
Slight Variations in the Sequence Downstream of the Polyadenylation Signal Significantly Increase Transgene Expression in HEK293T and CHO Cells
 , , ,
, , ,  and
and
Abstract
:1. Introduction
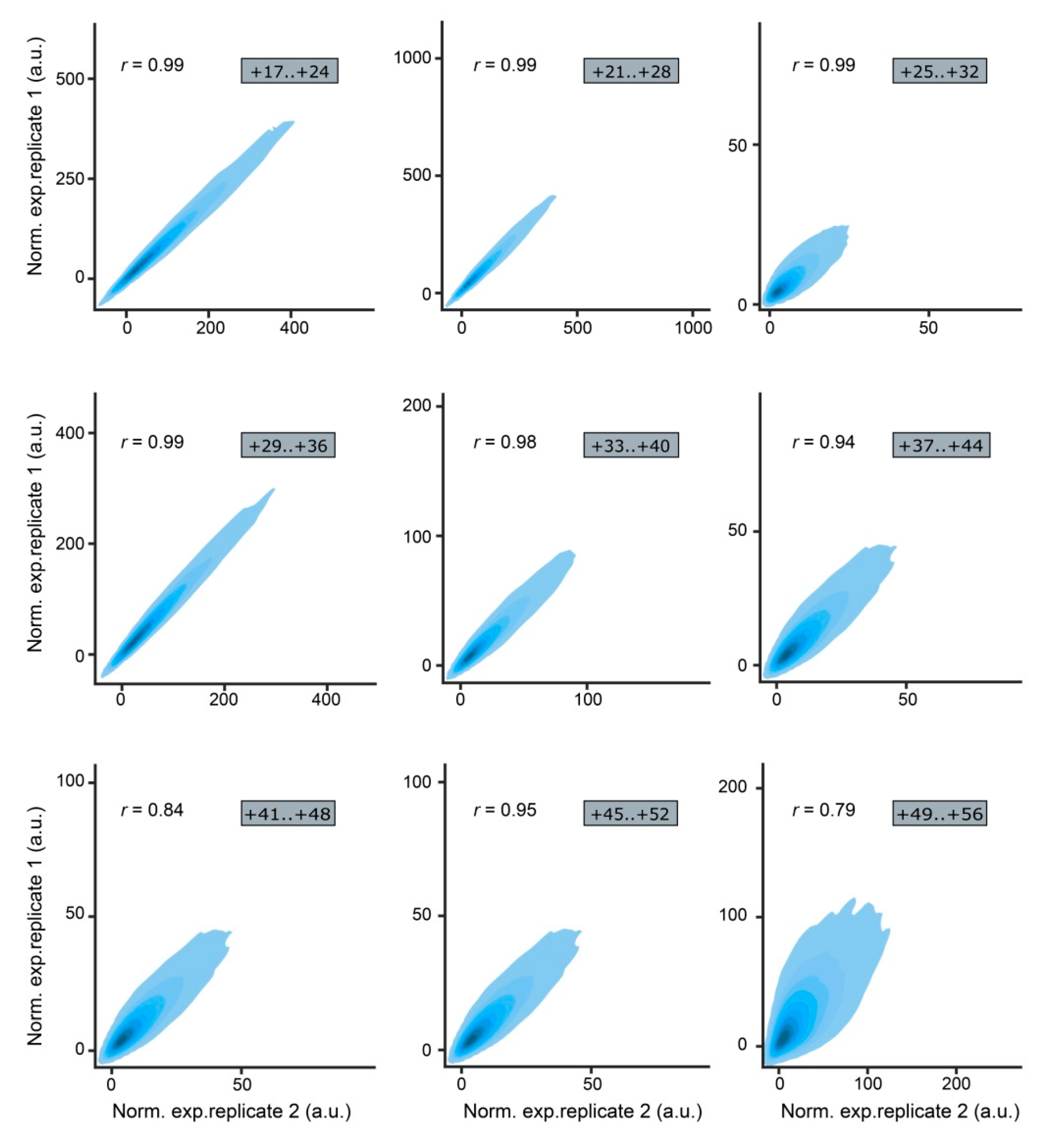
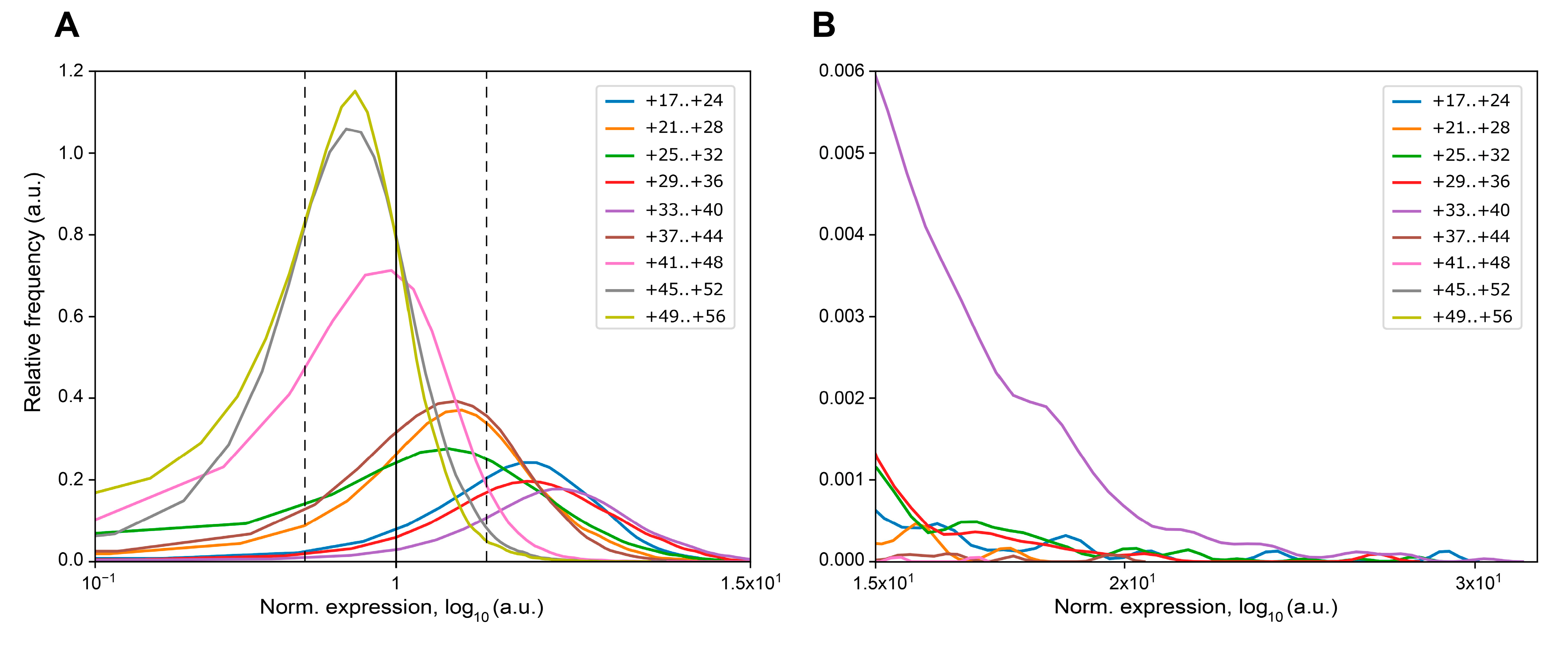
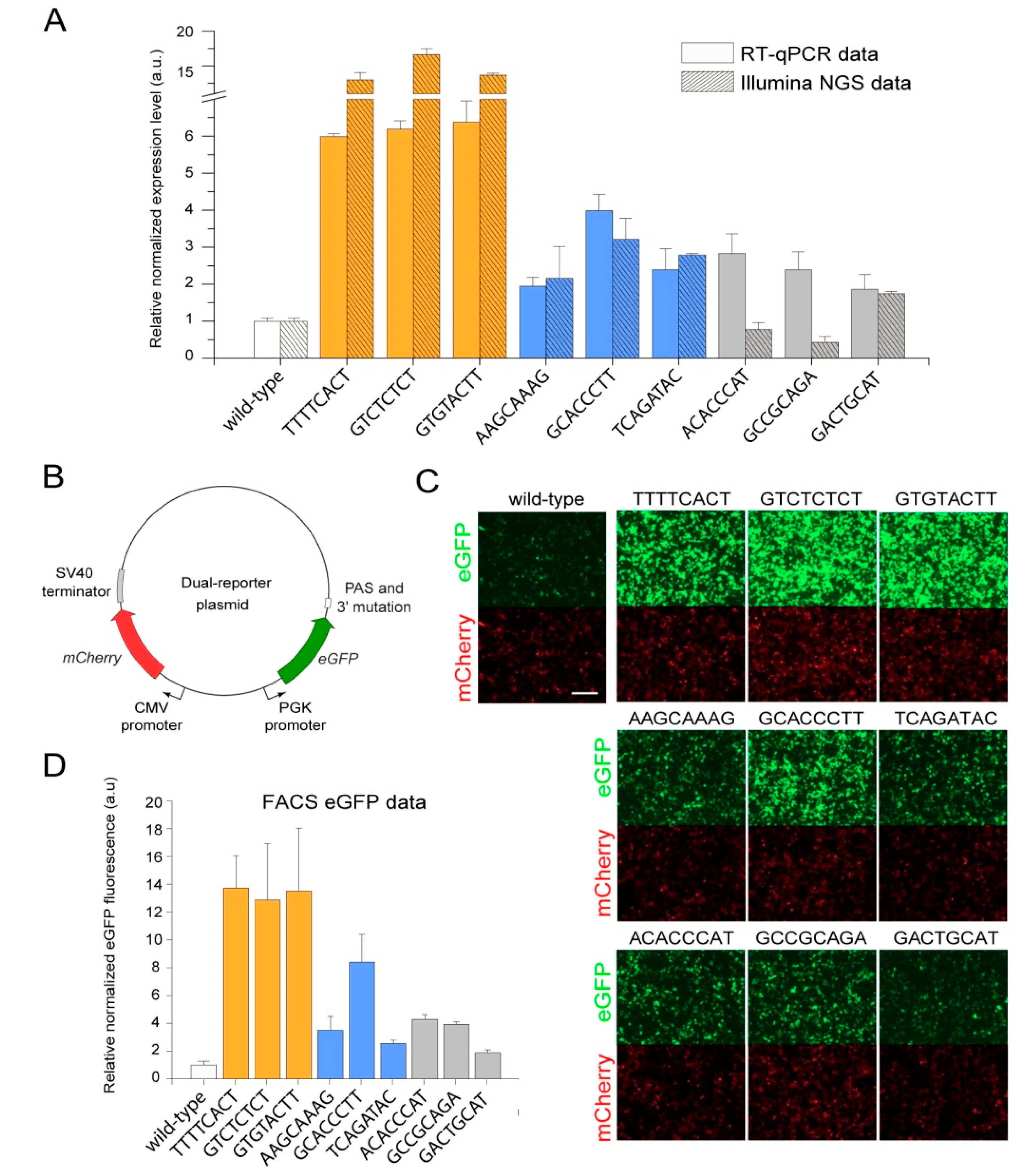
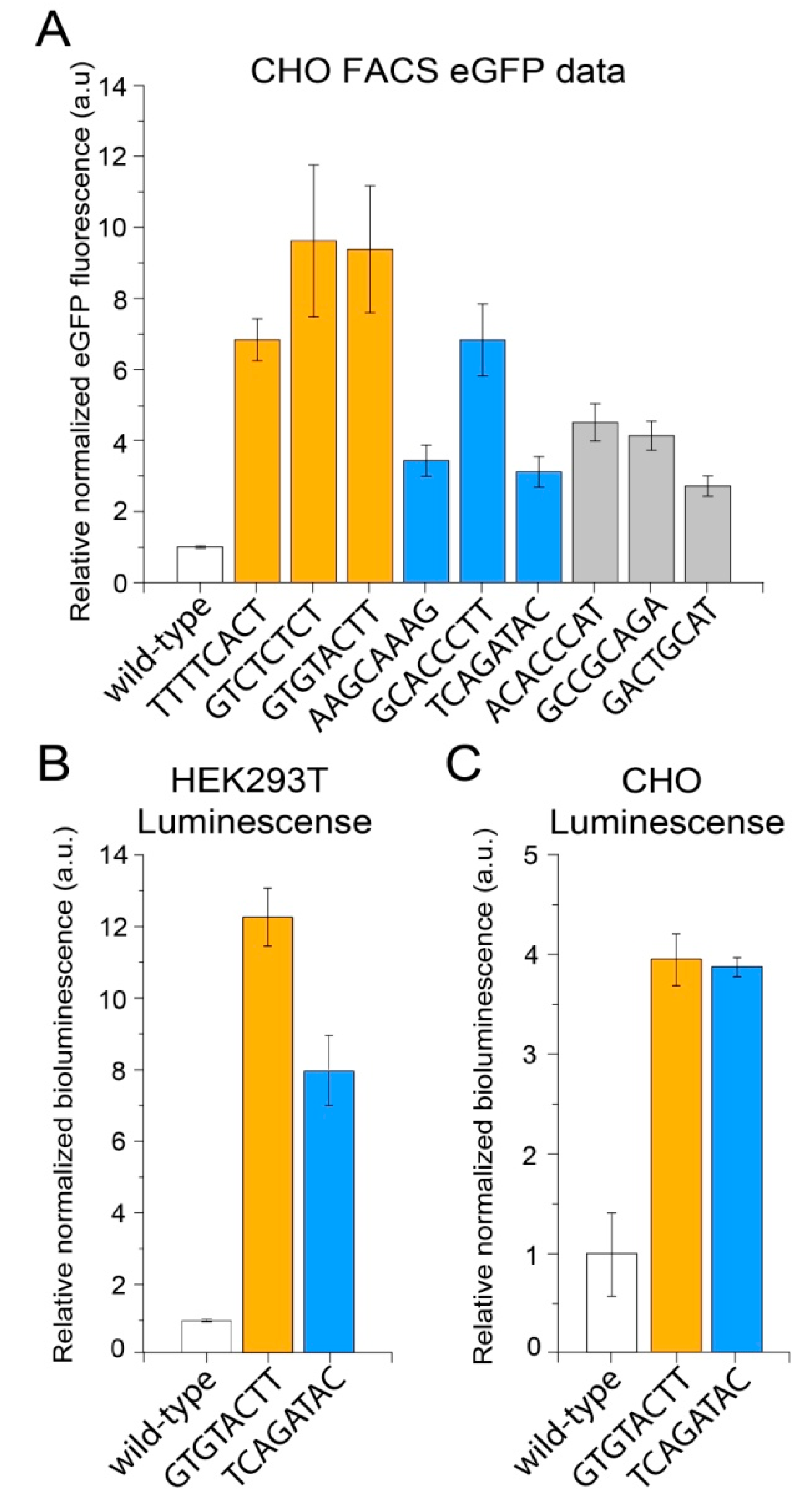
2. Results
3. Discussion
4. Materials and Methods
4.1. Gibson Assembly of MPRA Plasmid Libraries
4.2. Generation of Reference Barcoded Plasmid Constructs
4.3. Generation of Non-Barcoded Plasmid Constructs with Individual Mutations
4.4. Cell Lines and Transfections
4.5. MPRA Sample Preparation for NGS
4.6. Illumina NGS and Data Analysis
4.7. RNA Isolation and Quantitative Real-Time PCR
4.8. Fluorescence Microscopy
4.9. FACS Analysis
4.10. Luciferase Reporter Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Porrua, O.; Boudvillain, M.; Libri, D. Transcription termination: Variations on common themes. Trends Genet. 2016, 32, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, N.J. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 2016, 352, aad9926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neve, J.; Patel, R.; Wang, Z.; Louey, A.; Furger, A.M. Cleavage and polyadenylation: Ending the message expands gene regulation. RNA Biol. 2017, 14, 865–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porrua, O.; Libri, D. Transcription termination and the control of the transcriptome: Why, where and how to stop. Nat. Rev. Mol. Cell Biol. 2015, 16, 190–202. [Google Scholar] [CrossRef]
- Turner, R.E.; Pattison, A.D.; Beilharz, T.H. Alternative polyadenylation in the regulation and dysregulation of gene expression. Semin. Cell Dev. Biol. 2018, 75, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Masamha, C.P.; Wagner, E.J. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis 2018, 39, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, L.; Qiu, Q.; Zhou, Q.; Ding, J.; Lu, Y.; Liu, P. Alternative polyadenylation: Methods, mechanism, function, and role in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 51. [Google Scholar] [CrossRef]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Means to an end: Mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdiscip. Rev. RNA 2014, 5, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Rehfeld, A.; Plass, M.; Krogh, A.; Friis-Hansen, L. Alterations in polyadenylation and its implications for endocrine disease. Front. Endocrinol. 2013, 4, 53. [Google Scholar] [CrossRef]
- Curinha, A.; Oliveira Braz, S.; Pereira-Castro, I.; Cruz, A.; Moreira, A. Implications of polyadenylation in health and disease. Nucleus 2014, 5, 508–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmalingam, P.; Mahalingam, R.; Yalamanchili, H.K.; Weng, T.; Karmouty-Quintana, H.; Guha, A.; A Thandavarayan, R.A. Emerging roles of alternative cleavage and polyadenylation (APA) in human disease. J. Cell Physiol. 2022, 237, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.L. Rules of engagement: Co-transcriptional recruitment of pre-mRNA processing factors. Curr. Opin. Cell Biol. 2005, 17, 251–256. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Shi, Y.; Walz, T.; Tong, L. Structural insights into the human pre-mRNA 3′-end processing machinery. Mol. Cell 2020, 77, 800–809. [Google Scholar] [CrossRef]
