
Different Intermolecular Interactions Drive Nonpathogenic Liquid–Liquid Phase Separation and Potentially Pathogenic Fibril Formation by TDP-43


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. TDP-16 Phase Separation Is Very Sensitive to the Solution Conditions
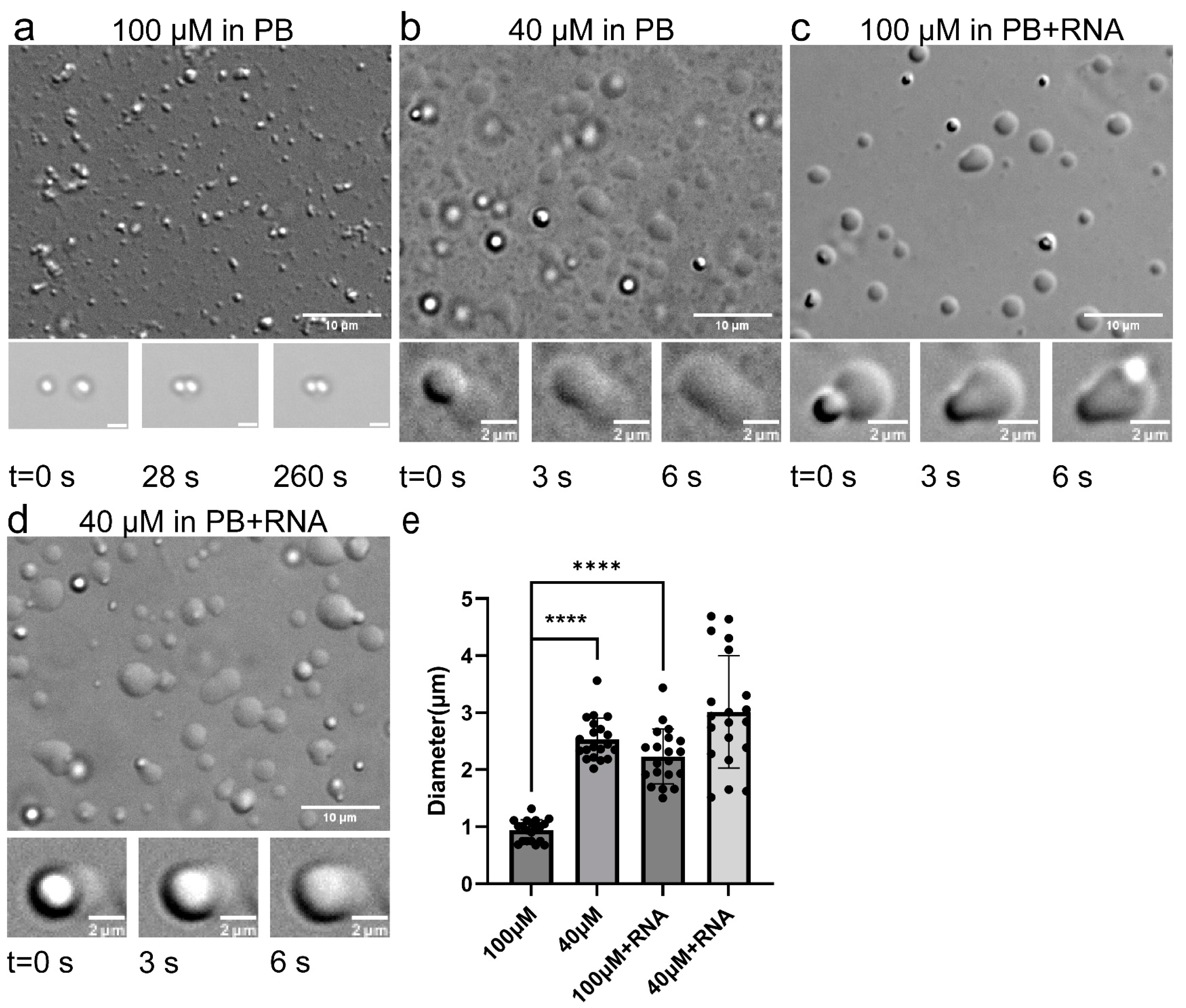
2.1.1. TDP-43 LCD Droplets Fusion Events Can Only Be Observed in a Narrow Concentration Range
2.1.2. RNA Enhanced TDP-43 LCD Droplet Fusion
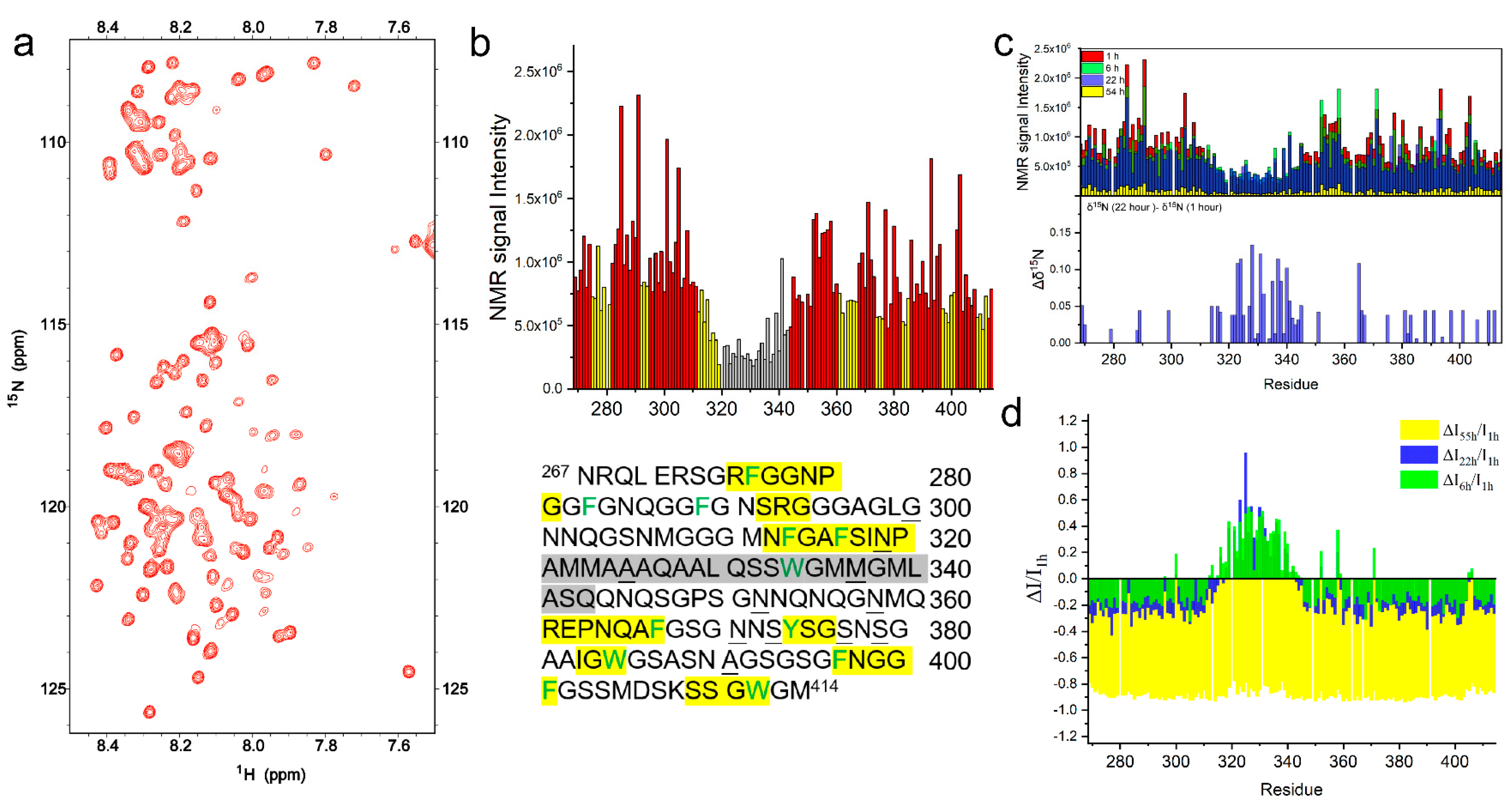
2.2. The Intermolecular Interactions of TDP-43 LCD Monitored by Solution NMR
2.2.1. TDP-43 LCD in Mature Droplets Is Still Mobile and Liquid-like
2.2.2. The Helical Intermolecular Interaction Was Reduced during TDP-43 LCD Fibrillation in PB
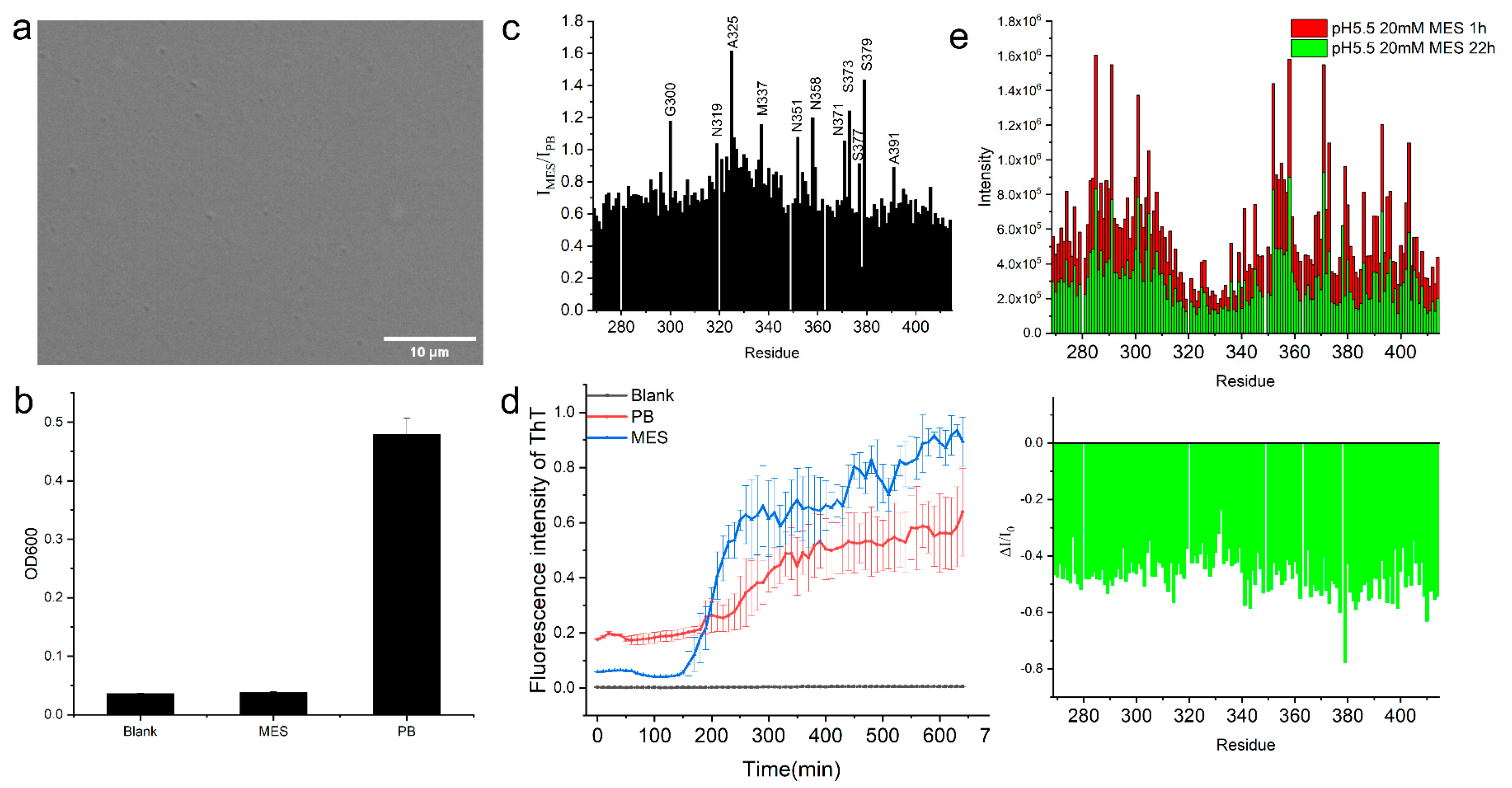
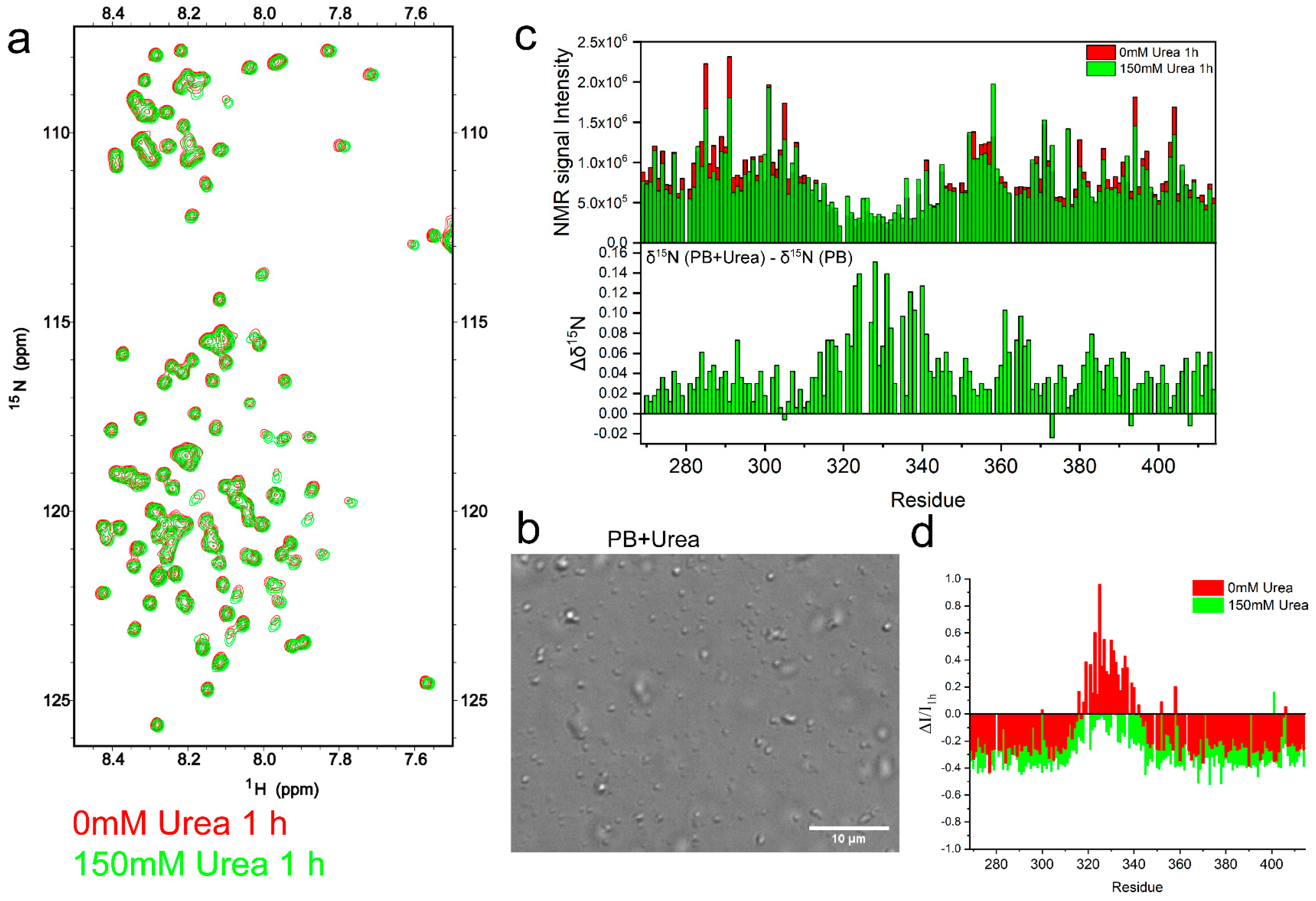
2.3. TDP-43 LCD Aggregation Was Enhanced by an Introduction of 150 mM Urea to the Solution
2.4. The Disordered Region Showed a Low LLPS Ability and a Slightly Slower Aggregation Rate
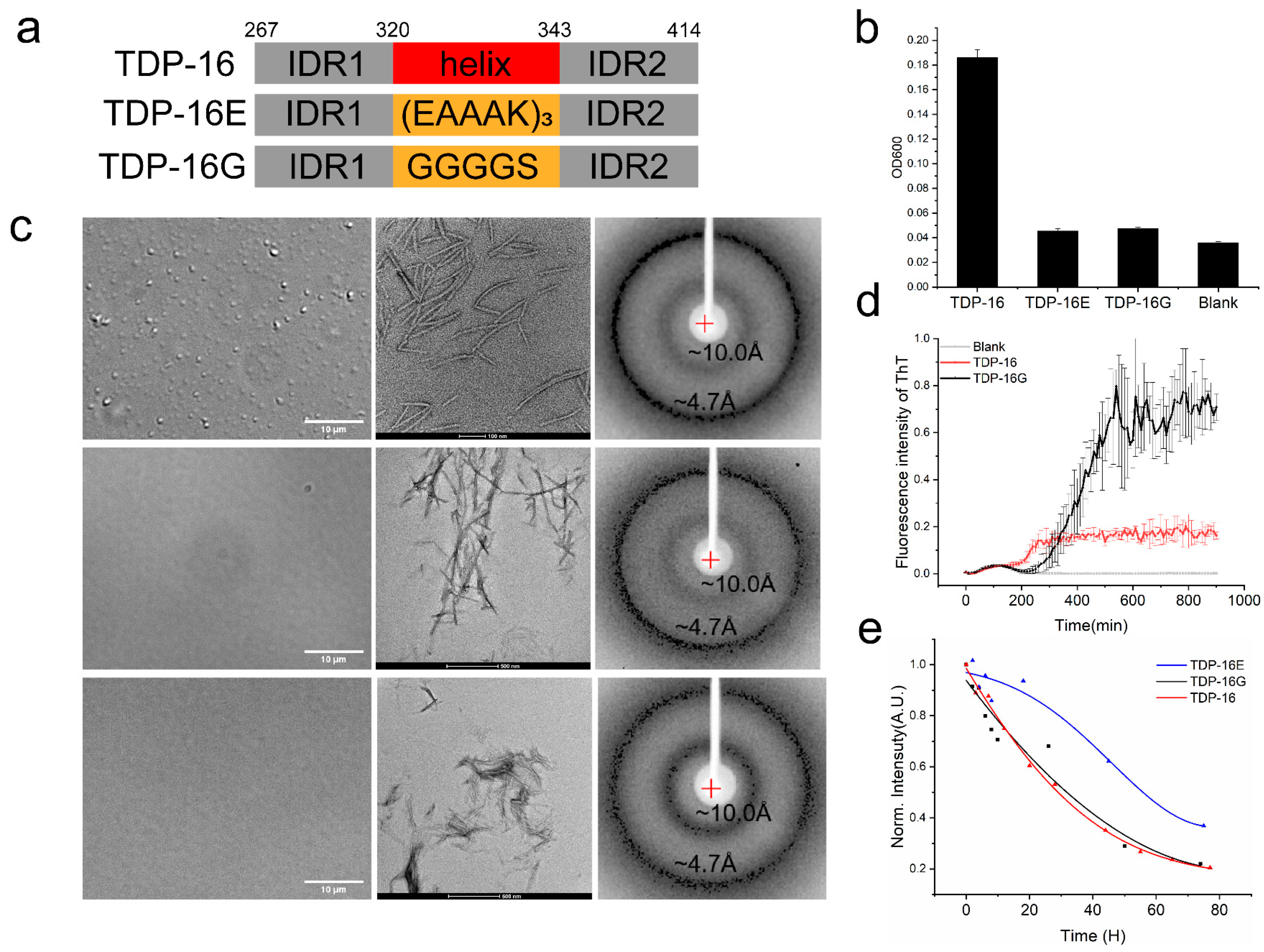
2.4.1. TDP-43 LCD Fragments without the Helices Have a Low LLPS Ability
