
Gold Nanosystems Covered with Doxorubicin/DNA Complexes: A Therapeutic Target for Prostate and Liver Cancer

 ,
,  ,
, 
 , ,
, , 
Abstract
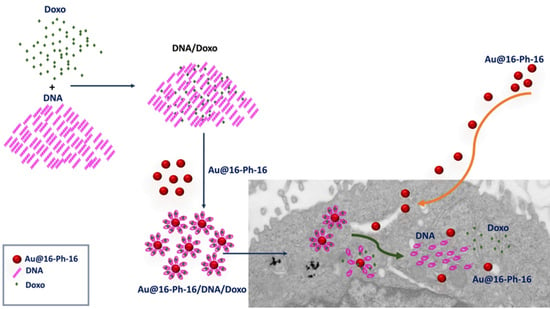
:
1. Introduction
2. Results and Discussion
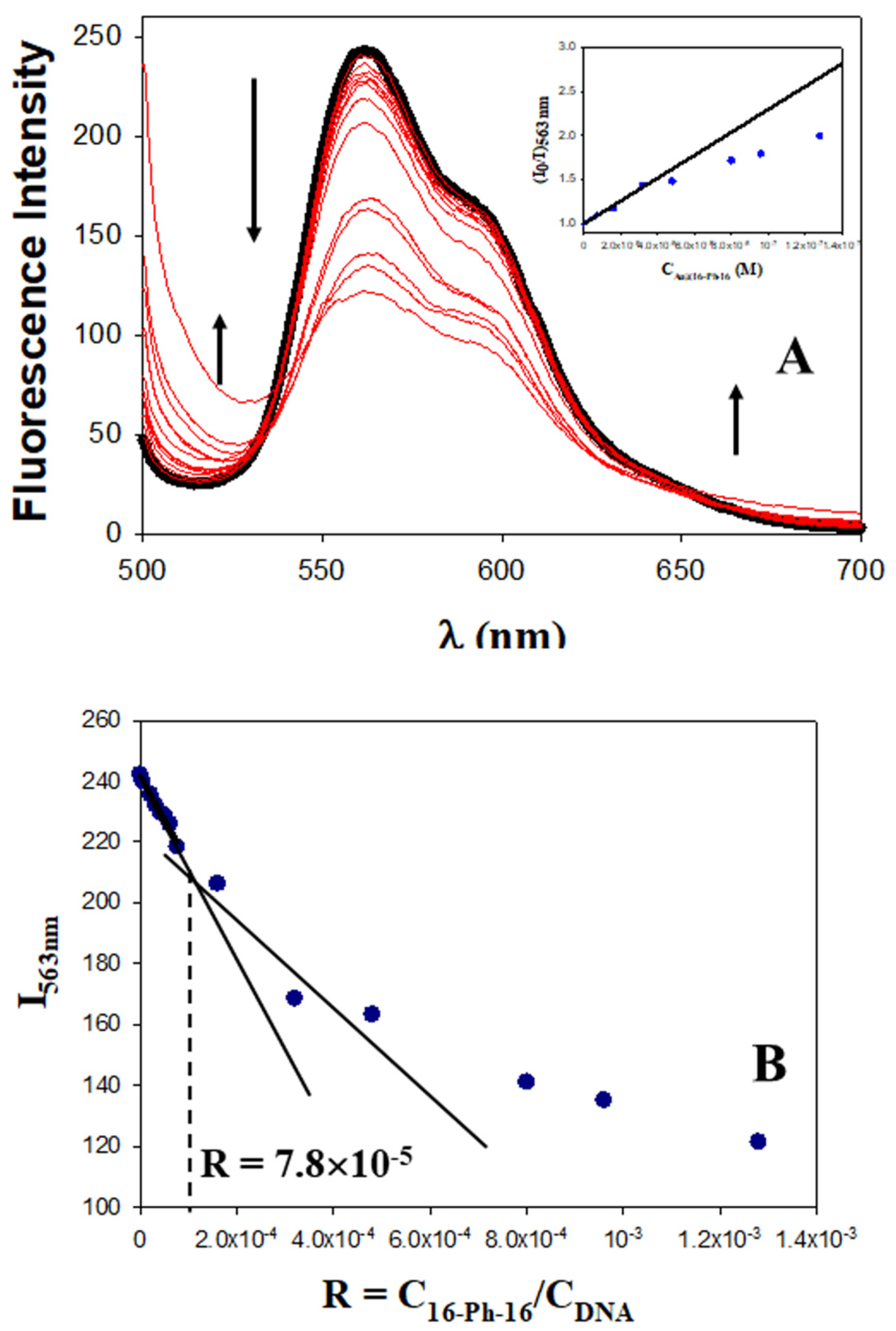