- Andersen, P.K.; Jensen, T.H.; Lykke-Andersen, S. Making ends meet: Coordination between RNA 3′-end processing and transcription initiation. Wiley Interdiscip. Rev. RNA 2013, 4, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Lepennetier, G.; Catania, F. Exploring the impact of cleavage and polyadenylation factors on pre-mRNA splicing across eukaryotes. G3 2017, 7, 2107–2114. [Google Scholar] [CrossRef]
- Zhao, J.; Hyman, L.; Moore, C. Formation of mRNA 3’ ends in eukaryotes: Mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev. 1999, 63, 405–445. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Proudfoot, N.J. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009, 136, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Rigo, F.; Martinson, H.G. Polyadenylation releases mRNA from RNA polymerase II in a process that is licensed by splicing. RNA 2009, 15, 823–836. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Hamilton, K.; Tong, L. Recent molecular insights into canonical pre-mRNA 3′-end processing. Transcription 2020, 11, 83–96. [Google Scholar] [CrossRef]
- Shi, Y.; Di Giammartino, D.C.; Taylor, D.; Sarkeshik, A.; Rice, W.J.; Yates, J.R., III; Frank, J.; Manley, J.L. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 2009, 33, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Veraldi, K.L.; Edwalds-Gilbert, G.; MacDonald, C.C.; Wallace, A.M.; Milcarek, C. Isolation and characterization of polyadenylation complexes assembled in vitro. RNA 2000, 6, 768–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, S.; Fong, N.; Yankulov, K.; Ballantyne, S.; Pan, G.; Greenblatt, J.; Patterson, S.D.; Wickens, M.; Bentley, D.L. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 1997, 385, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Manley, J.L. RNA polymerase II is an essential mRNA polyadenylation factor. Nature 1998, 395, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Fusby, B.; Kim, S.; Erickson, B.; Kim, H.; Peterson, M.L.; Bentley, D.L. Coordination of RNA polymerase II pausing and 3′ end processing factor recruitment with alternative polyadenylation. Mol. Cell Biol. 2016, 36, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Nag, A.; Narsinh, K.; Martinson, H.G. The poly(A)-dependent transcriptional pause is mediated by CPSF acting on the body of the polymerase. Nat. Struct. Mol. Biol. 2007, 14, 662–669. [Google Scholar] [CrossRef]
- Clerici, M.; Faini, M.; Aebersold, R.; Jinek, M. Structural insights into the assembly and polyA signal recognition mechanism of the human CPSF complex. Elife 2017, 6, e33111. [Google Scholar] [CrossRef]
- Salisbury, J.; Hutchison, K.W.; Graber, J.H. A multispecies comparison of the metazoan 3’-processing downstream elements and the CstF-64 RNA recognition motif. BMC Genomics 2006, 7, 55. [Google Scholar] [CrossRef]
- Chan, S.L.; Huppertz, I.; Yao, C.; Weng, L.; Moresco, J.J.; Yates, J.R., III; Ule, J.; Manley, J.L.; Shi, Y. CPSF30 and Wdr33 directly bind to AAUAAA in mammalian mRNA 3′ processing. Genes Dev. 2014, 28, 2370–2380. [Google Scholar] [CrossRef] [Green Version]