2.4.2. TDP-43 LCD Fragments without the Helices Still Form Fibril Aggregates
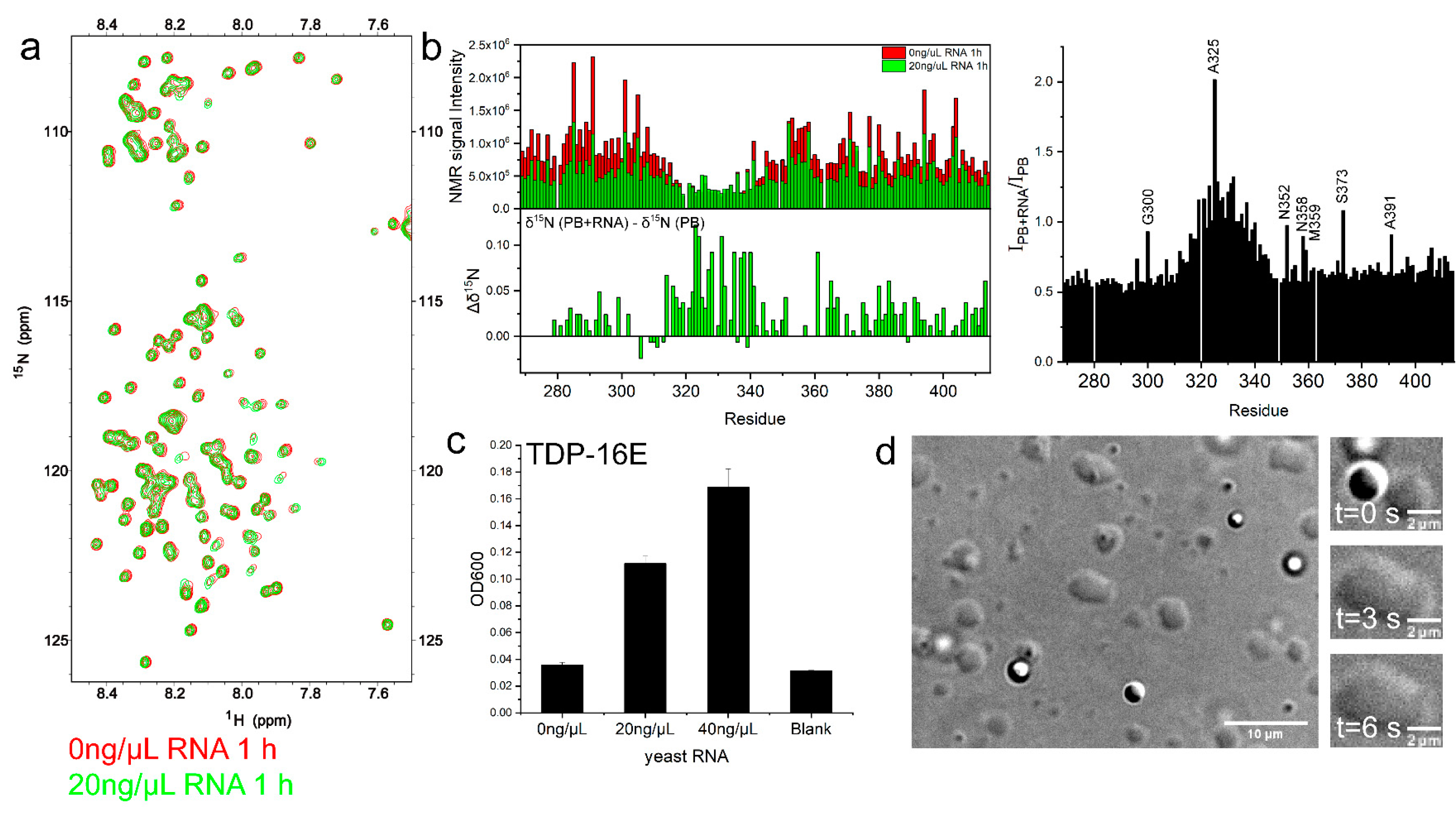
2.5. RNA Enhanced TDP-43 LCD LLPS Mainly by Mediating the Intermolecular Interactions with IDR1 and IDR2
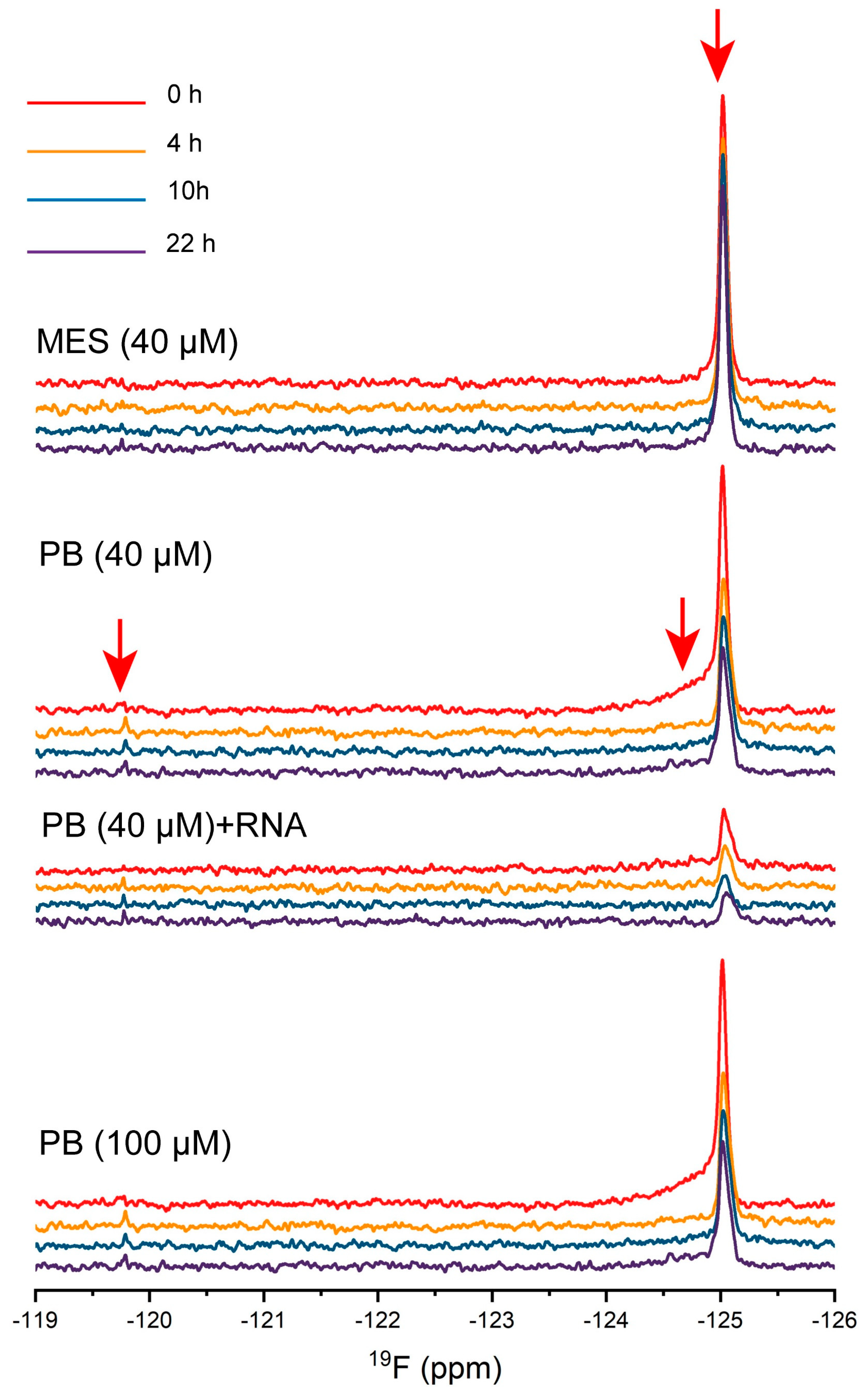
2.6. The Dynamic Changes of Protein Molecules Probed by 19F NMR
3. Discussion
3.1. A Fine Tune of the Different Interactions Affects the Protein LLPS Equilibrium and Droplet Sizes
3.2. The Molecular Status of Proteins in the Mature Droplets
3.3. TDP-43 LCD Aggregation Intermediate
3.4. The Polar, Aromatic Residue Rich Sequence of Low Complexity Domain
4. Materials and Methods
4.1. Expression and Purification
4.2. Turbidity Measurements
4.3. Thioflavin-T Assays
4.4. Intrinsic Fluorescence Spectroscopy
4.5. Differential Interference Contrast (DIC) Microscopy
4.6. Optical Tweezers
4.7. Negative-Staining Transmission Electron Microscope (TEM)
4.8. X-ray Diffraction (XRD)
4.9. Solution-State NMR
4.10. 19F NMR Spectroscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid-liquid phase separation in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 290. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Machyna, M.; Heyn, P.; Neugebauer, K.M. Cajal bodies: Where form meets function. Wiley Interdiscip. Rev. RNA 2013, 4, 17–34. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid-liquid phase separation as an organizing principle of intracellular space: Overview of the evolution of the cell compartmentalization concept. Cell. Mol. Life Sci. 2022, 79, 251. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Protein condensation diseases: Therapeutic opportunities. Nat. Commun. 2022, 13, 5550. [Google Scholar] [CrossRef]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Res. 2012, 1462, 16–25. [Google Scholar] [CrossRef]
- François-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Li, H.R.; Chen, T.C.; Hsiao, C.L.; Shi, L.; Chou, C.Y.; Huang, J.R. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta 2017, 1866, 214–223. [Google Scholar] [CrossRef]
- Jiang, L.L.; Che, M.X.; Zhao, J.; Zhou, C.J.; Xie, M.Y.; Li, H.Y.; He, J.H.; Hu, H.Y. Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 2013, 288, 19614–19624. [Google Scholar] [CrossRef]
- Sun, Y.; Chakrabartty, A. Phase to Phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet. 2015, 91, 1–53. [Google Scholar]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Reijns, M.A.; Alexander, R.D.; Spiller, M.P.; Beggs, J.D. A role for Q/N-rich aggregation-prone regions in P-body localization. J. Cell Sci. 2008, 121, 2463–2472. [Google Scholar] [CrossRef]
- Fan, H.C.; Ho, L.I.; Chi, C.S.; Chen, S.J.; Peng, G.S.; Chan, T.M.; Lin, S.Z.; Harn, H.J. Polyglutamine (PolyQ) diseases: Genetics to treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef]
- Mompeán, M.; Hervás, R.; Xu, Y.; Tran, T.H.; Guarnaccia, C.; Buratti, E.; Baralle, F.; Tong, L.; Carrión-Vázquez, M.; McDermott, A.E.; et al. Structural Evidence of Amyloid Fibril Formation in the Putative Aggregation Domain of TDP-43. J. Phys. Chem. Lett. 2015, 6, 2608–2615. [Google Scholar] [CrossRef]
- Murray, D.T.; Tycko, R. Side Chain Hydrogen-Bonding Interactions within Amyloid-like Fibrils Formed by the Low-Complexity Domain of FUS: Evidence from Solid State Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2020, 59, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Pantoja-Uceda, D.; Stuani, C.; Laurents, D.V.; McDermott, A.E.; Buratti, E.; Mompeán, M. Phe-Gly