Abstract
Rickettsia are obligate intracellular bacteria primarily carried by arthropod hosts. The genus Rickettsia contains several vertebrate pathogens vectored by hematophagous arthropods. Despite the potential for disease, our understanding of Rickettsias are limited by the difficulties associated with growing and manipulating obligate intracellular bacteria. To aid with this, our lab conducted an analysis of eight genomes and three plasmids from across the genus Rickettsia. Using OPT4e, a learning algorithm-based program designed to identify effector proteins secreted by the type 4 secretion system, we generated a putative effectome for the genus. We then consolidated effectors into homolog sets to identify effectors unique to Rickettsia with different life strategies or evolutionary histories. We also compared predicted effectors to non-effectors for differences in G+C content and gene splitting. Based on this analysis, we predicted 1571 effectors across the genus, resulting in 604 homolog sets. Each species had unique homolog sets, while 42 were present in all eight species analyzed. Effectors were flagged in association with pathogenic, tick and flea-borne Rickettsia. Predicted effectors also varied in G+C content and frequency of gene splitting as compared to non-effectors. Species effector repertoires show signs of expansion, degradation, and horizontal acquisition associated with lifestyle and lineage.
1. Introduction
The genus Rickettsia is comprised of obligate intracellular, α-proteobacteria from the family Rickettsiaceae. Bacteria in this genus colonize arthropods, for which they can be mutualistic, commensal, or pathogenic [1]. Many of their arthropod hosts are themselves parasites to plants and vertebrates. Over evolutionary time, Rickettsia species have adapted to take advantage of this exposure to a secondary host, leading to emerging vertebrate and plant diseases caused by Rickettsia [2]. The disease-causing Rickettsia were the first to be identified and have been the focus of scientific inquiry into the genus due to their pathogenetic nature. However, in recent years, additional Rickettsia species have been identified as exclusive endosymbionts to invertebrate hosts. This group includes close relatives of pathogenic Rickettsia, as well as more distantly related clades within the genus. This distribution suggests a pattern of pathogenesis gain and loss in the genus, potentially as a strategy for horizontal transmission between parasitic arthropod hosts.
Rickettsias have been separated into four groups based on phylogenetic relationship, antigenic profile, and arthropod host. These are the Spotted Fever group, the Typhus group, the Transitional group, and the Ancestral group [3]. The Spotted Fever group is a monophyletic group comprised of bacteria carried by ixodid ticks. These bacteria include pathogens of vertebrates, as well as tick endosymbionts. The monophyletic Transitional group is carried by both insects and mites and can cause disease in vertebrates. The Typhus group is comprised of two closely related species, carried by fleas and lice, which are both capable of causing human disease. Finally, the Ancestral group is a paraphyletic group comprised of Rickettsia that do not fit into the previous groups. It contains members that are carried by insects and arachnids. No member of this group has been shown to cause vertebrate disease, however, some members have reacted with antibodies from animals or humans [4,5]. There is an additional group referred to as the Torix group within the genus, which is ancestral to the Ancestral group, however, the lack of sequenced genomes prohibits their use in genomic analysis [1].
As is common for obligate intracellular bacteria, Rickettsia species have severely reduced genomes as compared to free living bacteria, with genome size ranging from 2.3 Mb (R. buchneri) to 1.1 Mb (R. typhi) [6,7]. The obligate intracellular nature of Rickettsias decreases opportunities for horizontal gene transfer (HGT), yet genome sequencing has revealed varying levels of genome degradation across the genus [8]. Notably, the more reduced Rickettsia genomes belong to the most virulent vertebrate pathogens suggesting the inverse relationship between virulence and genome size in Rickettsia [9]. Additionally, the larger genomes tend to have repetitive elements: R. felis, R. bellii, and R. buchneri have genomes with many transposases, Rickettsia palindromic elements, and short repeat sequences [10,11,12].
Though the basic host cell infection strategy is well established, little is known about the nuances of the Rickettsia interaction with their host cell. Rickettsia are capable of secreting effector proteins into the host through multiple secretion pathways including the Rickettsiales vir homolog type 4 secretion systems (T4SS) [13]. The T4SS is capable of secreting proteins through host membranes. This makes it ideal for manipulating a host cell during cell entry and for vacuole escape. Currently, fewer than 20 proteins have been identified as secreted effectors in Rickettsia. Of these, only 3 have been shown to be secreted through the T4SS [14,15,16]. The slow progress of this characterization is due to the limitation of bacterial culture techniques.
Currently, Rickettsias cannot be cultured in cell free media. Because of this, in vitro studies can only be conducted within host cells, and potential for genetic manipulation is severely limited. In addition, the T4SS is necessary for host infection, making T4SS knockout mutants impossible within Rickettsia. As a result, any understanding of Rickettsia-host interactions relies heavily on bioinformatic analysis and comparison to bacteria that can be grown outside of host cells.
To aid in effector identification, our lab has developed a trainable bioinformatic program to predict T4SS effectors [17,18]. This tool, called Optimal Prediction software for Type 4 effectors (OPT4e), takes advantage of three learning algorithms trained on data sets of known T4 effectors and non-effectors. In the present study, we used OPT4e to predict the T4 effectomes from eight species of Rickettsia, representing two species from each of the four phylogenetic groups. We compared the putative effector repertoires, identified homologs, identified a set of core effectors common to all species in the study, and examined domains of interest looking for trends that could explain lifestyle differences. This is the first large scale bioinformatic exploration of T4 effectors in the genus Rickettsia, and will provide a foundation for functional analyses of effector proteins.
2. Results and Discussion
Two fully sequenced Rickettsia genomes were selected from each of the four phylogenetic groups of Rickettsia (Figure 1). The Spotted Fever group included R. rickettsii str. Sheila Smith and R. montanensis str. OSU 85-930; the Typhus group included R. prowazekii str. Breinl, and R. typhi str. Wilmington, the Transitional group included R. felis str. URRWXCal2, R. akari str. Hartford, and the Ancestral group included R. bellii str. RML369-C and Rickettsia sp. MEAM1. Though the Ancestral group is paraphyletic, closely related Rickettsia were selected from the group to facilitate comparison with the other groups. The goal of the study was to examine and compare the effectomes of a set of organisms from the genus Rickettsia and identify commonalities and distinguishing hallmarks for the subgroups that might lead to a greater understanding of the relationship between host, pathogenicity, evolutionary history and the rickettsal effectome.