- Mandel, C.R.; Kaneko, S.; Zhang, H.; Gebauer, D.; Vethantham, V.; Manley, J.L.; Tong, L. Polyadenylation factor CPSF-73 is the pre-mRNA 3’-end-processing endonuclease. Nature 2006, 444, 953–956. [Google Scholar] [CrossRef]
- Venkataraman, K.; Brown, K.M.; Gilmartin, G.M. Analysis of a noncanonical poly(A) site reveals a tripartite mechanism for vertebrate poly(A) site recognition. Genes Dev. 2005, 19, 1315–1327. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Lutz, C.S.; Wilusz, J.; Tian, B. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 2005, 11, 1485–1493. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.M.; Gilmartin, G.M. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol. Cell 2003, 12, 1467–1476. [Google Scholar] [CrossRef]
- Yang, Q.; Gilmartin, G.M.; Doublié, S. The structure of human cleavage factor Im hints at functions beyond UGUA-specific RNA binding: A role in alternative polyadenylation and a potential link to 5′ capping and splicing. RNA Biol. 2011, 8, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Rüegsegger, U.; Beyer, K.; Keller, W. Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J. Biol. Chem. 1996, 271, 6107–6113. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tong, S.; Li, X.; Shi, H.; Ying, Z.; Gao, Y.; Ge, H.; Niu, L.; Teng, M. Structural basis of pre-mRNA recognition by the human cleavage factor Im complex. Cell Res. 2011, 21, 1039–1051. [Google Scholar] [CrossRef]
- Kubo, T.; Wada, T.; Yamaguchi, Y.; Shimizu, A.; Handa, H. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res. 2006, 34, 6264–6271. [Google Scholar] [CrossRef]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Cleavage factor Im is a key regulator of 3′UTR length. RNA Biol. 2012, 9, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Kyburz, A.; Sadowski, M.; Dichtl, B.; Keller, W. The role of the yeast cleavage and polyadenylation factor subunit Ydh1p/Cft2p in pre-mRNA 3′-end formation. Nucleic Acids Res. 2003, 31, 3936–3945. [Google Scholar] [CrossRef] [Green Version]
- Minvielle-Sebastia, L.; Preker, P.J.; Wiederkehr, T.; Strahm, Y.; Keller, W. The major yeast poly(A)-binding protein is associated with cleavage factor IA and functions in premessenger RNA 3′-end formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7897–7902. [Google Scholar] [CrossRef]
- Meinhart, A.; Cramer, P. Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 2004, 430, 223–226. [Google Scholar] [CrossRef]
- Noble, C.G.; Hollingworth, D.; Martin, S.R.; Ennis-Adeniran, V.; Smerdon, S.J.; Kelly, G.; Taylor, I.A.; Ramos, A. Key features of the interaction between Pcf11 CID and RNA polymerase II CTD. Nat. Struct. Mol. Biol. 2005, 12, 144–151. [Google Scholar] [CrossRef] [PubMed]
- West, S.; Proudfoot, N.J. Human Pcf11 enhances degradation of RNA polymerase II-associated nascent RNA and transcriptional termination. Nucleic Acids Res. 2008, 36, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Kamieniarz-Gdula, K.; Gdula, M.R.; Panser, K.; Nojima, T.; Monks, J.; Wiśniewski, J.R.; Riepsaame, J.; Brockdorff, N.; Pauli, A.; Proudfoot, N.J. Selective roles of vertebrate PCF11 in premature and full-length transcript termination. Mol. Cell 2019, 74, 158–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, H.; Rüegsegger, U.; Hübner, W.; Friedlein, A.; Langen, H.; Keller, W. Human pre-mRNA cleavage factor IIm contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J. 2000, 19, 5895–5904. [Google Scholar] [CrossRef] [Green Version]