motifs drive fibrillization of TDP-43’s prion-like domain condensates. PLoS Biol. 2021, 19, e3001198. [Google Scholar] [CrossRef] [PubMed]
- Pakravan, D.; Michiels, E.; Bratek-Skicki, A.; De Decker, M.; Van Lindt, J.; Alsteens, D.; Derclaye, S.; Van Damme, P.; Schymkowitz, J.; Rousseau, F.; et al. Liquid-Liquid Phase Separation Enhances TDP-43 LCD Aggregation but Delays Seeded Aggregation. Biomolecules 2021, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; Wei, Y.; Lu, Y.; Song, J. ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS Biol. 2016, 14, e1002338. [Google Scholar] [CrossRef] [PubMed]
- Mompeán, M.; Chakrabartty, A.; Buratti, E.; Laurents, D.V. Electrostatic Repulsion Governs TDP-43 C-terminal Domain Aggregation. PLoS Biol. 2016, 14, e1002447. [Google Scholar] [CrossRef]
- Guenther, E.L.; Cao, Q.; Trinh, H.; Lu, J.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Rodriguez, J.A.; Hughes, M.P.; Eisenberg, D.S. Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol. 2018, 25, 463–471. [Google Scholar] [CrossRef]
- Gianni, S.; Freiberger, M.I.; Jemth, P.; Ferreiro, D.U.; Wolynes, P.G.; Fuxreiter, M. Fuzziness and Frustration in the Energy Landscape of Protein Folding, Function, and Assembly. Acc. Chem. Res. 2021, 54, 1251–1259. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Sequence Determinants of the Aggregation of Proteins Within Condensates Generated by Liquid-liquid Phase Separation. J. Mol. Biol. 2022, 434, 167201. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Suzuki, Y.; Brender, J.R.; Soper, M.T.; Krishnamoorthy, J.; Zhou, Y.; Ruotolo, B.T.; Kotov, N.A.; Ramamoorthy, A.; Marsh, E.N.G. Resolution of oligomeric species during the aggregation of Abeta1-40 using (19)F NMR. Biochemistry 2013, 52, 1903–1912. [Google Scholar] [CrossRef]
- Larda, S.T.; Simonetti, K.; Al-Abdul-Wahid, M.S.; Sharpe, S.; Prosser, R.S. Dynamic equilibria between monomeric and oligomeric misfolded states of the mammalian prion protein measured by 19F NMR. J. Am. Chem. Soc. 2013, 135, 10533–10541. [Google Scholar] [CrossRef] [PubMed]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.E.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 alpha-helical structure tunes liquid-liquid phase separation and function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef] [PubMed]