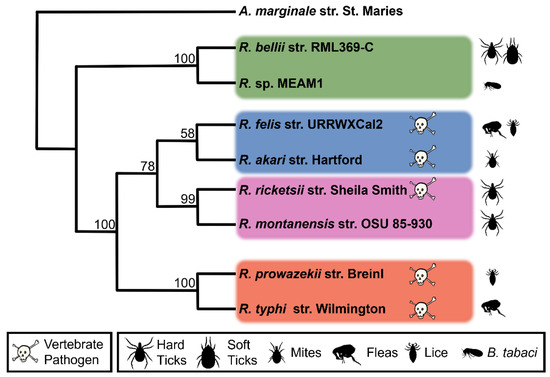
Figure 1.
Maximum likelihood phylogenetic tree of the eight species of Rickettsia used in this study. Rickettsia groups are demarcated by colored rectangles and vertebrate pathogens are marked with a skull and crossbones. Bootstrap values are given at corresponding nodes. Anaplasma marginale is used as an outgroup to root the tree. Silhouettes represent known arthropod hosts.
To supplement this comparison, available plasmids were analyzed. Three of the eight Rickettsia are known to contain plasmids. R. felis has two sequenced plasmids, one of which is a potential artifact of genome assembly [19]. As the second plasmid contains no effectors that are not present in the first (Figure 2), it does not have a significant effect on the analysis. R. bellii and R. akari have also been shown to have plasmids. However, only the plasmid for R. bellii is available on NCBI. This plasmid was included even though it is from a different strain of Rickettsia than was used for the rest of study.
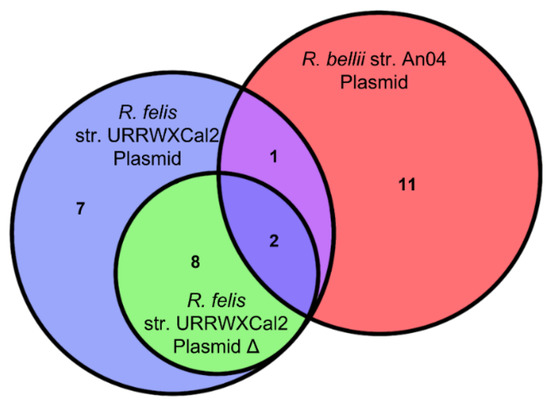
Figure 2.
The overlap of predicted effectors between the three analyzed rickettsial plasmids.
2.1. Effector Prediction Compared to Other Rickettsiales
The deduced proteomes from each genome were analyzed using OPT4e to predict putative effector repertoires. OPT4e predicted a total of 1577 effectors from the proteomes of the selected organisms, or a mean of 197 effectors per genome and a range of 84–345 (Table 1). When compared to other vector-borne organisms in the order such as Anaplasma marginale (32), Anaplasma phagocytophilum (48), Ehrlichia muris (89), and Ehrlichia ruminantium (109) only the Typhus group Rickettsia (101, 84) have a similar number of predicted effectors to the Ehrlichia organisms (Table S1) [18]. This trend was mirrored in by S4TE, an alternative T4 effector prediction program. Fewer effectors are predicted in Anaplasma species, while slightly more are predicted in Ehrlichia, and the most in Rickettsia (Table S2). While this pattern may be the result of OPT4e identifying motifs associated with host manipulation within the known effector list, and identifying effectors that are not secreted through the T4SS, different T4SS specific effector prediction programs produce the same trend, if not the same effector set. Moreover, there are distinctions between the two families that explain the large effectome in Rickettsia such as the rate of gene loss and the probability of lateral gene transfer for Rickettsiaceae as compared to Anaplasmataceae. The first difference between the groups is the capacity for conjugation and presence of plasmids in some of the Rickettsiaceae, which allows HGT between disparate strains and species within the family. This in turn could help in reducing gene loss and degradation associated with the obligate intracellular life cycle as compared to other Rickettsiales. Plasmids have been identified in members of every group of Rickettsia except for the Typhus group. Not surprisingly, this group also has the smallest genome, fewest coding sequences, and fewest predicted effectors of any of the Rickettsias. The two members of this group are the only Rickettsias with similar predicted effector counts to Ehrlichia spp. in the Anaplasmatacae. Secondly, analysis of R. bellii has revealed that ancestors of Rickettsia likely infected amoeba, allowing the common ancestor to acquire effectors from pathogens coinfecting the monocellular eukaryotes [12]. RalF, one of the three known T4 effectors in Rickettsia are most closely related to a T4 effector in Legionella pneumophila, a bacterial pathogen often found in free living amoeba [15]. Finally, many Rickettsia genomes are inundated with repetitive elements and mobile genetic elements [10,11,12]. These factors may have led to an increased abundance of effectors in the common ancestor of Rickettsia compared to that of Anaplasma, as well as an increase in the ability to maintain genes in the accessory genome and to duplicate and recombine sequences in proximity to repetitive elements and mobile genetic elements.

Table 1.
Predicted Effectors Compared to Coding Genes across the Genus Rickettsia.
2.2. Analysis of Sets of Homologous Effectors across Rickettsia
The predicted effectors were compared to the list of homologs (Table S3) generated from a BLASTp comparison of all coding sequences across the eight genomes resulting in 615 effector homolog sets (EHSs) which includes predicted effectors and homologous proteins even if they were not predicted by OPT4e. Eleven EHSs were removed from consideration as T4 effector sets (Table S4) based on their domain predictions, for a final list of 604 EHSs. Domains were predicted for all protein sequences in EHSs using the Pfam database [20] resulting in 458 domain predictions across 341 EHSs (Tables S4 and S5). After removing EHSs with domains associated with alternate secretion pathways, 449 domains remained across 330 EHSs. Consistent with other effector work, many EHSs contain predicted ankyrin repeats (49), tetracopeptide repeats (22), and leucine rich repeats (11) [21,22]. Although they can be found in other proteins across the tree of life, these motifs are overrepresented in known effectors, and perform essential functions in host manipulation [23,24,25]. Multiple transposase domains, phage-associated domains, and toxin-antitoxin domains are also present in predicted proteins. Proteins with toxin-antitoxin and phage-associated domains have been identified as secreted effectors [26,27].