- Preker, P.J.; Ohnacker, M.; Minvielle-Sebastia, L.; Keller, W. A multisubunit 3’ end processing factor from yeast containing poly(A) polymerase and homologues of the subunits of mammalian cleavage and polyadenylation specificity factor. EMBO J. 1997, 16, 4727–4737. [Google Scholar] [CrossRef] [Green Version]
- Mandel, C.R.; Bai, Y.; Tong, L. Protein factors in pre-mRNA 3’-end processing. Cell. Mol. Life Sci. 2008, 65, 1099–1122. [Google Scholar] [CrossRef] [Green Version]
- Dominski, Z. The hunt for the 3′ endonuclease. Wiley Interdiscip. Rev. RNA 2010, 1, 325–340. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Steiniger, M.; Marzluff, W.F. A core complex of CPSF73, CPSF100, and Symplekin may form two different cleavage factors for processing of poly(A) and histone mRNAs. Mol. Cell 2009, 34, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, Y.; Aik, W.S.; Yang, X.-C.; Marzluff, W.F.; Walz, T.; Dominski, Z.; Tong, L. Structure of an active human histone pre-mRNA 3′-end processing machinery. Science 2020, 367, 700–703. [Google Scholar] [CrossRef]
- Chan, S.; Choi, E.-A.; Shi, Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2011, 2, 321–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmonds, M. Polyadenylate polymerases. Methods Enzymol. 1990, 181, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Laishram, R.S.; Anderson, R.A. The poly A polymerase Star-PAP controls 3′-end cleavage by promoting CPSF interaction and specificity toward the pre-mRNA. EMBO J. 2010, 29, 4132–4145. [Google Scholar] [CrossRef] [Green Version]
- Takagaki, Y.; Ryner, L.C.; Manley, J.L. Separation and characterization of a poly(A) polymerase and a cleavage/specificity factor required for pre-mRNA polyadenylation. Cell 1988, 52, 731–742. [Google Scholar] [CrossRef]
- Laishram, R.S. Poly(A) polymerase (PAP) diversity in gene expression--star-PAP vs canonical PAP. FEBS Lett. 2014, 588, 2185–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Tian, B. RNA-binding proteins in regulation of alternative cleavage and polyadenylation. Adv. Exp. Med. Biol. 2014, 825, 97–127. [Google Scholar] [CrossRef]
- Shi, Y. Alternative polyadenylation: New insights from global analyses. RNA 2012, 18, 2105–2117. [Google Scholar] [CrossRef] [Green Version]
- Xiang, K.; Tong, L.; Manley, J.L. Delineating the structural blueprint of the pre-mRNA 3′-end processing machinery. Mol. Cell Biol. 2014, 34, 1894–1910. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.L.; Sharp, P.A. Accurate cleavage and polyadenylation of exogenous RNA substrate. Cell 1985, 41, 845–855. [Google Scholar] [CrossRef]