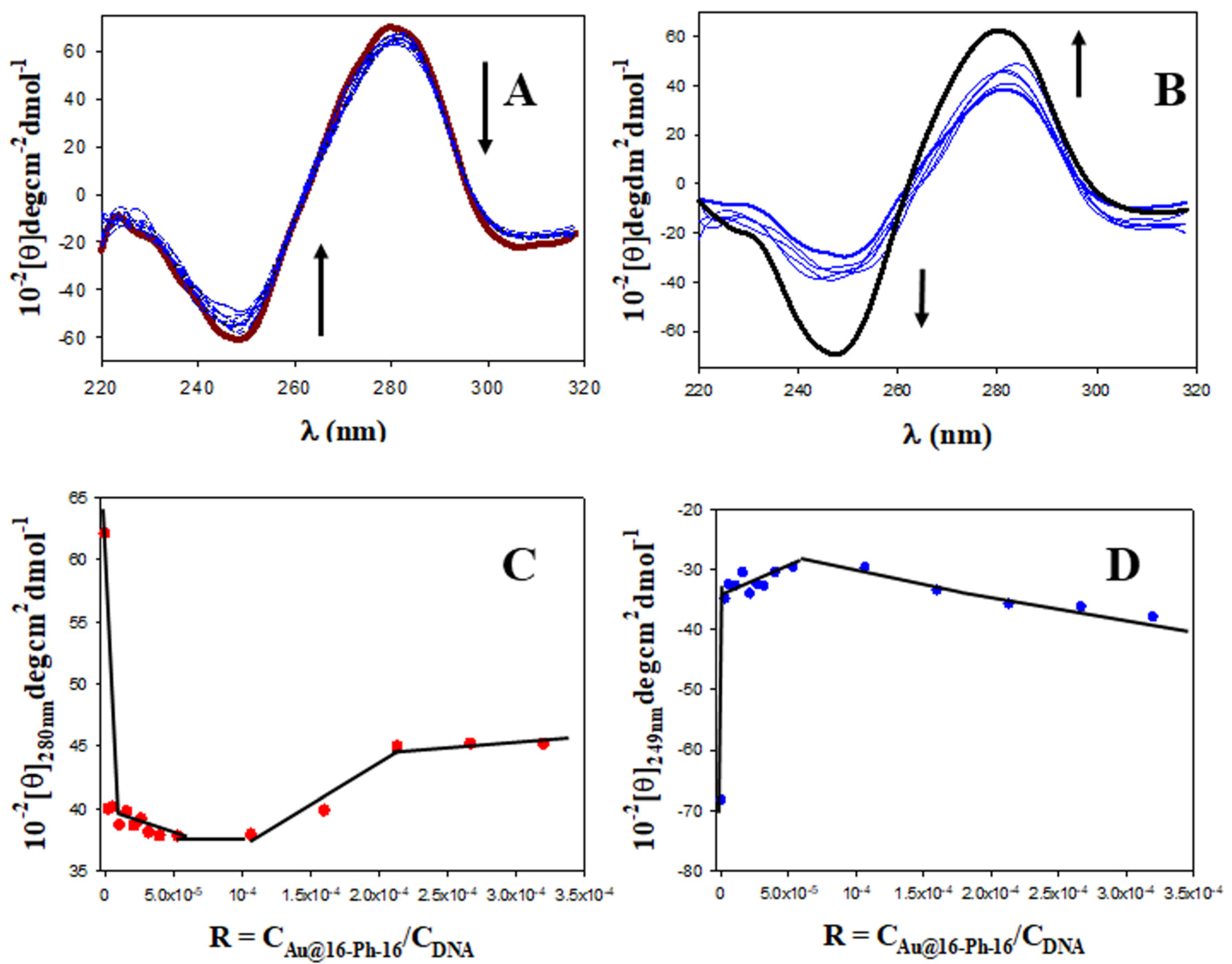
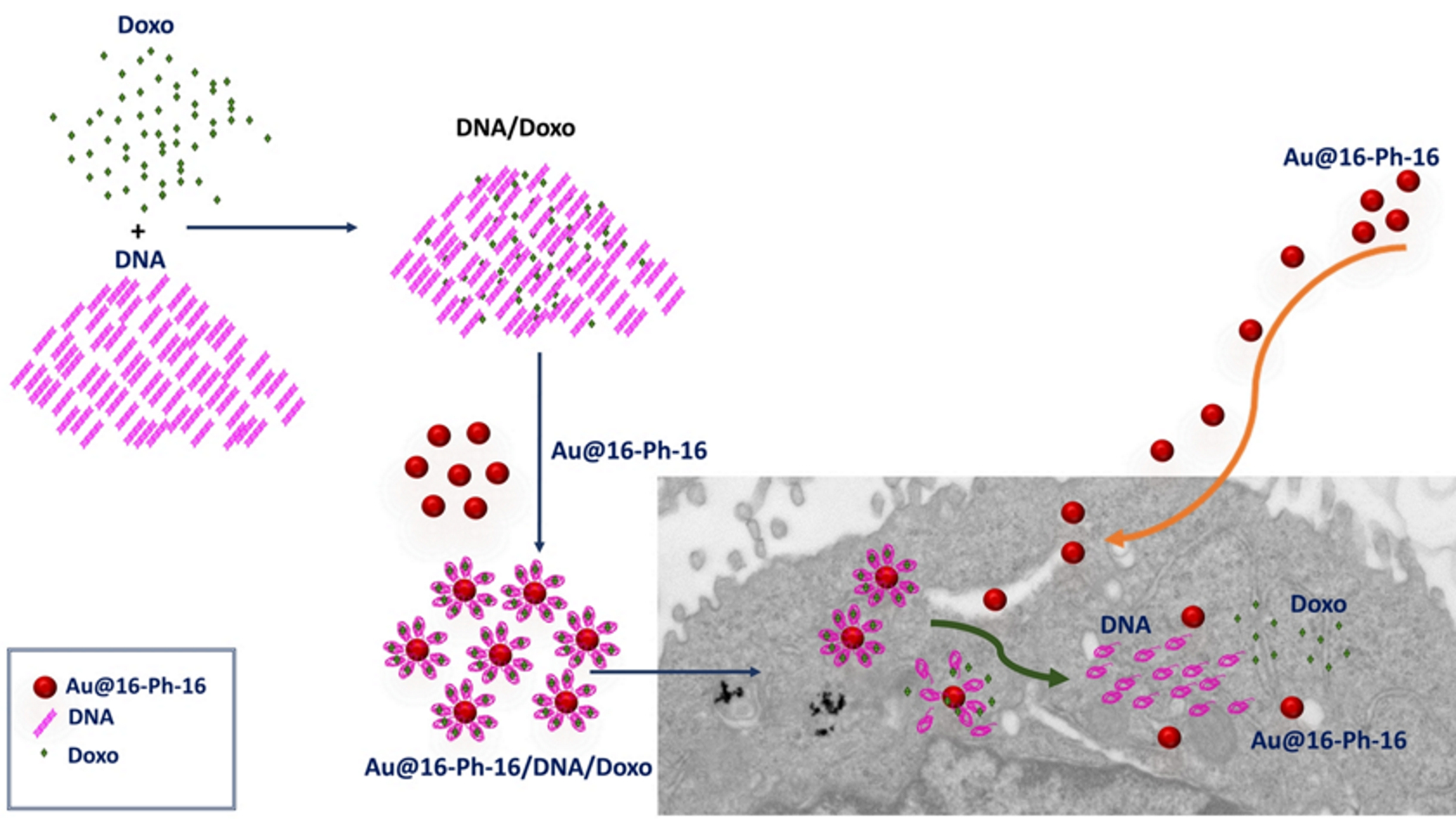
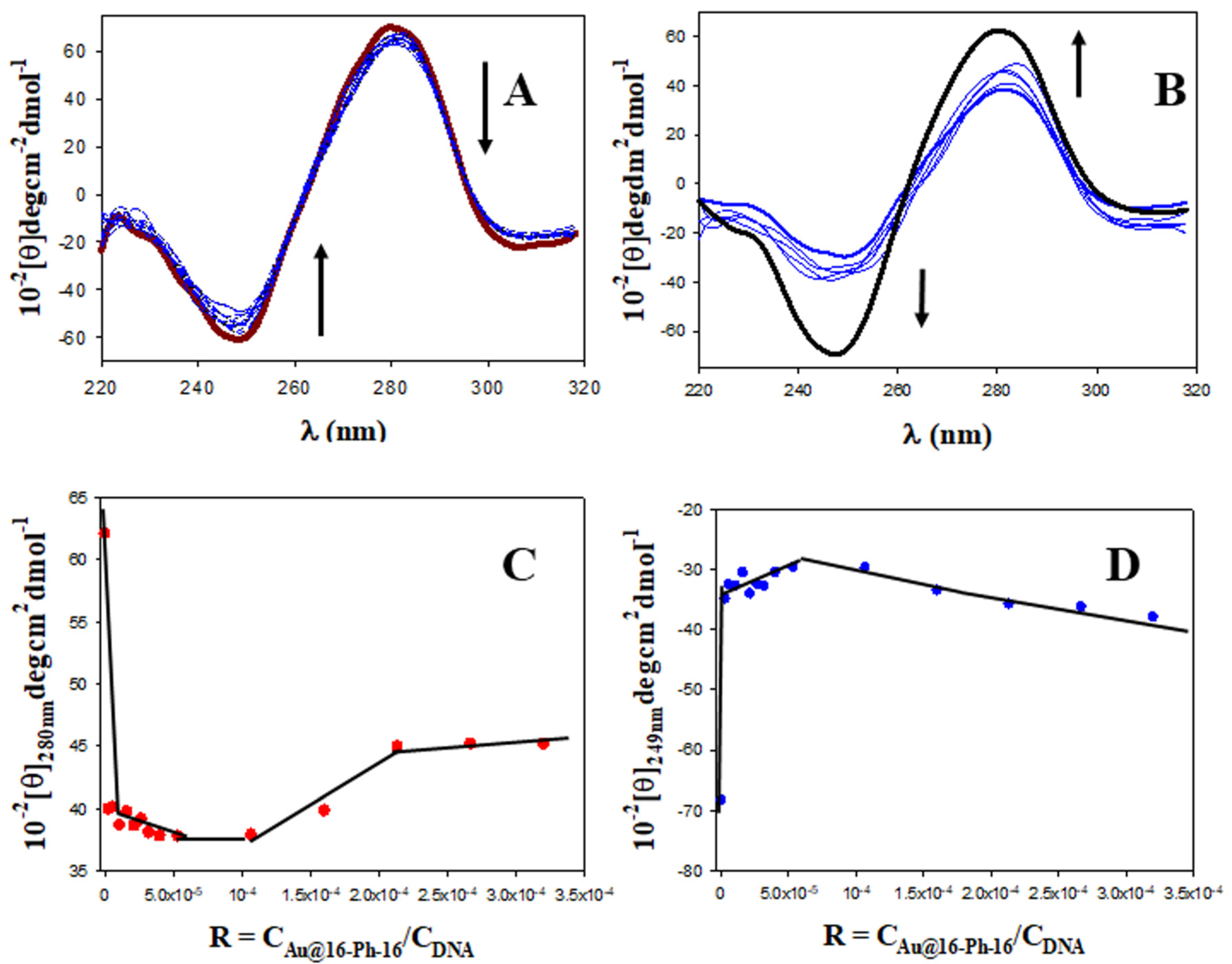
2.1. Conformational Changes