; Horvitz, H.R. Programmed cell death in Caenorhabditis elegans. Curr. Opin. Cell Biol. 1994, 4, 581–586. [Google Scholar] [CrossRef]
- Hengartner, M.O.; Horvitz, H.R. Activation of C. elegans cell death protein CED9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature 1994, 369, 318–320. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Edlich, F. BCL-2 proteins, and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef]
- Liang, H.; Fesik, S.W. Three-dimensional structures of proteins involved in programmed cell death. J. Mol. Biol. 1997, 274, 291–302. [Google Scholar] [CrossRef]
- Leist, M.; Jaattela, M. Four death, and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2001, 8, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, G.; London, L.; Hockenbery, D.; Alexander, M.; McKearn, J.P.; Korsmeyer, S.J. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoietic cell lines. J. Immunol. 1990, 144, 3602–3610. [Google Scholar] [PubMed]
- Nuñez, G.; Clarke, M.F. The Bcl-2 family of proteins: Regulators of cell death and survival. Trends Cell Biol. 1994, 4, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brumatti, G.; Salmanidis, M.; Ekert, P.G. Crossing paths: Interactions between the cell death machinery and growth factor survival signals. Cell. Mol. Life Sci. 2010, 67, 1619–1630. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatt, N.B.; Glick, G.D. Signaling pathways and effector mechanisms pre-programmed cell death. Bioorg. Med. Chem. 2001, 9, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Gambino, G.; Ferrini, F.; Alasia, S.; Merighi, A. Posttranslational regulation of BCL2 levels in cerebellar granule cells: A mechanism of neuronal survival. Dev. Neurobiol. 2009, 69, 855–870. [Google Scholar] [CrossRef]
- Lossi, L.; Gambino, G.; Salio, C.; Merighi, A. Autophagy regulates the post-translational cleavage of BCL-2 and promotes neuronal survival. Sci. World J. 2010, 10, 924–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felicella, M.M.; Jen, K.Y. Bcl-2 Gene Family. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 310–312. [Google Scholar]
- Alsop, A.E.; Fennell, S.C.; Bartolo, R.C.; Tan, I.K.; Dewson, G.; Kluck, R.M. Dissociation of Bak α1 helix from the core and latch domains is required for apoptosis. Nat. Commun. 2015, 6, 6841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for the normal development of multiple tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shindler, K.S.; Latham, C.B.; Roth, K.A. Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J. Neurosci. 1997, 17, 3112–3119. [Google Scholar] [CrossRef] [Green Version]
- Parsadanian, A.S.; Cheng, Y.; Keller-Peck, C.R.; Holtzman, D.M.; Snider, W.D. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J. Neurosci. 1998, 18, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelidis, T.M.; Sendtner, M.; Cooper, J.D.; Airaksinen, M.S.; Holtmann, B.; Meyer, M.; Thoenen, H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron 1996, 17, 75–89. [Google Scholar] [CrossRef]
- Tanabe, H.; Eguchi, Y.; Kamada, S.; Martinou, J.C.; Tsujimoto, Y. Susceptibility of cerebellar granule neurons derived from Bcl-2-deficient and transgenic mice to cell death. Eur. J. Neurosci. 1997, 9, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.; Du, C.; Wu, J.-W.; Kyin, S.; Wang, X.; Shi, Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000, 406, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, A.M.; Ekert, P.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Eckelman, B.P.; Salvesen, G.S. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem. 2006, 281, 3254–3260. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef]