There are 93 predicted domains, flagged across 81 EHSs, in which >95% of representative sequences are from eukaryotes. Of the original 449 predicted domains, 98 are eukaryotic-like domains (ELDs) from the Effective Database [28]. These domains were found to be associated with colonization of eukaryotic cells in a large-scale analysis of pathogenic bacteria. These two groups contain domains likely associated with host manipulation. Three EHSs from these groups have predicted actin binding domains. Two of them, RickA and Sca2, are known to be associated with actin-based motility [29], the third (RT4EHS_191) is a hypothetical protein only present in R. bellii, and R. rickettsii. In addition to actin-based motility, obligate intracellular bacteria manipulate actin during internalization, to support the bacteria-containing vacuole, to alter vesicular trafficking and pathogen dissemination [30]. Six EHSs were identified that contain Golgi associated and vesicle trafficking domain. Two of these (RalF and Risk1) are known effectors [15,16]. Since the Golgi is the sorting center of the cell, it is a common target in disease, and can result in altered glycosylation, and cellular trafficking [31]. Twelve EHSs can be linked to the eukaryotic cell cycle, containing predicted domains associated with the anaphase-promoting complex, kinetochore, and microtubule binding, or even DNA condensation and centromere attachment. Six EHSs have apoptosis-associated domains, and five EHSs have domains associated with arthropods. Identified domain associations are listed in the notes of Table S4.
2.3. Gene Fragment Analysis
As discussed above, rickettsial organisms have small and degraded genomes as compared to free living bacteria. This degradation leads to fragmentation and potentially pseudogenization of genes. Gene truncation, or fragmentation, does not necessarily preclude functionality, but the gene may no longer be functionally equivalent. To identify cases in which genes are homologous to portions of larger genes, we searched for instances in which smaller genes met all homolog criteria with a larger gene except the length criteria. In these cases, the smaller gene was designated as a gene fragment (Table S6). When the homolog sets for all genes in the eight Rickettsia genomes are analyzed for gene fragments, 1.33% to 21.44% of annotated coding sequences in the genome are flagged as fragments of larger genes (Table 2). This is a drastic variation between species, and may be a result of different algorithms being used when annotating the genomes. However, when all homolog sets smaller than 300 bp are excluded from the analysis, the variation is reduced to 0.9–9.6%. Interestingly, this correction also decreases the proportion of gene fragments in R. akari and R. rickettsii by 11.67% and 13.15%, respectively. The majority of analyzed species have between 6% and 10% of annotated coding sequences (larger than 300 bp) flagged as gene fragments (Table 2). The Typhus group, which also has the smallest genomes, has the smallest proportion of gene fragments. This could be a result of conservative CDS annotation. This is supported by the low proportion of gene fragments, even when CDSs smaller than 300 bp are included. It may also be associated with the small genome sizes in this group. The shrinking genomes likely lost gene fragments and pseudogenes more quickly than other Rickettsia species.
Table 2.
Gene Fragment Concentration in Different EHSs.
Effectors have a higher proportion of gene fragments than do other coding sequences. This is likely due to the nature of effectors. These genes are often part of the accessory genome which implies more rapid mutation and the potential for degradation. This change is the most evident in R. sp. MEAM1, where 25% of predicted effectors are gene fragments while only 7% of non-effectors from homolog sets that meet the size criteria are gene fragments.
2.4. G+C Content Analysis
Along with a higher rate of fragmentation, predicted effectors have a significantly different G+C content than the rest of the genome, with a mean consistently below that of other coding sequences (Figure 3). A similar variation in G+C content has been identified in T4 effectors of Legionella [32]. Regions of varying G+C content are often associated with HGT, and the low G+C content seen in Legionella effectors has been attributed to HGT, however, the lower G+C content of effectors in Rickettsia seems unlikely to be associated with HGT as Rickettsia already have very low G+C content and regions of other genomes with even lower G+C content would be uncommon.
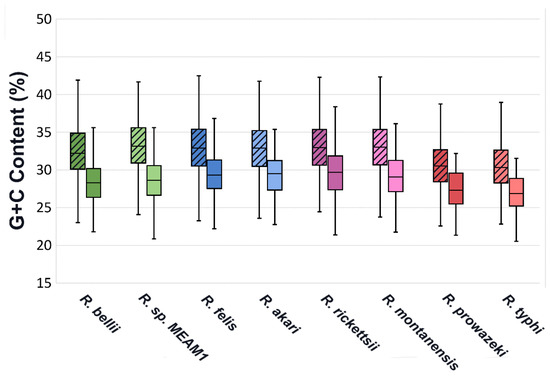
Figure 3.
G+C content range of EHSs for each species compared to all other coding sequences. Non-effector genes are represented by plots with diagonal hatching, while Effectors are in open plots. Effector sets are significantly different than Non-effectors in all species, with p values below 0.001 in all instances.
2.5. Effector Categorization
EHSs are separated into five groups (Figure 4 and Figure 5, Table S7). The Core Group consists of EHSs with at least one protein in every genome. Group Specific Effectors are only found in the two analyzed genomes from any of the four Rickettsia groups. Species Specific Effectors are only present in one species. Plasmid Specific effectors are only present on the analyzed plasmids. The Other group contains all EHSs that do not match any of the first four groups. Of the 604 EHSs, 42 are Core, 50 are Group Specific, 352 are Species Specific, 24 are Plasmid Specific, and 136 are Other. The EHSs in the Other group are split into three categories (GL1, GL2 and GL3) (Table S8). GL1 EHSs are present in species such that the pattern could be explained by a single gene gain or loss event across the phylogeny. GL2 contains gene patterns that are the result of at least two gene gain or loss events. GL3 contains EHSs showing a minimum of three gene gain or loss evens (Figure S1). A few possibilities of these patterns are illustrated in Figure 5B. GL1 contains 22 EHSs, GL2 contains 98, and GL3 contains 15. Though GL1 contains few EHSs compared to GL2, the EHSs used in this observation excluded Species Specific and Group Specific effectors, all of which could be explained by a single gene gain event.
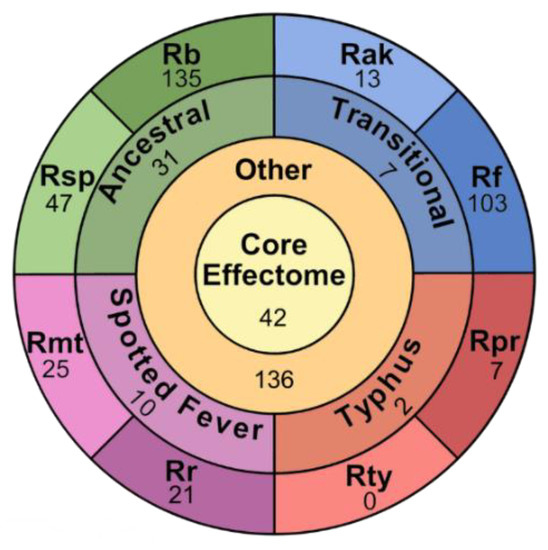
Figure 4.