in DNA/Doxo Complexes Induced by Au@16-Ph-16 Cationic Nanoparticles: Au@16-Ph-16/DNA–Doxo Complex Formation
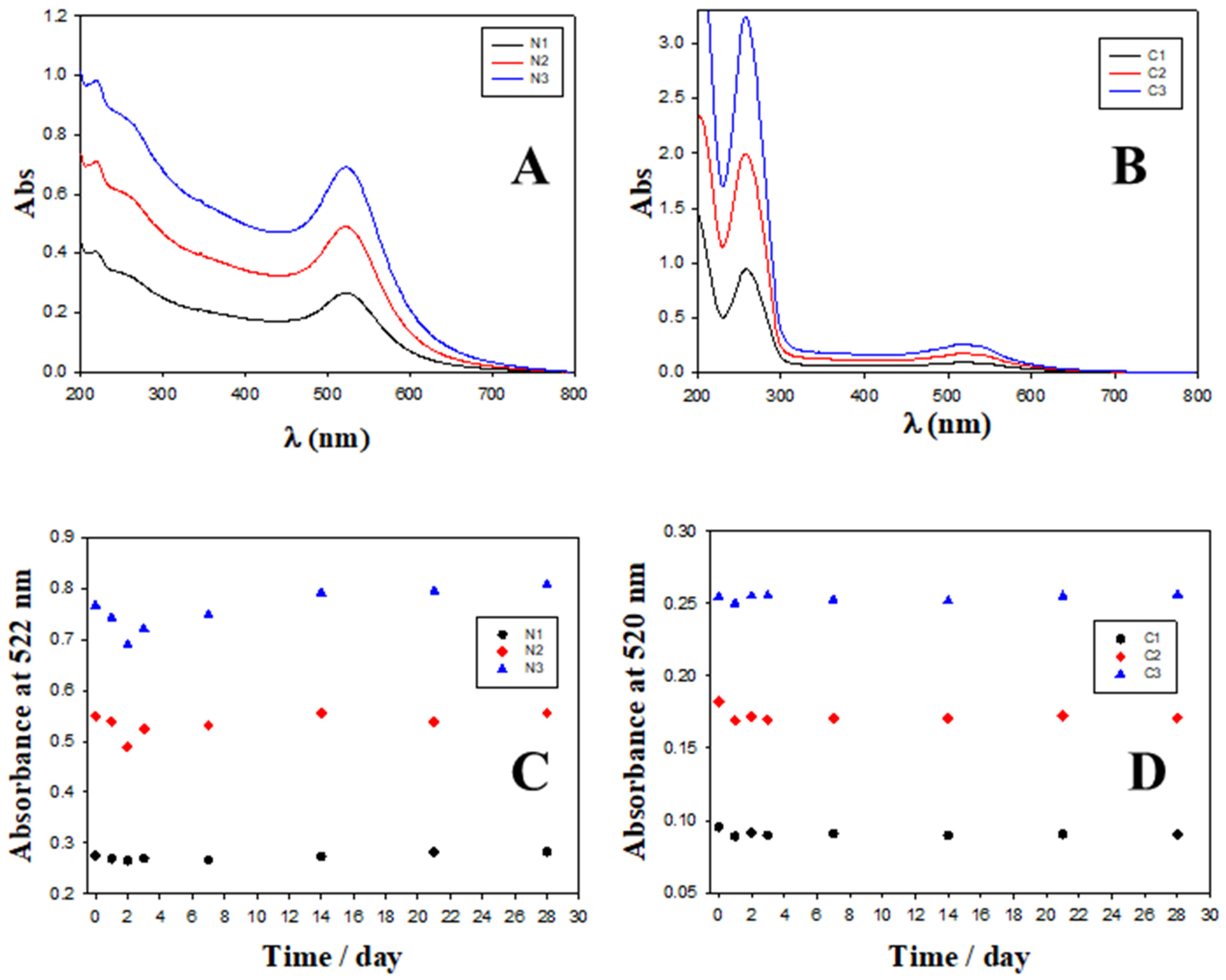