- Colgan, D.F.; Manley, J.L. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997, 11, 2755–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, J.; Falck-Pedersen, E.; Darnell, J.E., Jr.; Shenk, T. A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNA polymerase II in the mouseβmaj-globin gene. Proc. Natl. Acad. Sci. USA 1987, 84, 8306–8310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, P.; Manley, J.L. Transcription termination by nuclear RNA polymerases. Genes Dev. 2009, 23, 1247–1269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Rigo, F.; Martinson, H.G. Poly(A) signal-dependent transcription termination occurs through a conformational change mechanism that does not require cleavage at the poly(A) site. Mol. Cell 2015, 59, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gilmour, D.S. Pcf11 is a termination factor in Drosophila that dismantles the elongation complex by bridging the CTD of RNA polymerase II to the nascent transcript. Mol. Cell 2006, 21, 65–74. [Google Scholar] [CrossRef]
- West, S.; Gromak, N.; Proudfoot, N.J. Human 5′ --> 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 2004, 432, 522–525. [Google Scholar] [CrossRef]
- Tollervey, D. Molecular biology: Termination by torpedo. Nature 2004, 432, 456–457. [Google Scholar] [CrossRef]
- Eaton, J.D.; Davidson, L.; Bauer, D.L.V.; Natsume, T.; Kanemaki, M.T.; West, S. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 2018, 32, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Eaton, J.D.; Francis, L.; Davidson, L.; West, S. A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev. 2020, 34, 132–145. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Wilton, J.; Tellier, M.; Nojima, T.; Costa, A.M.; Oliveira, M.J.; Moreira, A. Simultaneous studies of gene expression and alternative polyadenylation in primary human immune cells. Methods Enzymol. 2021, 655, 349–399. [Google Scholar] [CrossRef]
- Manning, K.S.; Cooper, T.A. The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell Biol. 2017, 18, 102–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef]
- Eckmann, C.R.; Rammelt, C.; Wahle, E. Control of poly(A) tail length. Wiley Interdiscip. Rev. RNA 2011, 2, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Virtanen, A.; Kleiman, F.E. To polyadenylate or to deadenylate: That is the question. Cell Cycle 2010, 9, 4437–4449. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Manley, J.L. The end of the message: Multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015, 29, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Graber, J.H. Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip. Rev. RNA 2012, 3, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Ryner, L.C.; Takagaki, Y.; Manley, J.L. Sequences downstream of AAUAAA signals affect pre-mRNA cleavage and polyadenylation in vitro both directly and indirectly. Mol. Cell. Biol. 1989, 9, 1759–1771. [Google Scholar] [CrossRef]
- Gruber, A.J.; Schmidt, R.; Gruber, A.R.; Martin, G.; Ghosh, S.; Belmadani, M.; Keller, W.; Zavolan, M. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016, 26, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef]