Categorization of 580 chromosomal EHSs. Each section contains a number corresponding to the number of EHSs found in the category. The center circle labeled “core effectome” represents all chromosomal EHSs conserved in all analyzed species of Rickettsia. The “other” ring represents the EHSs that are not core, but also not specific to a single group or species. The “group” ring is split into four segments, representing the four groups of Rickettsia analyzed in this study. Finally, the “species” ring is split into eight segments, representing each species used in this study. The 2–3 letter labels correspond to the species name with Rb standing in for R. bellii str. RML396-C, Rsp for R. sp. MEAM1, Rf for R. felis str. URRWXCal2, Rak for R. akari str. Hartford, Rr for R. rickettsia str. Sheila Smith, Rmt for R. montanensis str. OSU 85-930, Rpr for R. prowazekii str. Breinl, and Rt for R. typhi str. Wilmington.
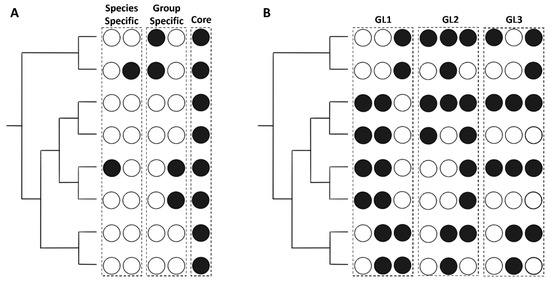
Figure 5.
Examples of the categorization scheme used for the EHSs. Circles correspond to the species at the tips of the phylogenies to the right of the figure. Black circles indicate the presence of a homolog in the species, while white circles represent an absence. Instances that meet the conditions for a specific category are grouped within a dashed box and the corresponding group is labeled above. (A) shows the instances in which EHSs would be categorized into Species Specific, Group Specific, or Core. (B) focuses on the Other group, and gives examples of instances that would be categorized as GL1, GL2, or GL3.
2.5.1. Core Effectors
Of the 42 Core EHSs, 28 contain sequences that are predicted by OPT4e in every species. Although 14 of the predictions are not consistent across all homologs, we think it unlikely that effectors conserved across all species would be secreted using different systems. Seventeen core EHSs contain domains found primarily in eukaryotes or flagged as ELDs. Two proteins that stand out in this list are Sca4 and Patatin 1 (RT4EHS_043, RT4EHS_051). These two proteins are known effectors in Rickettsia with no identified secretion system [33,34]. In addition to being maintained on the Rickettsia chromosomes, Patatin 1 is present on all 3 plasmids analyzed, although just over half of the homologs of Patatin1 are predicted by OPT4e. Several core homologs contain domains associated with eukaryotic cytoskeleton interaction, and host replication (RT4EHS_002, RT4EHS_013, RT4EHS_014, RT4EHS_016, RT4EHS_043, RT4EHS_061), which could play a role in bacterial motility or vesicle trafficking. Two of the core proteins predicted are DNA polymerase components. Though these cannot be eliminated out of hand, to our knowledge, there are no known cases of DNA polymerase subunits being secreted by bacteria, making it unlikely that these conserved proteins moonlight as effectors in Rickettsia.
2.5.2. Group Specific Effectors
Group specific effectors comprise 52 of the EHSs. Most of them are found in the Ancestral group. As indicated by the name, this group is ancestral to the other Rickettsia, the high number of group specific effectors is therefore not surprising. In contrast the Typhus group has only 3 group specific effectors, the lowest number of any of the groups, despite being less closely related to the Spotted Fever and Transitional groups than they are to each other. R. typhi and R. prowazekii have the most reduced genomes of any of the Rickettsia, which may be reflected in the low number of group specific effectors.
2.5.3. Species Specific Effectors
The Species Specific EHSs show the same trends as the overall OPT4e predictions. R. bellii has the highest number, followed by R. felis. The Typhus group has the fewest, and the other Rickettsia fall somewhere in between (Figure 4). In R. bellii and R. felis, species-specific effectors also comprise 45.6%, and 44.6% of their total predicted effectors, respectively. This is higher than any of the other species. R. sp. MEAM1 comes the closest with 29.6% of effectors being species-specific. All other species have putative effectomes comprised of less than 18% species specific effectors.
2.5.4. Plasmid-Borne Effectors
Of the 26 EHSs found on the 3 plasmids analyzed, Patatin1 (RT4EHS_051) and RT4EHS_17 are maintained in the genomes of at least one species of Rickettsia and RT4EHS_594 is closely related to chromosomal EHSs. These proteins are addressed in either the core effectors section or host specific section. Of the remaining 23, 8 have predicted domains, all but one of which are associated with DNA binding, or antibiotic resistance. That one exception is an ankyrin repeat containing protein (RT4EHS_587) and was the only plasmid exclusive EHS containing either ELDs or eukaryotic domains.
2.5.5. Other Effectors
The 136 EHSs classified as “Other”, are uniquely capable of illuminating trends within the genus. This group contains proteins that were lost in evolutionary lineages or gained within a single clade, Genes that were lost or gained multiple times, or genes that were potentially spread through HGT between Rickettsia. The separation of these EHSs into the 3 groups described above is intended to indicate patterns of gene loss and gain. GL3 is the most likely to have occurred by HGT between different clades of Rickettsia, GL2 by either HGT between Rickettsia or through differential selection, and GL1 being instances of gene loss or gain at a single node in the evolutionary tree (Figure 5).
Of the 22 GL1 EHSs, seven were found in all members of the Spotted Fever group and Transitional group. One, three, and four EHSs were absent only in the Spotted Fever, Ancestral, and Typhus group, respectively and eight were absent in only a single species (Figure 6A). R. bellii and R. felis have the highest number of GL2 effectors (63 in both). Interestingly, they share 43 of these, 20 of which are only present in these two species (Figure 6B). In comparison, only 31 EHSs are shared exclusively between R. bellii and its group member R. sp. MEAM, and only 7 EHSs are exclusive to R. felis and R. akari (Figure 4). In the GL3 effectors, R. bellii, R. felis, and R. montanensis each contain 13 EHSs, most of which they have in common with each other (Figure 6C). The large number of genes shared between R. bellii and R. felis has been noted before [3]. It is, however, interesting to note that this trend is maintained, and even exaggerated, in predicted effectors.
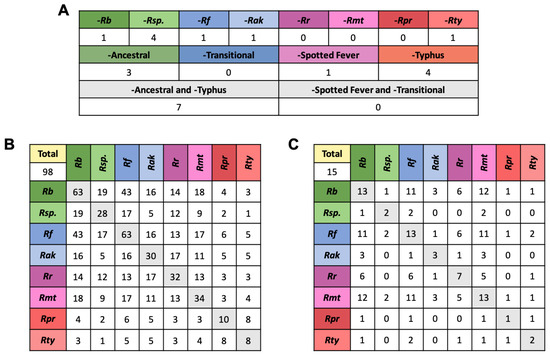
Figure 6.