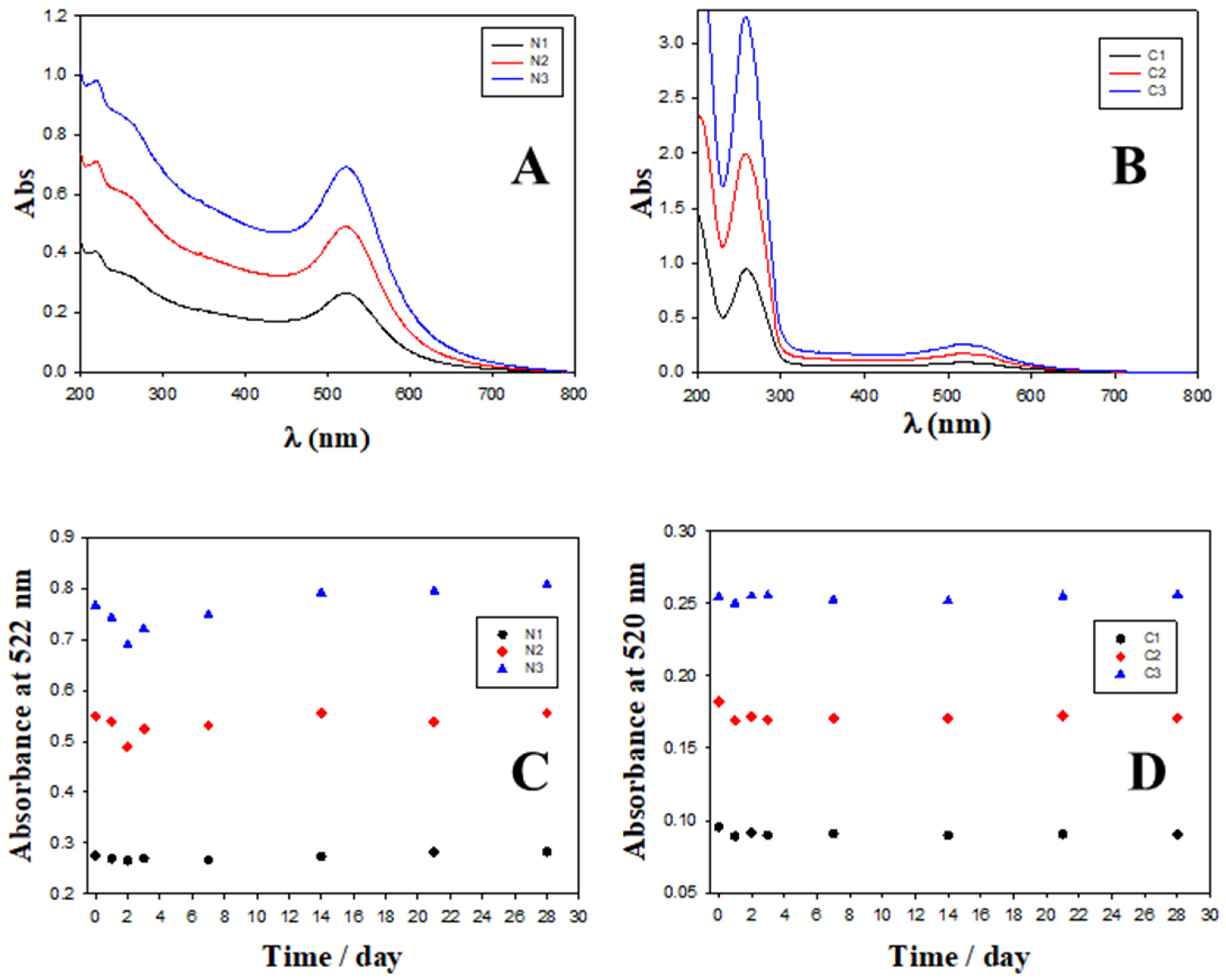
2.2. Charge, Size and Stability of Au@16-Ph-16 and Au@16-Ph-16/DNA–Doxo Nanosystems