- Ditzel, M.; Broemer, M.; Tenev, T.; Bolduc, C.; Lee, T.V.; Rigbolt, K.T.; Elliott, R.; Zvelebil, M.; Blagoev, B.; Bergmann, A.; et al. Inactivation of effector caspases through nondegradative polyubiquitylation. Mol. Cell 2008, 32, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Morizane, Y.; Honda, R.; Fukami, K.; Yasuda, H. X-linked inhibitor of apoptosis functions as ubiquitin ligase toward mature caspase-9 and cytosolic Smac/DIABLO. J. Biochem. 2005, 137, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1, and c-IAP2 are critical mediators of tumor necrosis factor-alpha (TNF alpha)-induced NF-kappaB activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibert, B.; Mehlen, P. Dependence receptors, and cancer: Addiction to trophic ligands. Cancer Res. 2015, 75, 5171–5175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.R.; Stillman, D.J.; Thorburn, A. Regulation of Fas-associated Death Domain Interactions by the Death Effector Domain Identified by a Modified Reverse Two-hybrid Screen. J. Biol. Chem. 2002, 277, 34343–34348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Bonsignore, F.; Gobbo, F.; Amodeo, R.; Calvello, M.; Jacob, A.; Signore, G.; Schirripa Spagnolo, C.; Porciani, D.; Mainardi, M.; et al. Fast-diffusing p75(NTR) monomers support apoptosis and growth cone collapse by neurotrophin ligands. Proc. Natl. Acad. Sci. USA 2019, 116, 21563–21572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guicciardi, M.E.; Gores, G.J. Life, and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Bridgham, J.T.; Wilder, J.A.; Hollocher, H.; Johnson, A.L. All in the family: Evolutionary and functional relationships among death receptors. Cell Death Differ. 2003, 10, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.-M.; Li, Y.; Lu, A.; Li, Z.; Vajjhala, P.R.; Cruz, A.C.; Srivastava, D.B.; DiMaio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrik, I.N. Systems biology of death receptor networks: Live and let die. Cell Death Dis. 2014, 5, e1259. [Google Scholar] [CrossRef] [Green Version]
- Yeh, W.-C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.-B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V.; et al. Requirement for casper (c-FLIP) in the regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Fricker, N.; Beaudouin, J.; Richter, P.; Eils, R.; Krammer, P.H.; Lavrik, I.N. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J. Cell Biol. 2010, 190, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Majkut, J.; Sgobba, M.; Holohan, C.; Crawford, N.; Logan, A.E.; Kerr, E.; Higgins, C.A.; Redmond, K.L.; Riley, J.S.; Stasik, I.; et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat. Commun. 2014, 5, 3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortner, C.D.; Oldenburg, N.B.; Cidlowski, J.A. The role of DNA fragmentation in apoptosis. Trends Cell Biol 1995, 5, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Arur, S.; Uche, U.E.; Rezaul, K.; Fong, M.; Scranton, V.; Cowan, A.E.; Mohler, W.; Han, D.K. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell 2003, 4, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lossi, L.; Alasia, S.; Salio, C.; Merighi, A. Cell death, and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog. Neurobiol. 2009, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Suzuki, J.; Denning, D.P.; Imanishi, E.; Horvitz, H.R.; Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013, 341, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Imanishi, E.; Nagata, S. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 9509–9514. [Google Scholar] [CrossRef] [Green Version]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [Green Version]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the dogma—Phosphatidylserine in non-apoptotic cell death. Cell Commun. Signal. 2019, 17, 139. [Google Scholar] [CrossRef] [Green Version]
- Ozören, N.; El-Deiry, W.S. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 2002, 4, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 type I/type II model. Semin. Immunol. 2003, 15, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Harris, A.W.; Huang, D.C.; Krammer, P.H.; Cory, S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995, 14, 6136–6147. [Google Scholar] [CrossRef] [PubMed]