- Wang, R.; Zheng, D.; Yehia, G.; Tian, B. A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res. 2018, 28, 1427–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, N.M.; Li, W.; Tian, B.; Furger, A. A functional human poly(A) site requires only a potent DSE and an A-rich upstream sequence. EMBO J. 2010, 29, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Beisang, D.; Bohjanen, P.R. Perspectives on the ARE as it turns 25 years old. Wiley Interdiscip. Rev. RNA 2012, 3, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.; Kamen, R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 1986, 46, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.A.; Shyu, A.-B. Selective degradation of early-response-gene mRNAs: Functional analyses of sequence features of the AU-rich elements. Mol. Cell. Biol. 1994, 14, 8471–8482. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Z.; Di Marco, S.; Gallouzi, I.; Rola-Pleszczynski, M.; Radzioch, D. RNA-binding protein HuR is required for stabilization of SLC11A1 mRNA and SLC11A1 protein expression. Mol. Cell. Biol. 2005, 25, 8139–8149. [Google Scholar] [CrossRef] [Green Version]
- Beaudoing, E.; Freier, S.; Wyatt, J.R.; Claverie, J.-M.; Gautheret, D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000, 10, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, W.; de Jong, J.; Pindyurin, A.V.; Pagie, L.; Meuleman, W.; de Ridder, J.; Berns, A.; Wessels, L.F.A.; van Lohuizen, M.; van Steensel, B. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 2013, 154, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Boldyreva, L.V.; Yarinich, L.A.; Kozhevnikova, E.N.; Ivankin, A.V.; Lebedev, M.O.; Pindyurin, A.V. Fine gene expression regulation by minor sequence variations downstream of the polyadenylation signal. Mol. Biol. Rep. 2021, 48, 1539–1547. [Google Scholar] [CrossRef]
- Komura, R.; Aoki, W.; Motone, K.; Satomura, A.; Ueda, M. High-throughput evaluation of T7 promoter variants using biased randomization and DNA barcoding. PLoS ONE 2018, 13, e0196905. [Google Scholar] [CrossRef]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F.; et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Omelina, E.S.; Ivankin, A.V.; Letiagina, A.E.; Pindyurin, A.V. Optimized PCR conditions minimizing the formation of chimeric DNA molecules from MPRA plasmid libraries. BMC Genomics 2019, 20, 536. [Google Scholar] [CrossRef] [PubMed]
- Letiagina, A.E.; Omelina, E.S.; Ivankin, A.V.; Pindyurin, A.V. MPRAdecoder: Processing of the raw MPRA data with a priori unknown sequences of the region of interest and associated barcodes. Front. Genet. 2021, 12, 618189. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kapoor, A.; Lee, C.; Mudgett, M.; Beer, M.A.; Chakravarti, A. Sequence-based correction of barcode bias in massively parallel reporter assays. Genome Res. 2021, 31, 1638–1645. [Google Scholar] [CrossRef]
- Ashuach, T.; Fischer, D.S.; Kreimer, A.; Ahituv, N.; Theis, F.J.; Yosef, N. MPRAnalyze: Statistical framework for massively parallel reporter assays. Genome Biol. 2019, 20, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalb, B.; Michel, M.; Zacher, B.; Frühauf, K.; Demel, C.; Tresch, A.; Gagneur, J.; Cramer, P. TT-seq maps the human transient transcriptome. Science 2016, 352, 1225–1228. [Google Scholar] [CrossRef]