Representation of GL1,2,3 EHSs across Rickettsia. (A) shows the GL1 genes that are absent in a single species, a single group, or a combination of groups, indicating the evolutionary node at which a gain or loss event likely occurred. (B,C) show the number of putative effectors from the GL2 and GL3 group, respectively, that are present in every species, as well as how many they share with other species. This shows patterns of shared genes between distantly related species. For an example of how homologs are placed in these tables see Figure S1. The 2–3 letter labels correspond to species with Rb standing in for R. bellii str. RML396-C, Rsp. for R. sp. MEAM1 (B. tabaci), Rf for R. felis str. URRWXCal2, Rak for R. akari str. Hartford, Rr for R. rickettsia str. Sheila Smith, Rmt for R. montanensis str. OSU 85-930, Rpr for R. prowazekii str. Breinl, and Rt for R. typhi str. Wilmington.
2.6. Effectors in Pathogenc and Non-Pathogec Rickettsia
One effector was identified in all Rickettsia known to be vertebrate pathogens. RT4EHS_031 is a 241–319 amino acid hypothetical protein found in R. felis, akari, rickettsii, prowazekii, and typhi. HMMer analysis predicts a SieB domain in this protein, which is associated with phage superinfection exclusion. Effector, RT4EHS_196, is found only in the Transitional group and R. Rickettsia. This protein is annotated as hypothetical and has no recognized domains (Table S9).
There are no effector homologs present in all non-pathogenic Rickettsia species. However, seven effectors are shared between R. bellii and R. montanensis. One of these genes is a gene fragment of a larger gene in R. sp. MEAM1 (RT4EHS_146, RT4EHS_416), the only other non-pathogenic Rickettsia. HMMer predictions for this EHS included multiple ELDs as well as a eukaryote specific domain found in vesicle trafficking proteins (SNAP) (Tables S5 and S7).
2.7. Arthropod Host Associated Effectors
Some effectors are associated with colonization of specific arthropod hosts. Two effectors (RT4EHS_129, RT4EHS_163) were found only in tick-borne Rickettsia (Table S9). RT4EHS_129 has multiple eukaryotic domains predicted, including a clavanin domain, which can be associated with innate immune system modulation, as well as a domain associated with exocytosis, and another associated with histone modification. In addition, transcription of this gene is upregulated at low temperatures, indicating a possible association with the arthropod host [35]. RT4EHS_163 has a Fic domain which was first identified in bacterial toxins, members of the group have been identified as effectors [36]. An additional four EHSs were shared by all tick specific Rickettsias as well as R. felis. Though the primary vector for R. felis is the cat flea (Ctenocephalides felis), it has been detected in wild ticks, and shown to be capable of vertical transmission in at least one tick species [37,38]. Of these, two EHSs have eukaryotic domains. One contains both a helix turn helix domain (HTH3) and an A+T rich interaction domain (ARID) found in eukaryotic transcription factors, while the other contains a nucleotide diphosphate cleavage domain (NUDIX) and a repeat domain (WD40_alt) that is specific to eukaryotes.
No putative effectors were conserved across the genomes of flea-borne Rickettsias in this homolog analysis. However, the two putative effectors identified on the plasmid of Rickettsia felis (RT4EHS_017, RT4EHS_594) could be associated with colonization of the flea host. Interestingly, along with the low identity match between RT4EHS_594 and RT4EHS_018, a third, much shorter gene in R. sp. MEAM1 (RT4EHS_371) has identity to the two genes. Even though it is classified as a gene fragment, RT4EHS_371 matches to both RT4EHS_594 and RT4EHS_018, and is predicted to be an effector. Although not homologous by our definition, these three genes contain a region conserved in all insect-borne Rickettsia. The presence of both genes on a plasmid in Rickettsia felis also highlights the potential for host switching mediated by HGT within Rickettsia.
2.8. The Relationship between R. bellii and R. felis
Although R. bellii and R. felis belong to different clades in the phylogeny of the Rickettsias they have the highest numbers of predicted effectors at 296 and 251 effectors included in the final set of EHSs, respectively. Compared to R. bellii and R. felis, R. sp. MEAM1, the other member of the Ancestral group, and R. akari, from the Transitional group, have much smaller numbers of putative effectors, suggesting that R. belli and R. felis may have undergone a genome expansion that included expansion of the effector repertoire. These two organisms also have a notably higher proportion of mobile genetic elements and repeat sequences compared to other Rickettsia species, which may correspond to genomic rearrangement leading to a decrease in synteny between these organisms and their closely related Rickettsia [3]. This could have facilitated expansion of the effectome through gene duplication and shuffling. However, even with signs of genomic rearrangement, these bacteria do not have a higher proportion of gene fragments than the other analyzed species. In R. bellii, 9.46% of genes longer than 300 bp are categorized as gene fragments, while in R. felis 13.20% fall into this category (Table 2). These values are lower than all other Rickettsia except for the Typhus group, discussed above. The remarkable number of effectors in the “Other” category shared by these two species also suggest either convergent evolution or HGT between these organisms, potentially associated with host adaptation (Figure 6B,C).
Even though R. bellii is a tick-borne Rickettsia, and the primary arthropod host for R. felis is the cat flea, these two Rickettsia have a larger potential host pool than the other species in this study. R. bellii has been identified in more than 25 species of hard and soft ticks [39], while R. felis has been found in, and can persist in hard ticks, fleas, lice, and mosquitos [40,41]. Their large and partially convergent effector repertoires may be an important factor in this broad arthropod host range.
3. Materials and Methods
3.1. Sequences
Eight fully sequenced Rickettsia genomes were selected which are available at NCBI. Genomes used included: R. bellii str. RML369-C (CP000087.1), R. sp. MEAM1 (CP016305.1), R. felis str. URRWXCal2 (CP000053.1), R. akari str. Hartford (CP000847.1), R. rickettsii str. Sheila Smith (CP000848.1), R. montanensis str. OSU 85-930 (CP003340.1), R. prowazekii str. Breinl (CP004889.1), and R. typhi str. Wilmington (AE017197.1). It became evident that in R. sp. MEAM1 many genes annotated as pseudogenes were nearly full-length reading frames with homologs in the other species. Therefore, we generated coding sequences for R. sp. MEAM1 pseudogenes. If multiple ORFs were present within the gene, they were assigned the locus tag corresponding to the pseudogene followed by a letter to denote the ORF. All manually curated coding sequences can be found in supplementary data (File S1). We included plasmid sequences in our analyses when available. The R. bellii isolate AN04 (CP015011.1) and R. felis URRWXCal2 (CP000054.1, CP000055.1) were used.