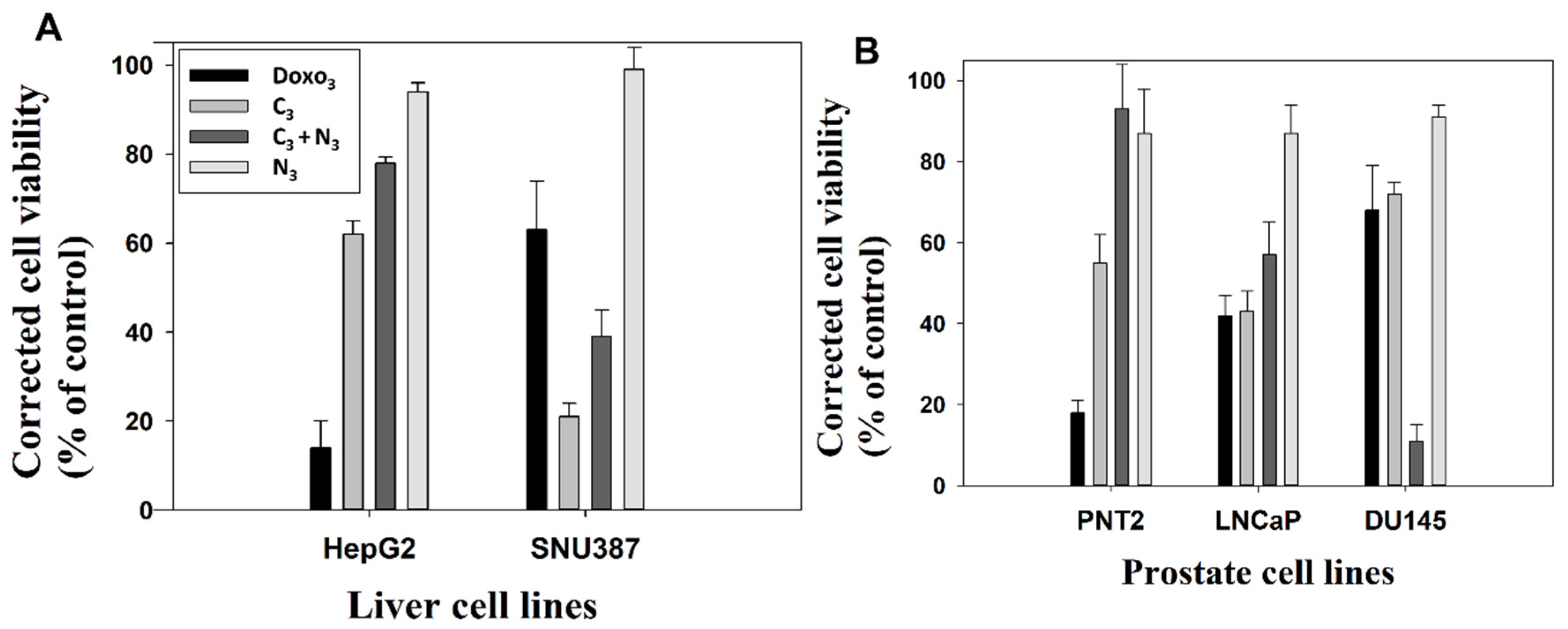
2.3. In Vitro Biocompatibility of Gold Nanosystems
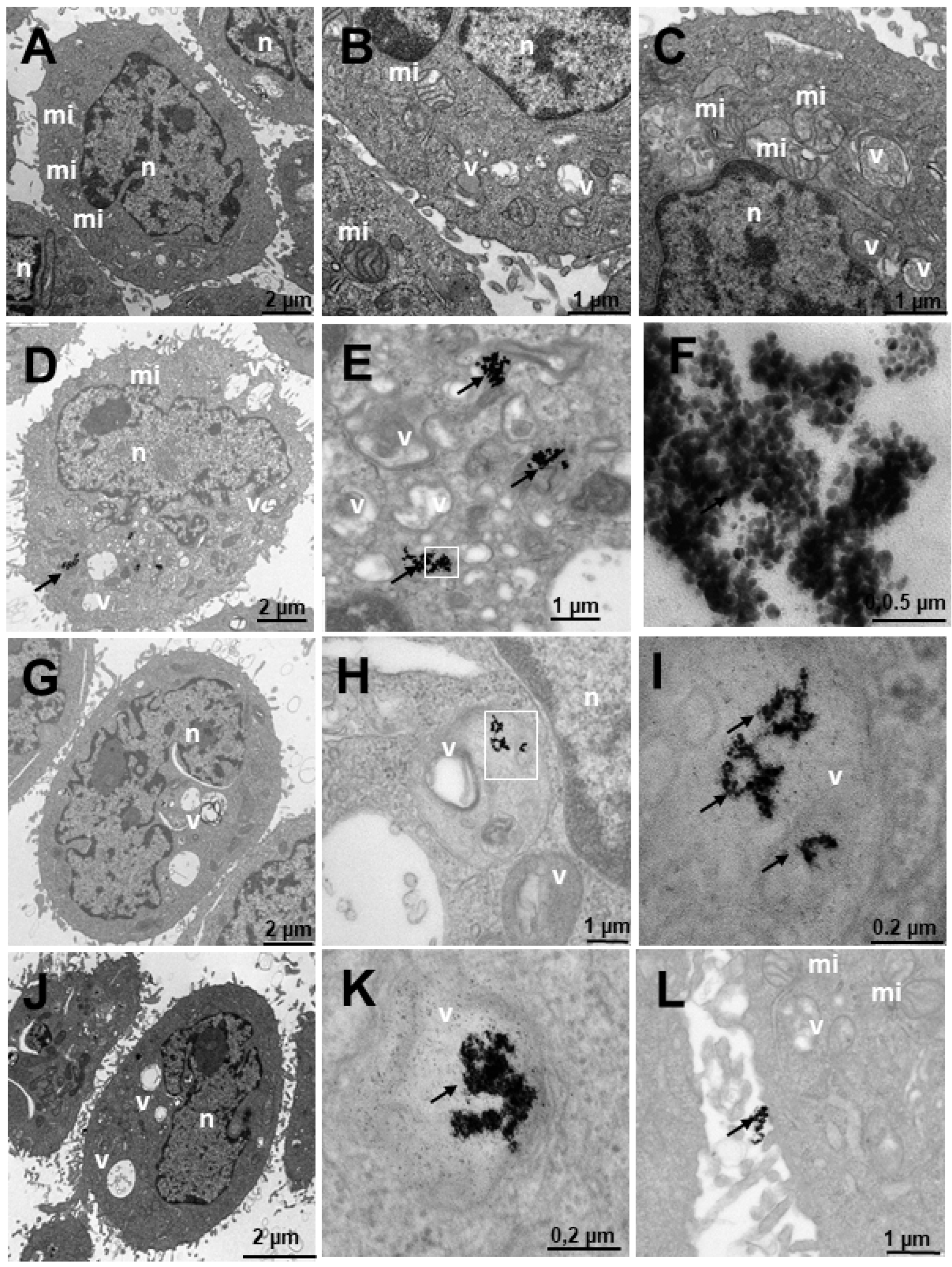