- Baejen, C.; Andreani, J.; Torkler, P.; Battaglia, S.; Schwalb, B.; Lidschreiber, M.; Maier, K.C.; Boltendahl, A.; Rus, P.; Esslinger, S.; et al. Genome-wide analysis of RNA polymerase II termination at protein-coding genes. Mol. Cell 2017, 66, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the transcriptome through RNA structure. Nat Rev Genet 2011, 12, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Eisen, T.J.; Li, J.J.; Bartel, D.P. The interplay between translational efficiency, poly(A) tails, microRNAs, and neuronal activation. RNA 2022, 28, 808–831. [Google Scholar] [CrossRef]
- Rabani, M. Massively parallel analysis of regulatory RNA sequences. Methods Mol. Biol. 2021, 2218, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Mapendano, C.K.; Jensen, T.H. An ending is a new beginning: Transcription termination supports re-initiation. Cell Cycle 2011, 10, 863–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, Y.; Wei, J.; Li, Y.-Y. Transcriptional regulation and spatial interactions of head-to-head genes. BMC Genomics 2014, 15, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.; Chin, C.S.H.; Lim, Z.F.S.; Ng, S.K. HEK293 cell line as a platform to produce recombinant proteins and viral vectors. Front. Bioeng. Biotechnol. 2021, 9, 796991. [Google Scholar] [CrossRef]
- Abaandou, L.; Quan, D.; Shiloach, J. Affecting HEK293 cell growth and production performance by modifying the expression of specific genes. Cells 2021, 10, 1667. [Google Scholar] [CrossRef]
- Chin, C.L.; Goh, J.B.; Srinivasan, H.; Liu, K.I.; Gowher, A.; Shanmugam, R.; Lim, H.L.; Choo, M.; Tang, W.Q.; Tan, A.H.-M.; et al. A human expression system based on HEK293 for the stable production of recombinant erythropoietin. Sci. Rep. 2019, 9, 16768. [Google Scholar] [CrossRef] [Green Version]
- König, J.; Hust, M.; van den Heuvel, J. Validation of the production of antibodies in different formats in the HEK 293 transient gene expression system. Methods Mol. Biol. 2021, 2247, 59–76. [Google Scholar] [CrossRef]
- Heng, Z.S.-L.; Yeo, J.Y.; Koh, D.W.-S.; Gan, S.K.-E.; Ling, W.-L. Augmenting recombinant antibody production in HEK293E cells: Optimizing transfection and culture parameters. Antib. Ther. 2022, 5, 30–41. [Google Scholar] [CrossRef]
- Jäger, V.; Büssow, K.; Wagner, A.; Weber, S.; Hust, M.; Frenzel, A.; Schirrmann, T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol. 2013, 13, 52. [Google Scholar] [CrossRef]
- Ahmadi, S.; Davami, F.; Davoudi, N.; Nematpour, F.; Ahmadi, M.; Ebadat, S.; Azadmanesh, K.; Barkhordari, F.; Mahboudi, F. Monoclonal antibodies expression improvement in CHO cells by PiggyBac transposition regarding vectors ratios and design. PLoS ONE 2017, 12, e0179902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zúñiga, R.A.; Gutiérrez-González, M.; Collazo, N.; Sotelo, P.H.; Ribeiro, C.H.; Altamirano, C.; Lorenzo, C.; Aguillón, J.C.; Molina, M.C. Development of a new promoter to avoid the silencing of genes in the production of recombinant antibodies in chinese hamster ovary cells. J. Biol. Eng. 2019, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPRA Library ID | Number of Unique Mutations | Mutations Associated with 1 BC | Mutations Associated with 2 BCs | Mutations Associated with ≥3 BCs | |||
|---|---|---|---|---|---|---|---|
| Number | % | Number | % | Number | % | ||
| +17..+24 | 8380 | 7567 | 90.3 | 731 | 8.7 | 82 | 1.0 |