3.2. Phylogenetic Analysis
All phylogenetic analysis was conducted using MEGAX [42]. Protein sequences for FtsZ, GltA, GroEL, and rpoB were acquired from the eight Rickettsia genomes, and A. marginale str. St. Maries, which was used as the out group. Sequences were concatenated and aligned using MUSCLE alignment under base parameters. The start sequences of all concatenated genes were examined, and any alignment overlap from one protein sequence to another was manually corrected. The phylogenetic tree was constructed using the JTT maximum likelihood model with 10,000 replicates.
3.3. Effector Prediction
We used the OPT4e program to predict effectors from the deduced proteomes. Three sequences of known type 4 secreted effectors from Rickettsia (RT0135, RT0362, RT0600) to the default training set [14,15,16]. Though homologs to these proteins are found in multiple species, only the protein sequences from R. typhi were used. Protein sequences with internal stop codons cannot be run through this program, so sequences with internal stop codons were run as the open reading frame before the first internal stop codon. Outputs were then used for further analysis. In addition to the above, OPT4e was also used to predict effectors in A. marginale str. St. Maries (CP000030.1), E. ruminantium str. Springbokfontein7 (CP040111.1), and E. muris str. AS145 (CP006917.1) using the original training data set without the addition of T4 effectors from Rickettsia. We used published OPT4e prediction data for A. phagocytophilum from Ashari et al., 2019.
Effector prediction was conducted using S4TE 2.0 [43]. Anaplasma and Ehrlichia species listed above were accessed via the publicly available data set provided by S4TE 2.0, while R. bellii, R. montanensis, and R. typhi were uploaded as genbank files and analyzed. As E. ruminantium str. Springbokfontein7 was not available, str. Gardel was used in its place.
3.4. Homolog Identification
To generate a homolog set across the eight genomes, we used an all versus all BlastP search of all proteins within the genomes to identify regions of high similarity between protein sequences. This search was executed with a local download of Blast+ version 2.10.1 [44]. For protein comparison, we only included blast hits that had an E value of less than 1 × 10−15 and a percent identity greater than 40%. If the combined length of all blast hits between two proteins, excluding overlap between blast hits, was greater than 60% of the length of both proteins, these were considered homologs and used in further analysis. If the length of all blast hits was greater than 60% of one protein and less than 60% of the other, the shorter protein was designated as a gene fragment and analyzed separately. All other hits were discarded. All homolog sets in which the longest gene was shorter than 300 bp were removed from the data set. When generating the effector homolog list, all homolog sets in which less than 50% of the homologs were predicted to be secreted through the T4SS were eliminated from the final list.
3.5. Protein Domain Identification
To characterize proteins, we used HMMER3.3.1 to check protein sequences against the PfamA database [20,45]. All identified domains above the default threshold for the program were used for further analysis.
3.6. Domain of Interest Identification
To identify domains of interest, we compared the list of Pfam domains predicted by HMMer to the data set of Eukaryotic Like Domains (ELDs) available on effectivedb.org [28]. To identify additional eukaryotic domains, we looked at each domain manually on the Pfam website and identified the origin of the sample sequences for the domain on the tree of life. Domains in which >95% of the sequences originated from eukaryotes were designated as predominantly eukaryotic domains.
3.7. Comparison of G+C Content
Using the genomes listed above, the G+C content for all genes was calculated and genes were separated into effectors (genes found in the final effector list), and non-effectors (genes that did not fall into the final effector list). These two groups were then evaluated for normality using the Shapiro–Wilks test with an alpha value of 0.05, and the populations were compared by t-test or Mann–Whitney test based on the results.
4. Conclusions
OPT4e predicts a large number of T4 effectors across the genus Rickettsia. This result is particularly striking when compared to the lower prediction counts of closely related vacuole-bound intracellular bacteria. R. bellii and R. felis, the largest Rickettsia genomes in this study, have the highest number of effectors, the highest proportion of effectors, and the most effectors in common, despite their distance on the phylogenetic tree and difference in lifestyle. Of the 604 predicted EHSs, more than 60% were found in only a single species. The core and group-specific effectors comprise an additional 15%. The remaining 135 EHSs comprise the GL1, GL2, and GL3 groups. The GL1 group shows patterns of gene gain or loss within clades, however, the GL2 and GL3 groups show indications of HGT and convergent evolution. In the GL2 group, with 98 EHSs, 44% are shared between R. bellii and R. felis, while in the GL3 group, 11–12 of the 15 EHSs are shared between R. bellii, R. felis, and R. montanensis. This indicates a pattern of HGT between the separate groups of Rickettsia. Interestingly, two of the three species that stand out in this effector analysis are not known to be vertebrate pathogens. This suggests that these common effectors may be associated with colonization of arthropods rather than vertebrates.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232415513/s1.
Author Contributions
Conceptualization, J.A.A. and K.A.B.; methodology, J.A.A. and K.A.B.; validation, J.A.A. and K.A.B.; formal analysis, J.A.A.; investigation, J.A.A.; data curation, J.A.A.; writing—original draft preparation, J.A.A.; writing—review and editing, J.A.A. and K.A.B.; visualization, J.A.A. and K.A.B.; supervision, K.A.B.; funding acquisition, K.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by Agriculture & Food Research Initiative Competitive Grant no. 2018-67015-28304 from the USDA National Institute of Food and Agriculture.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Publicly available data were analyzed in this study. All data were downloaded from NCBI (www.ncbi.nlm.nih.gov/genome) on 14 May 2020. Specific accession numbers are listed Section 3.1.