- Hough, L.E.; Dutta, K.; Sparks, S.; Temel, D.B.; Kamal, A.; Tetenbaum-Novatt, J.; Rout, M.P.; Cowburn, D. The molecular mechanism of nuclear transport revealed by atomic-scale measurements. Elife 2015, 4, e10027. [Google Scholar] [CrossRef]
- Zhuo, X.F.; Wang, J.; Zhang, J.; Jiang, L.L.; Hu, H.Y.; Lu, J.X. Solid-State NMR Reveals the Structural Transformation of the TDP-43 Amyloidogenic Region upon Fibrillation. J. Am. Chem. Soc. 2020, 142, 3412–3421. [Google Scholar] [CrossRef]
- Ladokhin, A.S. Fluorescence Spectroscopy in Peptide and Protein Analysis. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2000; pp. 1–18. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10, 2006. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Ma, C.; Lu, J. Hydroxyapatite Formation Coexists with Amyloid-like Self-Assembly of Human Amelogenin. Int. J. Mol. Sci. 2020, 21, 2946. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.-T.; Bi, L.-L.; Zhuo, X.-F.; Yang, L.-Y.; Sun, B.; Lu, J.-X. Different Intermolecular Interactions Drive Nonpathogenic Liquid–Liquid Phase Separation and Potentially Pathogenic Fibril Formation by TDP-43. Int. J. Mol. Sci. 2022, 23, 15227. https://doi.org/10.3390/ijms232315227