- Urresti, J.; Ruiz-Meana, M.; Coccia, E.; Arévalo, J.C.; Castellano, J.; Fernández-Sanz, C.; Galenkamp, K.M.; Planells-Ferrer, L.; Moubarak, R.S.; Llecha-Cano, N.; et al. Lifeguard Inhibits Fas Ligand-mediated Endoplasmic Reticulum-Calcium Release Mandatory for Apoptosis in Type II Apoptotic Cells. J. Biol. Chem. 2016, 291, 1221–1234. [Google Scholar] [CrossRef] [Green Version]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.S.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009, 460, 1035–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lossi, L.; Cocito, C.; Alasia, S.; Merighi, A. Ex vivo imaging of active caspase 3 by a FRET-based molecular probe demonstrates the cellular dynamics and localization of the protease in cerebellar granule cells and its regulation by the apoptosis-inhibiting protein survivin. Mol. Neurodegen. 2016, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Bougie, P.; Halliez, M.; Moreau, P.; Pellat-Deceunynck, C.; Amiot, M. Repression of Mcl-1 and disruption of the Mcl-1/Bak interaction in myeloma cells couple ER stress to mitochondrial apoptosis. Cancer Lett. 2016, 383, 204–211. [Google Scholar] [CrossRef]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic reticulum stress-induced apoptosis: Multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Shu, C.W.; Xu, W.; Shiau, C.W.; Grant, D.; Vasile, S.; Cosford, N.D.; Reed, J.C. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 2009, 284, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar] [CrossRef] [Green Version]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.G.; Yu, Y.; Weiss, R.M.; Felder, R.B. Endoplasmic reticulum stress increases brain MAPK signaling, inflammation, and renin-angiotensin system activity and sympathetic nerve activity in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H871–H880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Nolan, K.; Walter, F.; Tuffy, L.P.; Poeschel, S.; Gallagher, R.; Haunsberger, S.; Bray, I.; Stallings, R.L.; Concannon, C.G.; Prehn, J.H. Endoplasmic reticulum stress-mediated upregulation of miR-29a enhances sensitivity to neuronal apoptosis. Eur. J. Neurosci. 2016, 43, 640–652. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Jia, J.; Zhang, Y.; Li, J.; Zhu, Z.; Wang, H.; Tang, J.; Hu, J. Transmembrane E3 ligase RNF183 mediates ER stress-induced apoptosis by degrading Bcl-xL. Proc. Natl. Acad. Sci. USA 2018, 115, E2762–E2771. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.G.H. Developmental cell death: Morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar] [CrossRef]
- Lossi, L.; Castagna, C.; Merighi, A. Neuronal cell death: An overview of its different forms in central and peripheral neurons. In Neuronal Cell Death; Lossi, L., Merighi, A., Eds.; Springer: New York, NY, USA, 2015; pp. 1–18. [Google Scholar]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. MicroRNAs in apoptosis, autophagy, and necroptosis. Oncotarget 2015, 6, 8474–8490. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Fan, H.; Tang, H.B.; Kang, J.; Shan, L.; Song, H.; Zhu, K.; Wang, J.; Ju, G.; Wang, Y.Z. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience 2015, 311, 362–373. [Google Scholar] [CrossRef]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [Green Version]
- Zakeri, Z.; Lockshin, R.A. Cell death during development. J. Immunol. Methods 2002, 265, 3–20. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Caspase-independent cell deaths. Curr. Opin. Cell Biol. 2002, 14, 727–733. [Google Scholar] [CrossRef]
- Zakeri, Z.; Bursch, W.; Tenniswood, M.; Lockshin, R.A. Cell death, programmed apoptosis, necrosis, or other. Cell Death Differ. 1995, 2, 87–96. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology, and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol. Cancer Res. MCR 2018, 16, 1073–1076. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jehan, C.; Cartier, D.; Bucharles, C.; Anouar, Y.; Lihrmann, I. Emerging roles of ER-resident selenoproteins in brain physiology and physiopathology. Redox Biol. 2022, 55, 102412. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of calcium homeostasis and flux between the endoplasmic reticulum and the cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Lee, C.-H.; Poburko, D.; Kuo, K.-H.; Seow, C.Y.; Breemen, C.V. Ca2+ oscillations, gradients, and homeostasis in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1571–H1583. [Google Scholar] [CrossRef]