Acknowledgments
We acknowledge Alyssa Aspinwall for her help with figure design, and Jason Park for his expertise and insight.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Perlman, S.J.; Hunter, M.S.; Zchori-Fein, E. The Emerging Diversity of Rickettsia. Proc. R. Soc. B 2006, 273, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.E.; Martijn, J.; Vosseberg, J.; Köstlbacher, S.; Ettema, T.J.G. The Evolutionary Origin of Host Association in the Rickettsiales. Nat. Microbiol. 2022, 7, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.J.; Williams, K.; Shukla, M.; Snyder, E.E.; Nordberg, E.K.; Ceraul, S.M.; Dharmanolla, C.; Rainey, D.; Soneja, J.; Shallom, J.M.; et al. Rickettsia Phylogenomics: Unwinding the Intricacies of Obligate Intracellular Life. PLoS ONE 2008, 3, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Bozeman, M.F.; Elisberg, B.L.; Humphries, J.W.; Runcik, K.; Palmer, D.B. Serologic Evidence of Rickettsia canada Infection of Man. J. Infect. Dis. 1970, 121, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.B.; Cunha-Filho, N.A.; Pacheco, R.C.; Ferreira, F.; Pappen, F.G.; Farias, N.A.R.; Larsson, C.E.; Labruna, M.B. Canine Infection by Rickettsiae and Ehrlichiae in Southern Brazil. Am. J. Trop. Med. 2008, 79, 102–108. [Google Scholar] [CrossRef]
- Rao, Q.; Rollat-Farnier, P.-A.; Zhu, D.-T.; Santos-Garcia, D.; Silva, F.J.; Moya, A.; Latorre, A.; Klein, C.C.; Vavre, F.; Sagot, M.-F.; et al. Genome Reduction and Potential Metabolic Complementation of the Dual Endosymbionts in the Whitefly Bemisia tabaci. BMC Genom. 2015, 16, 226:1–226:13. [Google Scholar] [CrossRef]
- Zomorodipour, A.; Andersson, S.G.E. Obligate Intracellular Parasites: Rickettsia prowazekii and Chlamydia trachomatis. FEBS Lett. 1999, 452, 11–15. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Verma, C.; Chow, V.T.K. Comparative Study of Overlapping Genes in Bacteria, with Special Reference to Rickettsia prowazekii and Rickettsia conorii. Int. J. Syst. Evol. Microbiol. 2005, 55, 1205–1209. [Google Scholar] [CrossRef]
- Fournier, P.-E.; El Karkouri, K.; Leroy, Q.; Robert, C.; Giumelli, B.; Renesto, P.; Socolovschi, C.; Parola, P.; Audic, S.; Raoult, D. Analysis of the Rickettsia africae Genome Reveals That Virulence Acquisition in Rickettsia Species May Be Explained by Genome Reduction. BMC Genom. 2009, 10, 166:1–166:15. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Joardar, V.; Williams, K.P.; Driscoll, T.; Hostetler, J.B.; Nordberg, E.; Shukla, M.; Walenz, B.; Hill, C.A.; Nene, V.M.; et al. A Rickettsia Genome Overrun by Mobile Genetic Elements Provides Insight into the Acquisition of Genes Characteristic of an Obligate Intracellular Lifestyle. J. Bacteriol. 2012, 194, 376–394. [Google Scholar] [CrossRef]
- Ogata, H.; Renesto, P.; Audic, S.; Robert, C.; Blanc, G.; Fournier, P.-E.; Parinello, H.; Claverie, J.-M.; Raoult, D. The Genome Sequence of Rickettsia felis Identifies the First Putative Conjugative Plasmid in an Obligate Intracellular Parasite. PLoS Biol. 2005, 3, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; La Scola, B.; Audic, S.; Renesto, P.; Blanc, G.; Robert, C.; Fournier, P.-E.; Claverie, J.-M.; Raoult, D. Genome Sequence of Rickettsia bellii Illuminates the Role of Ancestral Protozoa in Gene Exchanges between Intracellular Pathogens. PLoS Genet. 2006, 2, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.J.; Phan, I.Q.H.; Driscoll, T.P.; Guillotte, M.L.; Lehman, S.S.; Rennoll-Bankert, K.E.; Subramanian, S.; Beier-Sexton, M.; Myler, P.J.; Rahman, M.S.; et al. The Rickettsia Type IV Secretion System: Unrealized Complexity Mired by Gene Family Expansion. Pathog. Dis. 2016, 74, ftw058:1–ftw058:13. [Google Scholar] [CrossRef] [PubMed]
- Lehman, S.S.; Noriea, N.F.; Aistleitner, K.; Clark, T.R.; Dooley, C.A.; Nair, V.; Kaur, S.J.; Rahman, M.S.; Gillespie, J.J.; Azad, A.F.; et al. The Rickettsial Ankyrin Repeat Protein 2 Is a Type IV Secreted Effector That Associates with the Endoplasmic Reticulum. mBio 2018, 9, e00975-18:1–e00975-18:15. [Google Scholar] [CrossRef] [PubMed]
- Rennoll-Bankert, K.E.; Rahman, M.S.; Gillespie, J.J.; Guillotte, M.L.; Kaur, S.J.; Lehman, S.S.; Beier-Sexton, M.; Azad, A.F. Which Way In? The RalF Arf-GEF Orchestrates Rickettsia Host Cell Invasion. PLOS Pathog. 2015, 11, e1005115:1–e1005115:28. [Google Scholar] [CrossRef] [PubMed]
- Voss, O.H.; Gillespie, J.J.; Lehman, S.S.; Rennoll, S.A.; Beier-Sexton, M.; Rahman, M.S.; Azad, A.F. Risk1, a Phosphatidylinositol 3-Kinase Effector, Promotes Rickettsia typhi Intracellular Survival. mBio 2020, 11, e00820-20:1–e00820-20:22. [Google Scholar] [CrossRef]
- Ashari, Z.E.; Dasgupta, N.; Brayton, K.A.; Broschat, S.L. An Optimal Set of Features for Predicting Type IV Secretion System Effector Proteins for a Subset of Species Based on a Multi-Level Feature Selection Approach. PLoS ONE 2018, 13, e0197041:1–e0197041:16. [Google Scholar] [CrossRef]
- Ashari, Z.E.; Brayton, K.A.; Broschat, S.L. Prediction of T4SS Effector Proteins for Anaplasma phagocytophilum Using OPT4e, a New Software Tool. Front. Microbiol. 2019, 10, 1391:1–1391:10. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Beier, M.S.; Rahman, M.S.; Ammerman, N.C.; Shallom, J.M.; Purkayastha, A.; Sobral, B.S.; Azad, A.F. Plasmids and Rickettsial Evolution: Insight from Rickettsia felis. PLoS ONE 2007, 2, e266:1–e266:17. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Burstein, D.; Amaro, F.; Zusman, T.; Lifshitz, Z.; Cohen, O.; Gilbert, J.A.; Pupko, T.; Shuman, H.A.; Segal, G. Genomic Analysis of 38 Legionella Species Identifies Large and Diverse Effector Repertoires. Nat. Genet. 2016, 48, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Noroy, C.; Meyer, D.F. The Super Repertoire of Type IV Effectors in the Pangenome of Ehrlichia Spp. Provides Insights into Host-Specificity and Pathogenesis. PLOS Comput. Biol. 2021, 17, e1008788:1–e1008788:26. [Google Scholar] [CrossRef]