| +21..+28 | 8266 | 7347 | 88.9 | 797 | 9.6 | 122 | 1.5 |
| +25..+32 | 39,705 | 21,267 | 53.6 | 11,018 | 27.7 | 7420 | 18.7 |
| +29..+36 | 10,084 | 8830 | 87.5 | 944 | 9.4 | 310 | 3.1 |
| +33..+40 | 27,232 | 19,346 | 71.0 | 5807 | 21.3 | 2079 | 7.7 |
| +37..+44 | 33,866 | 19,721 | 58.2 | 8544 | 25.2 | 5601 | 16.6 |
| +41..+48 | 56,470 | 20,208 | 35.8 | 15,077 | 26.7 | 21,185 | 37.5 |
| +45..+52 | 25,814 | 18,373 | 71.2 | 5501 | 21.3 | 1940 | 7.5 |
| +49..+56 | 17,938 | 11,888 | 66.3 | 3489 | 19.5 | 2561 | 14.2 |
| MPRA Library ID | Number of Unique Mutations | Number of Mutations with Zero Counts in Expression Samples | Max Increase of eGFP Expression * | Max Decrease of eGFP Expression * | Mutations Leading to the ≥2-fold Increase of eGFP mRNA * | Mutations Leading to the ≥2-fold Decrease of eGFP mRNA * | ||
|---|---|---|---|---|---|---|---|---|
| Number | % | Number | % | |||||
| +17..+24 | 8380 | 5 | 28.15 | 0.19 | 6961 | 83.07 | 22 | 0.26 |
| +21..+28 | 8266 | 6 | 17.33 | 0.14 | 4474 | 54.13 | 79 | 0.96 |
| +25..+32 | 39,705 | 160 | 27.38 | 0.05 | 23,204 | 58.44 | 1276 | 3.21 |
| +29..+36 | 10,084 | 0 | 26.09 | 0.24 | 8724 | 86.51 | 7 | 0.07 |
| +33..+40 | 27,232 | 11 | 29.45 | 0.23 | 25,323 | 92.99 | 25 | 0.09 |
| +37..+44 | 33,866 | 58 | 19.67 | 0.07 | 16,304 | 48.14 | 746 | 2.20 |
| +41..+48 | 56,470 | 277 | 16.74 | 0.01 | 6216 | 11.01 | 6187 | 10.96 |
| +45..+52 | 25,814 | 25 | 7.59 | 0.01 | 1036 | 4.01 | 3771 | 14.61 |
| +49..+56 | 17,938 | 249 | 6.74 | 0.02 | 518 | 2.89 | 3196 | 17.82 |
| Mutation Sequence | Normalization Replicate 1, Read Count | Normalization Replicate 2, Read Count | Increase of eGFP Expression, Fold * | Group |
|---|---|---|---|---|
| TTTTCACT | 3 | 15 | 12.99 | High expression |
| GTCTCTCT | 11 | 11 | 16.63 | |
| GTGTACTT | 450 | 382 | 13.68 | |
| AAGCAAAG | 3 | 4 | 2.16 | Medium expression |
| GCACCCTT | 9 | 11 | 3.21 | |
| TCAGATAC | 281 | 264 | 2.78 | |
| ACACCCAT | 3 | 10 | 0.77 | Low expression |
| GCCGCAGA | 11 | 15 | 0.43 | |
| GACTGCAT | 92 | 64 | 1.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omelina, E.S.; Letiagina, A.E.; Boldyreva, L.V.; Ogienko, A.A.; Galimova, Y.A.; Yarinich, L.A.; Pindyurin, A.V.; Andreyeva, E.N. Slight Variations in the Sequence Downstream of the Polyadenylation Signal Significantly Increase Transgene Expression in HEK293T and CHO Cells. Int. J. Mol. Sci. 2022, 23, 15485. https://doi.org/10.3390/ijms232415485
Omelina ES, Letiagina AE, Boldyreva LV, Ogienko AA, Galimova YA, Yarinich LA, Pindyurin AV, Andreyeva EN. Slight Variations in the Sequence Downstream of the Polyadenylation Signal Significantly Increase Transgene Expression in HEK293T and CHO Cells. International Journal of Molecular Sciences. 2022; 23(24):15485. https://doi.org/10.3390/ijms232415485
Chicago/Turabian StyleOmelina, Evgeniya S., Anna E. Letiagina, Lidiya V. Boldyreva, Anna A. Ogienko, Yuliya A. Galimova, Lyubov A. Yarinich, Alexey V. Pindyurin, and Evgeniya N. Andreyeva. 2022. "Slight Variations in the Sequence Downstream of the Polyadenylation Signal Significantly Increase Transgene Expression in HEK293T and CHO Cells" International Journal of Molecular Sciences 23, no. 24: 15485. https://doi.org/10.3390/ijms232415485