- Putney, J.W., Jr.; Broad, L.M.; Braun, F.-J.; Lievremont, J.-P.; Bird, G.S.J. Mechanisms of capacitative calcium entry. J. Cell Sci. 2001, 114, 2223–2229. [Google Scholar] [CrossRef]
- Schuster, S.; Marhl, M.; Höfer, T. Modelling of simple and complex calcium oscillations from single-cell responses to intercellular signalling. Eur. J. Biochem. 2002, 269, 1333–1355. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. Calcium signalling: A historical account, recent developments, and future perspectives. Cell. Mol. Life Sci. 2000, 57, 354–370. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Baumann, O.; Walz, B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 2001, 205, 149–214. [Google Scholar] [PubMed]
- Fill, M.; Copello, J.A. Ryanodine receptor calcium release channels. Physiol. Rev. 2002, 82, 893–922. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002, 1, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Mioletti, S.; Merighi, A. Synapse-independent and synapse-dependent apoptosis of cerebellar granule cells in postnatal rabbits occur at two subsequent but partly overlapping developmental stages. Neuroscience 2002, 112, 509–523. [Google Scholar] [CrossRef]
- Hallak, H.; Ramadan, B.; Rubin, R. Tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1) by oxidant stress in cerebellar granule neurons: Modulation by N-methyl-D-aspartate through calcineurin activity. J. Neurochem. 2001, 77, 63–70. [Google Scholar] [CrossRef]
- Sato, M.; Suzuki, K.; Yamazaki, H.; Nakanishi, S. A pivotal role of calcineurin signaling in development and maturation of postnatal cerebellar granule cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5874–5879. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, S.; Okazawa, M. Membrane potential-regulated Ca2+ signalling in development and maturation of mammalian cerebellar granule cells. J. Physiol. 2006, 575, 389–395. [Google Scholar] [CrossRef]
- Yao, C.J.; Lin, C.W.; Lin-Shiau, S.Y. Astrocytes modulate thapsigargin-induced changes in calcium concentration and neuronal survival. Proc. Natl. Sci. Counc. Repub. China B 2000, 24, 81–87. [Google Scholar]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Gaston, B.; Reilly, J.; Drazen, J.M.; Fackler, J.; Ramdev, P.; Arnelle, D.; Mullins, M.E.; Sugarbaker, D.J.; Chee, C.; Singel, D.J.; et al. Endogenous nitrogen oxides, and bronchodilator S-nitrosothiols in human airways. Proc. Natl. Acad. Sci. USA 1993, 90, 10957–10961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Kang, H.; Yan, F.; Chen, C. The endoplasmic reticulum-related events in S-nitrosoglutathione-induced neurotoxicity in cerebellar granule cells. Brain Res. 2004, 1015, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Brewster, J.L.; Linseman, D.A.; Bouchard, R.J.; Loucks, F.A.; Precht, T.A.; Esch, E.A.; Heidenreich, K.A. Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase-3β and caspase-9-dependent apoptosis in cerebellar granule neurons. Mol. Cell. Neurosci. 2006, 32, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, Y.; Sakikubo, T.; Ishige, K.; Ito, Y. Comparative study of endoplasmic reticulum stress-induced neuronal death in rat cultured hippocampal and cerebellar granule neurons. Neurochem. Int. 2006, 49, 285–293. [Google Scholar] [CrossRef]
- Ishige, K.; Takagi, N.; Imai, T.; Rausch, W.D.; Kosuge, Y.; Kihara, T.; Kusama-Eguchi, K.; Ikeda, H.; Cools, A.R.; Waddington, J.L.; et al. Role of caspase-12 in amyloid beta-peptide-induced toxicity in organotypic hippocampal slices cultured for long periods. J. Pharmacol. Sci. 2007, 104, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, B.; Fan, Z.; Shi, X.; Ke, Z.J.; Luo, J. Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience 2007, 144, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ma, C.; Bower, K.A.; Shi, X.; Ke, Z.; Luo, J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: Involvement of oxidative stress. J. Neurosci. Res. 2008, 86, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Chavis, P.; Fagni, L.; Lansman, J.B.; Bockaert, J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature 1996, 382, 719–722. [Google Scholar] [CrossRef]