- Bröms, J.E.; Edqvist, P.J.; Forsberg, Å.; Francis, M.S. Tetratricopeptide Repeats Are Essential for PcrH Chaperone Function in Pseudomonas aeruginosa Type III Secretion. FEMS Microbiol. Lett. 2006, 256, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Haraga, A.; Miller, S.I. A Salmonella enterica Serovar Typhimurium Translocated Leucine-Rich Repeat Effector Protein Inhibits NF- κB-Dependent Gene Expression. Infect. Immun. 2003, 71, 4052–4058. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Luhrmann, A.; Satoh, A.; Laskowski-Arce, M.A.; Roy, C.R. Ankyrin Repeat Proteins Comprise a Diverse Family of Bacterial Type IV Effectors. Science 2008, 320, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Campanacci, V.; Mukherjee, S.; Roy, C.R.; Cherfils, J. Structure of the Legionella Effector AnkX Reveals the Mechanism of Phosphocholine Transfer by the FIC Domain. EMBO J. 2013, 32, 1469–1477. [Google Scholar] [CrossRef]
- Triplett, L.R.; Shidore, T.; Long, J.; Miao, J.; Wu, S.; Han, Q.; Zhou, C.; Ishihara, H.; Li, J.; Zhao, B.; et al. AvrRxo1 Is a Bifunctional Type III Secreted Effector and Toxin-Antitoxin System Component with Homologs in Diverse Environmental Contexts. PLoS ONE 2016, 11, e0158856:1–e0158856:16. [Google Scholar] [CrossRef]
- Eichinger, V.; Nussbaumer, T.; Platzer, A.; Jehl, M.-A.; Arnold, R.; Rattei, T. EffectiveDB—Updates and Novel Features for a Better Annotation of Bacterial Secreted Proteins and Type III, IV, VI Secretion Systems. Nucleic Acids Res. 2015, 44, D669–D674. [Google Scholar] [CrossRef]
- Reed, S.C.O.; Lamason, R.L.; Risca, V.I.; Abernathy, E.; Welch, M.D. Rickettsia Actin-Based Motility Occurs in Distinct Phases Mediated by Different Actin Nucleators. Curr. Biol. 2014, 24, 98–103. [Google Scholar] [CrossRef]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking Host Cell Highways: Manipulation of the Host Actin Cytoskeleton by Obligate Intracellular Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 107:1–107:8. [Google Scholar] [CrossRef]
- Aistleitner, K.; Clark, T.; Dooley, C.; Hackstadt, T. Selective Fragmentation of the trans-Golgi Apparatus by Rickettsia rickettsii. PLOS Pathog. 2020, 16, e1008582. [Google Scholar] [CrossRef] [PubMed]
- Burstein, D.; Zusman, T.; Degtyar, E.; Viner, R.; Segal, G.; Pupko, T. Genome-Scale Identification of Legionella pneumophila Effectors Using a Machine Learning Approach. PLOS Pathog. 2009, 5, e1000508:1–e1000508:12. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Gillespie, J.J.; Kaur, S.J.; Sears, K.T.; Ceraul, S.M.; Beier-Sexton, M.; Azad, A.F. Rickettsia typhi Possesses Phospholipase A2 Enzymes That Are Involved in Infection of Host Cells. PLOS Pathog. 2013, 9, e1003399:1–e1003399:1. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sears, K.T.; Ceraul, S.M.; Gillespie, J.J.; Allen, E.D.; Popov, V.L.; Ammerman, N.C.; Rahman, M.S.; Azad, A.F. Surface Proteome Analysis and Characterization of Surface Cell Antigen (Sca) or Autotransporter Family of Rickettsia typhi. PLOS Pathog. 2012, 8, e1002856:1–e1002856:17. [Google Scholar] [CrossRef]
- Ellison, D.W.; Clark, T.R.; Sturdevant, D.E.; Virtaneva, K.; Hackstadt, T. Limited Transcriptional Responses of Rickettsia rickettsii Exposed to Environmental Stimuli. PLoS ONE 2009, 4, e5612:1–e5612:11. [Google Scholar] [CrossRef]
- Luong, P.; Kinch, L.N.; Brautigam, C.A.; Grishin, N.V.; Tomchick, D.R.; Orth, K. Kinetic and Structural Insights into the Mechanism of AMPylation by VopS Fic Domain. J. Biol. Chem. 2010, 285, 20155–20163. [Google Scholar] [CrossRef]
- Harris, E.K.; Verhoeve, V.I.; Banajee, K.H.; Macaluso, J.A.; Azad, A.F.; Macaluso, K.R. Comparative Vertical Transmission of Rickettsia by Dermacentor variabilis and Amblyomma maculatum. Ticks Tick-borne Dis. 2017, 8, 598–604. [Google Scholar] [CrossRef]
- Roth, T.; Lane, R.S.; Foley, J. A Molecular Survey for Francisella tularensis and Rickettsia Spp. In Haemaphysalis leporispalustris (Acari: Ixodidae) in Northern California. J. Med. Entomol. 2016, 54, 492–495. [Google Scholar] [CrossRef]
- Krawczak, F.S.; Labruna, M.B.; Hecht, J.A.; Paddock, C.D.; Karpathy, S.E. Genotypic Characterization of Rickettsia bellii Reveals Distinct Lineages in the United States and South America. BioMed Res. Int. 2018, 2018, 8505483:1–8505483:8. [Google Scholar] [CrossRef]
- Behar, A.; McCormick, L.J.; Perlman, S.J. Rickettsia felis Infection in a Common Household Insect Pest, Liposcelis bostrychophila (Psocoptera: Liposcelidae). Appl. Environ. Microbiol. 2010, 76, 2280–2285. [Google Scholar] [CrossRef]
- Fongsaran, C.; Jirakanwisal, K.; Tongluan, N.; Latour, A.; Healy, S.; Christofferson, R.C.; Macaluso, K.R. The Role of Cofeeding Arthropods in the Transmission of Rickettsia felis. PLOS Negl. Trop. Dis. 2022, 16, e0010576:1–e0010576:12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Noroy, C.; Lefrançois, T.; Meyer, D.F. Searching Algorithm for Type IV Effector Proteins (S4TE) 2.0: Improved Tools for Type IV Effector Prediction, Analysis and Comparison in Proteobacteria. PLOS Comput. Biol. 2019, 15, e1006847:1–e1006847:12. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421:1–421:9. [Google Scholar] [CrossRef] [PubMed]
- HMMER. Available online: http://hmmer.org/ (accessed on 25 July 2020).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).