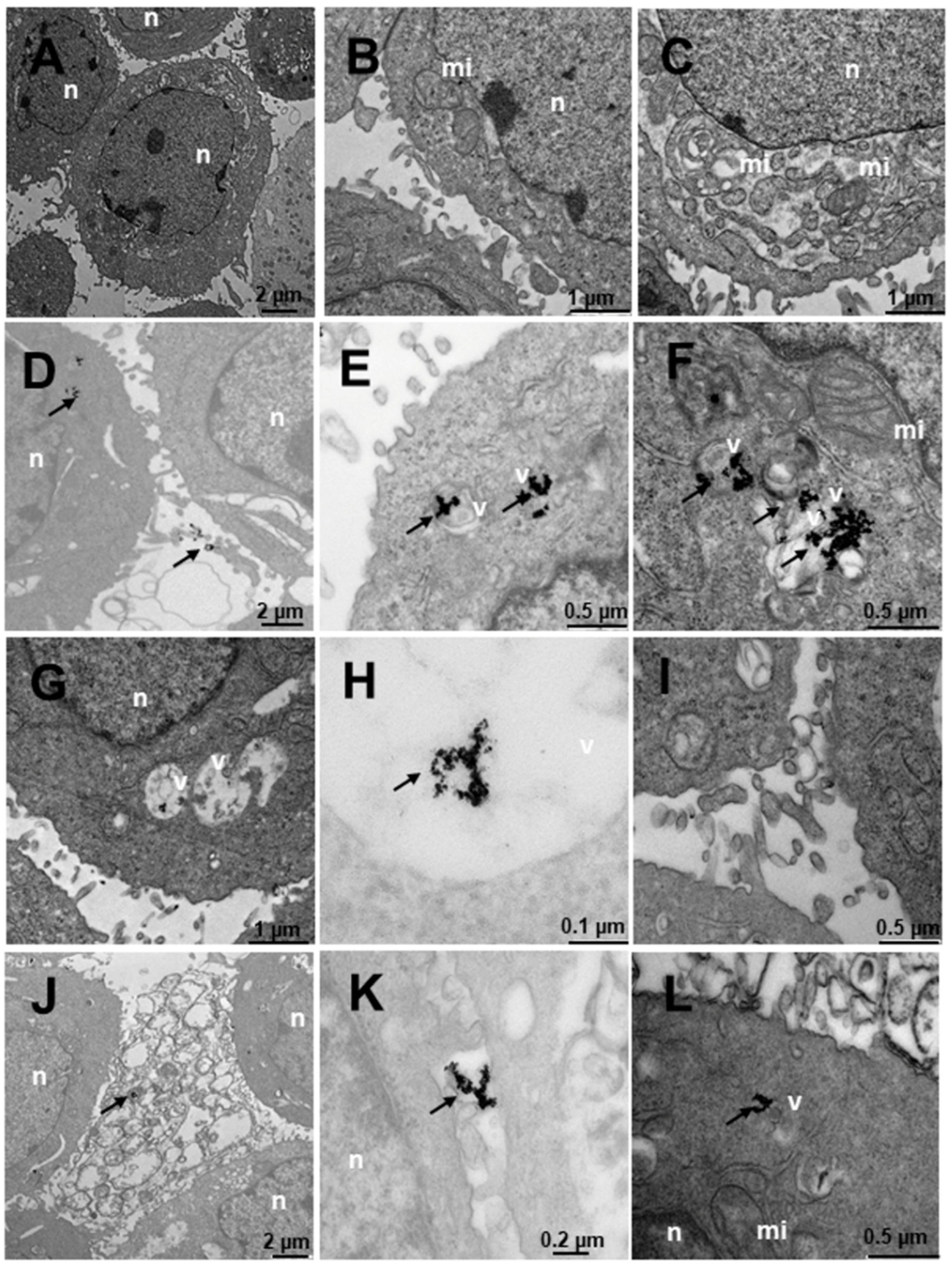
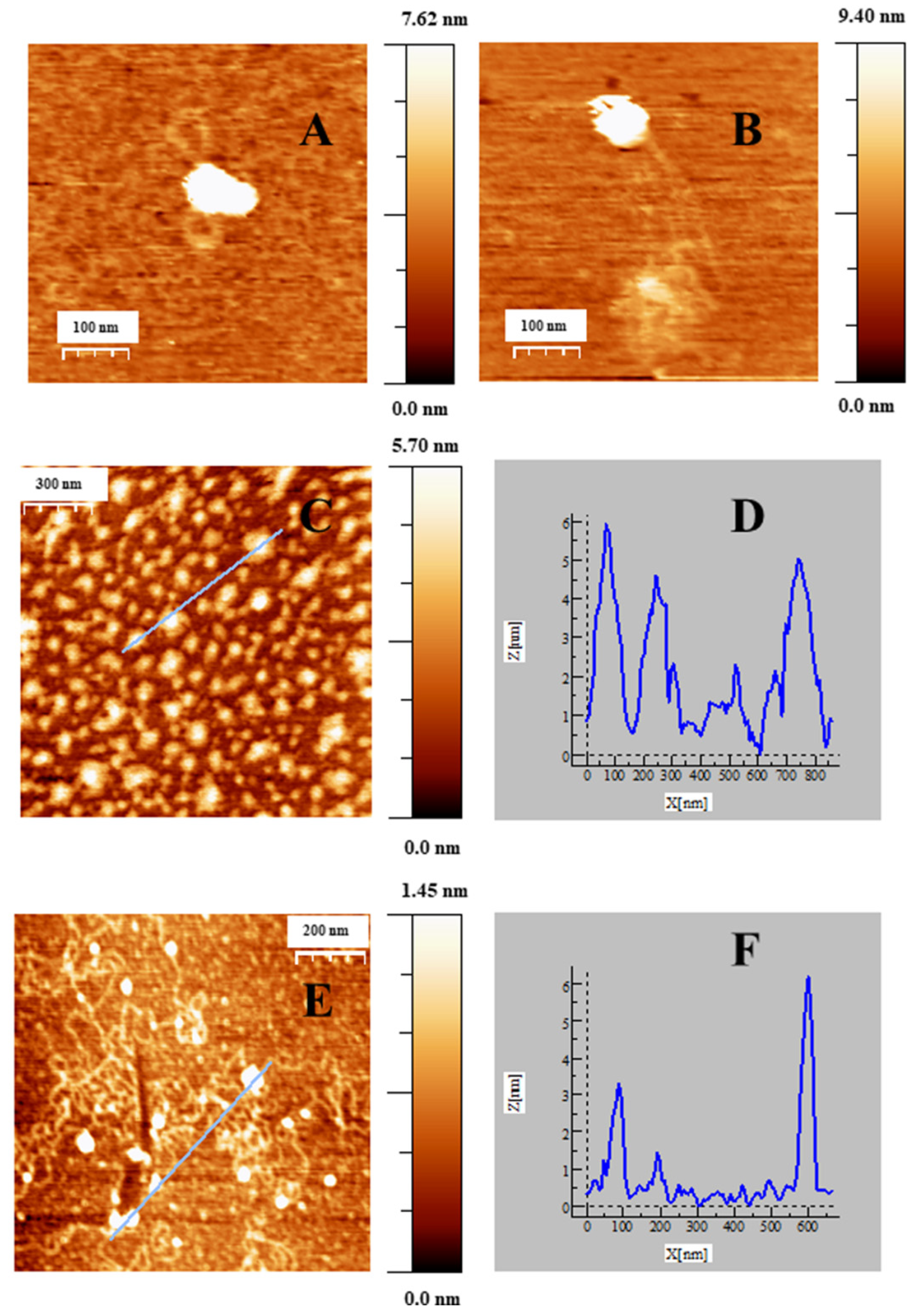
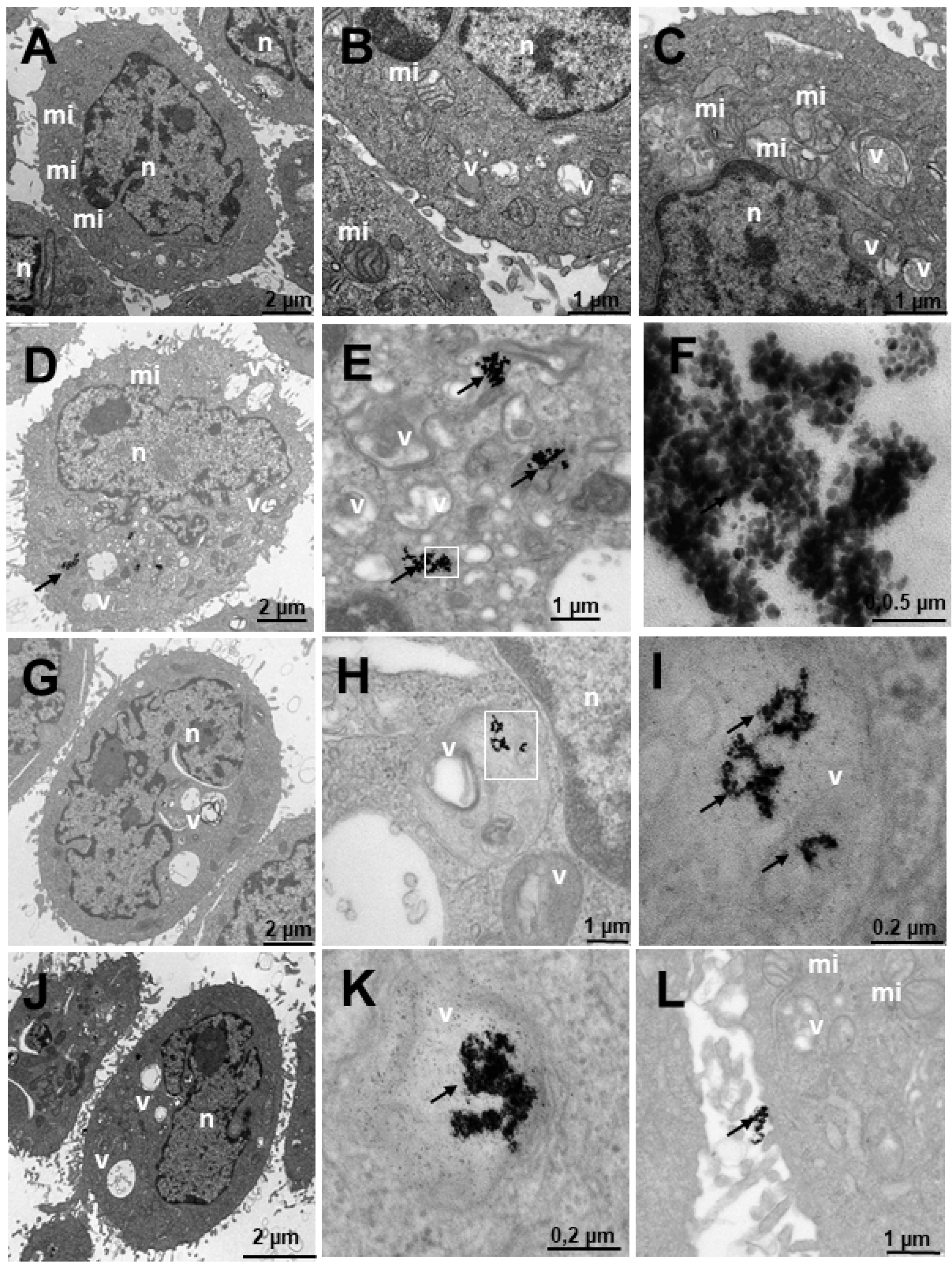
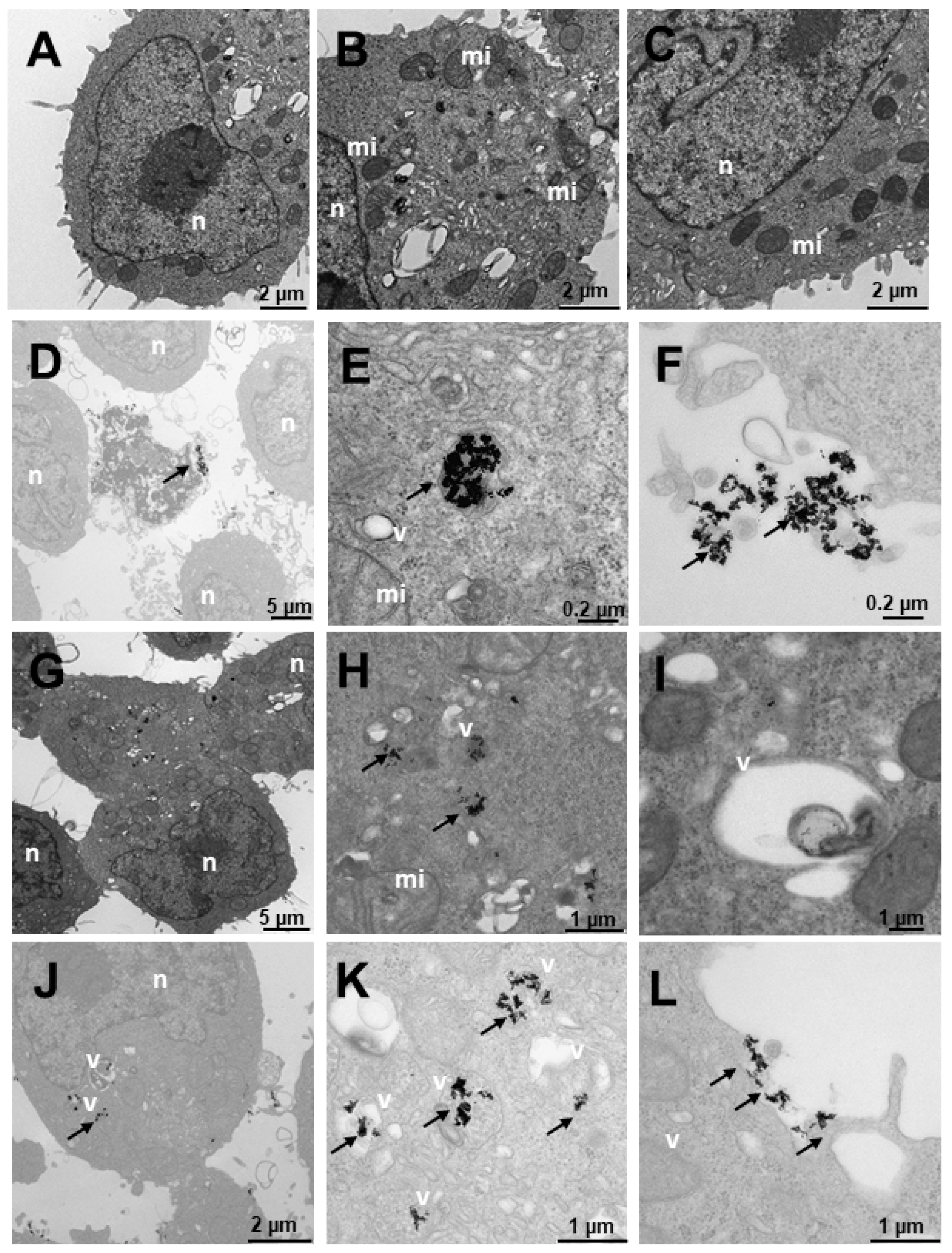