- Higo, T.; Hamada, K.; Hisatsune, C.; Nukina, N.; Hashikawa, T.; Hattori, M.; Nakamura, T.; Mikoshiba, K. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 2010, 68, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yi, P.; Zhang, B.; Xu, C.; Liu, Q.; Pi, Z.; Xu, X.; Chevet, E.; Liu, J. Differences in endoplasmic reticulum stress signalling kinetics determine cell survival outcome through activation of MKP-1. Cell. Signal. 2011, 23, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, Y.; Zhou, K.; Freeze, H.H.; Zhang, Y.W.; Xu, H. Insufficient ER-stress response causes selective mouse cerebellar granule cell degeneration resembling that seen in congenital disorders of glycosylation. Mol. Brain 2013, 6, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benítez-Rangel, E.; Olguín-Albuerne, M.; López-Méndez, M.C.; Domínguez-Macouzet, G.; Guerrero-Hernández, A.; Morán, J. Caspase-3 Activation Correlates With the Initial Mitochondrial Membrane Depolarization in Neonatal Cerebellar Granule Neurons. Front. Cell Dev. Biol. 2020, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Naughton, M.; McMahon, J.; Healy, S.; FitzGerald, U. Profile of the unfolded protein response in rat cerebellar cortical development. J. Comp. Neurol. 2019, 527, 2910–2924. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; DiFiglia, M.; Heintz, N.; Nixon, R.A.; Qin, Z.H.; Ravikumar, B.; Stefanis, L.; Tolkovsky, A. Autophagy and its possible roles in nervous system diseases, damage, and repair. Autophagy 2005, 1, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.; Unsicker, K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience 2006, 138, 1055–1065. [Google Scholar] [CrossRef]
- Zhai, H.; Nakade, K.; Oda, M.; Mitsumoto, Y.; Akagi, M.; Sakurai, J.; Fukuyama, Y. Honokiol-induced neurite outgrowth promotion depends on activation of extracellular signal-regulated kinases (ERK1/2). Eur. J. Pharmacol. 2005, 516, 112–117. [Google Scholar] [CrossRef]
- Liu, J.; Yang, R.; Meng, H.; Zhou, T.; He, Q. In vitro treatment of 3 T3-L1 adipocytes with recombinant Calcium/calmodulin-dependent Protein Kinase IV (CaMKIV) limits ER stress and improves insulin sensitivity through inhibition of autophagy via the mTOR/CREB signaling pathway. BMC Endocr. Disord. 2020, 20, 104. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Dai, C.; Tang, S.; Yang, X.; Li, D.; Zhao, K.; Xiao, X. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol. Toxicol. 2016, 32, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.; Hadi-Alijanvand, H.; Sabbaghian, M.; Kiaei, M.; Khodagholi, F. Interaction of 2-APB, dantrolene, and TDMT with IP3R and RyR modulates ER stress-induced programmed cell death I and II in neuron-like PC12 cells: An experimental and computational investigation. J. Biomol. Struct. Dyn. 2014, 32, 1211–1230. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Hoozemans, J.J.M. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Kaibuchi, K. Neuronal polarity: From extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 2007, 8, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Cajigas, I.J.; Will, T.; Schuman, E.M. Protein homeostasis and synaptic plasticity. EMBO J. 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merighi, A.; Lossi, L. Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. Int. J. Mol. Sci. 2022, 23, 15186. https://doi.org/10.3390/ijms232315186
Merighi A, Lossi L. Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. International Journal of Molecular Sciences. 2022; 23(23):15186. https://doi.org/10.3390/ijms232315186
Chicago/Turabian StyleMerighi, Adalberto, and Laura Lossi. 2022. "Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death" International Journal of Molecular Sciences 23, no. 23: 15186. https://doi.org/10.3390/ijms232315186
APA StyleMerighi, A., & Lossi, L. (2022). Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. International Journal of Molecular Sciences, 23(23), 15186. https://doi.org/10.3390/ijms232315186