Zeng Y-T, Bi L-L, Zhuo X-F, Yang L-Y, Sun B, Lu J-X. Different Intermolecular Interactions Drive Nonpathogenic Liquid–Liquid Phase Separation and Potentially Pathogenic Fibril Formation by TDP-43. International Journal of Molecular Sciences. 2022; 23(23):15227. https://doi.org/10.3390/ijms232315227
Chicago/Turabian StyleZeng, Yu-Teng, Lu-Lu Bi, Xiao-Feng Zhuo, Ling-Yun Yang, Bo Sun, and Jun-Xia Lu. 2022. "Different Intermolecular Interactions Drive Nonpathogenic Liquid–Liquid Phase Separation and Potentially Pathogenic Fibril Formation by TDP-43" International Journal of Molecular Sciences 23, no. 23: 15227. https://doi.org/10.3390/ijms232315227
APA StyleZeng, Y.-T., Bi, L.-L., Zhuo, X.-F., Yang, L.-Y., Sun, B., & Lu, J.-X. (2022). Different Intermolecular Interactions Drive Nonpathogenic Liquid–Liquid Phase Separation and Potentially Pathogenic Fibril Formation by TDP-43. International Journal of Molecular Sciences, 23(23), 15227. https://doi.org/10.3390/ijms232315227