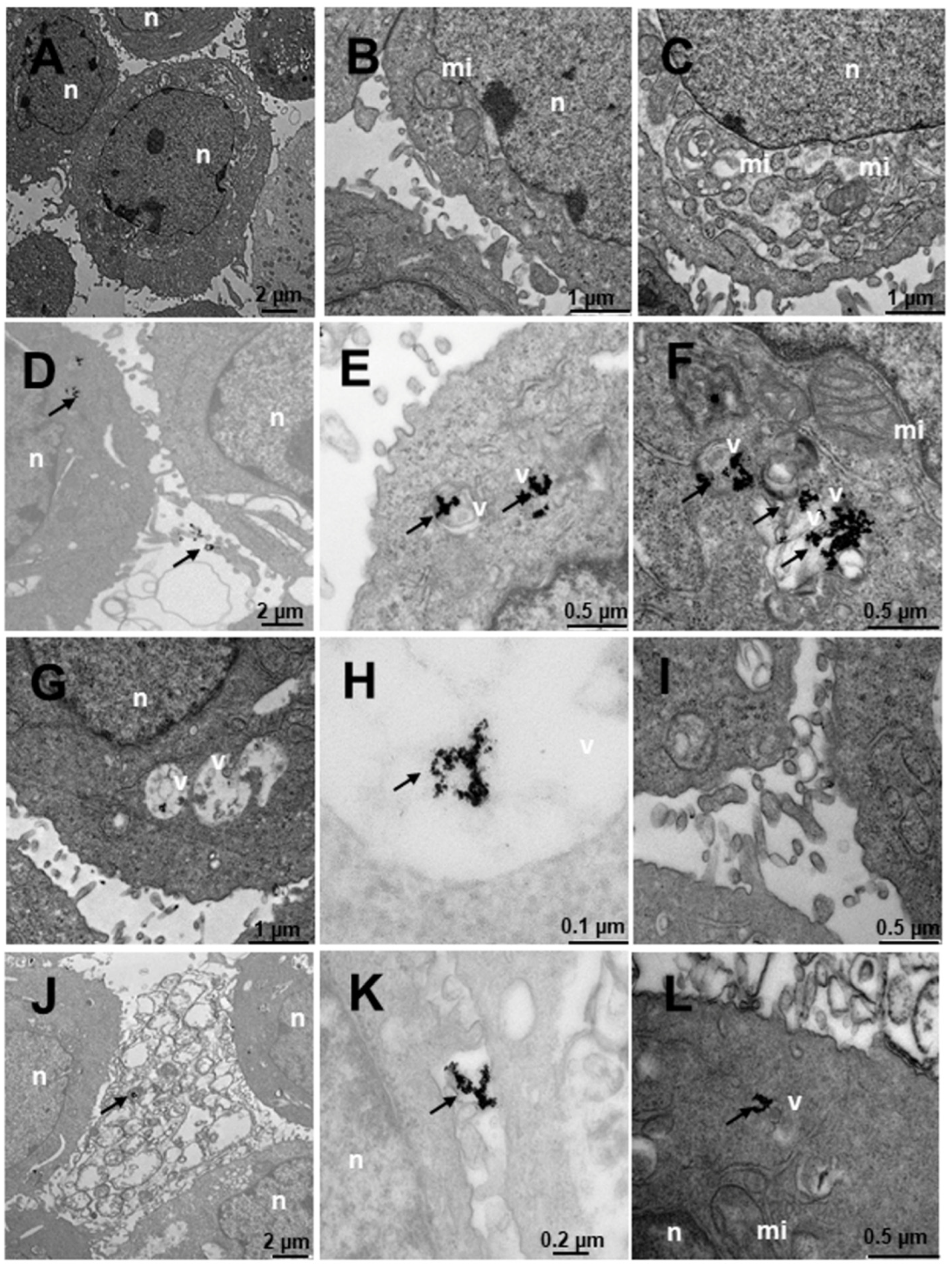
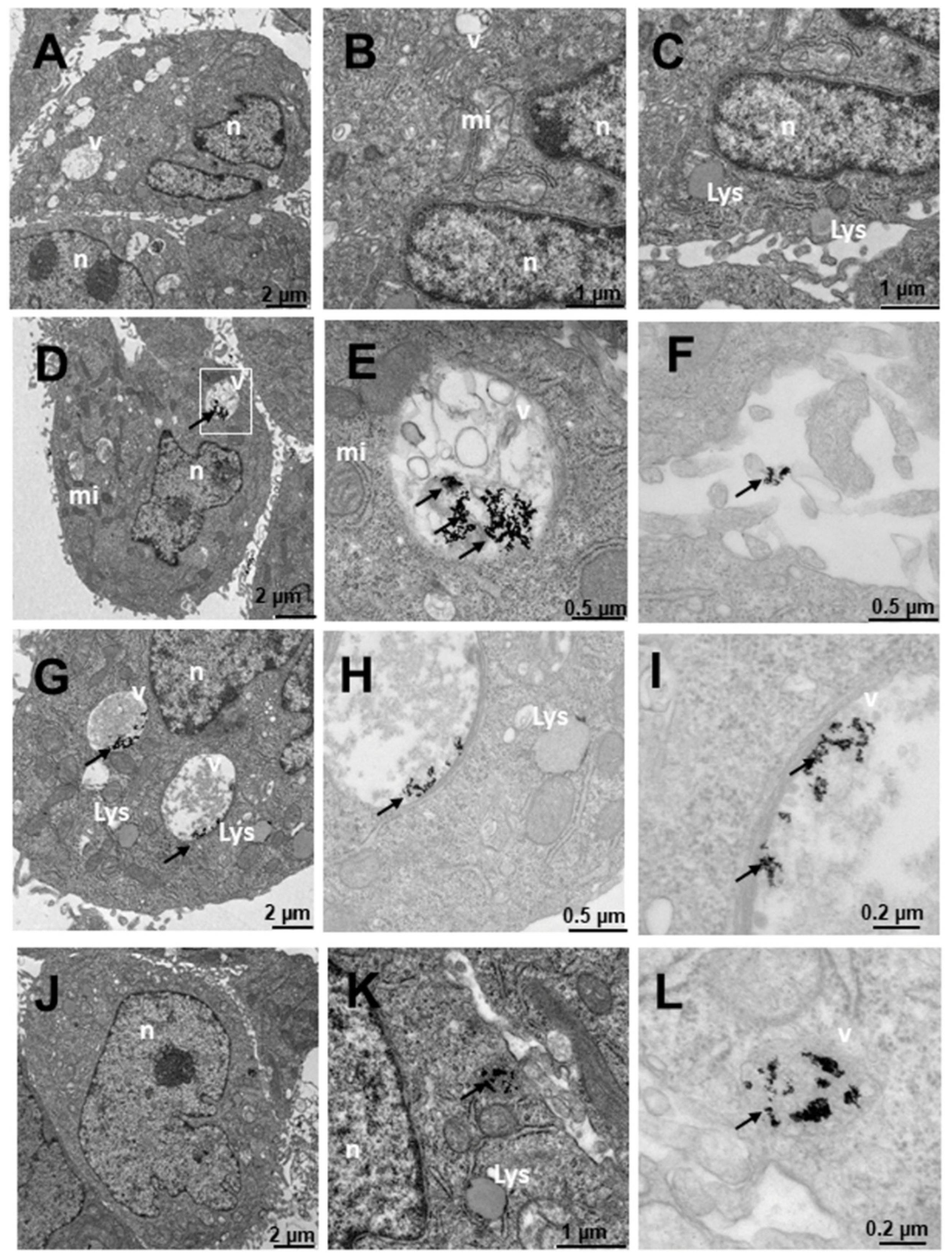
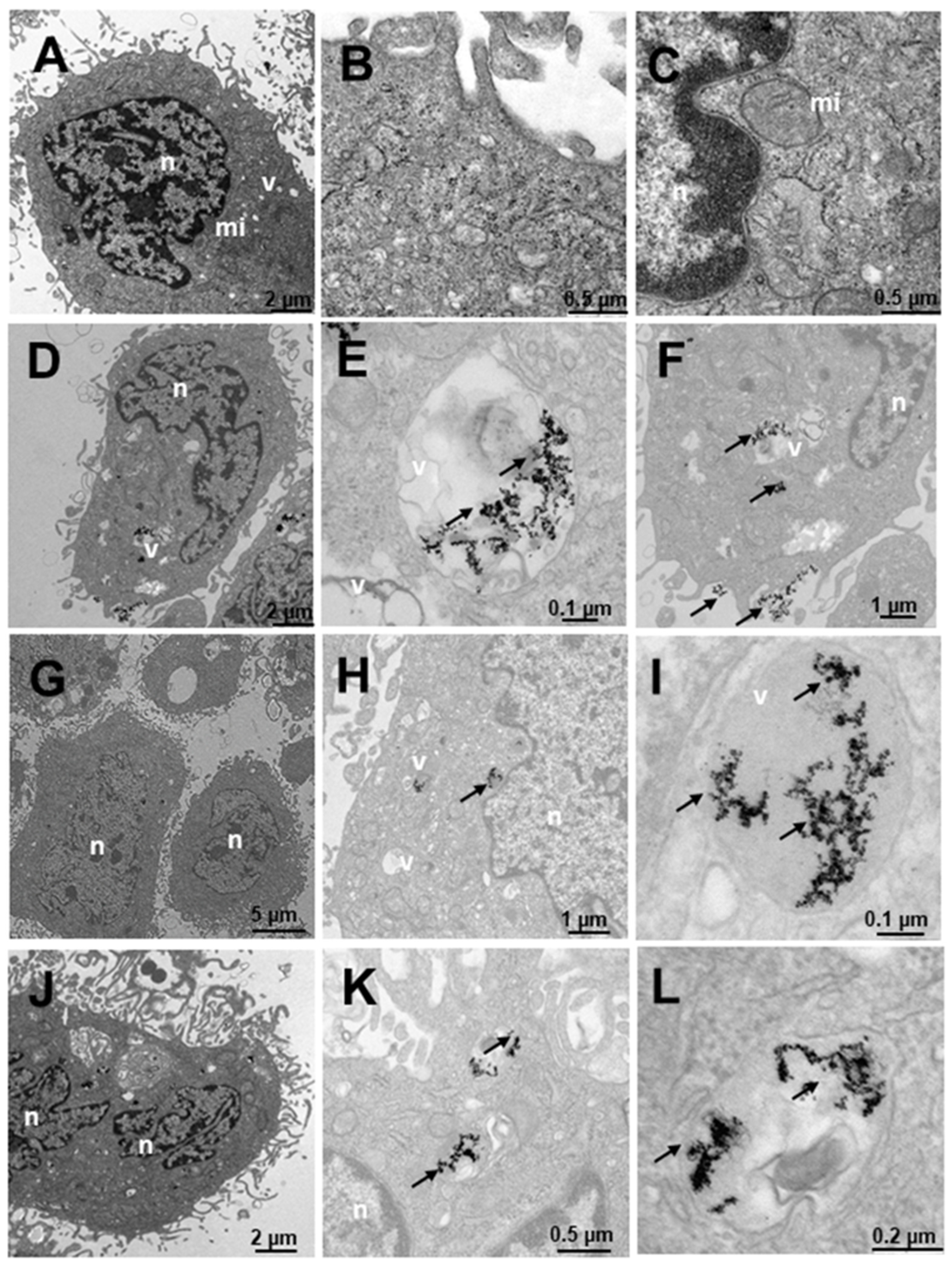
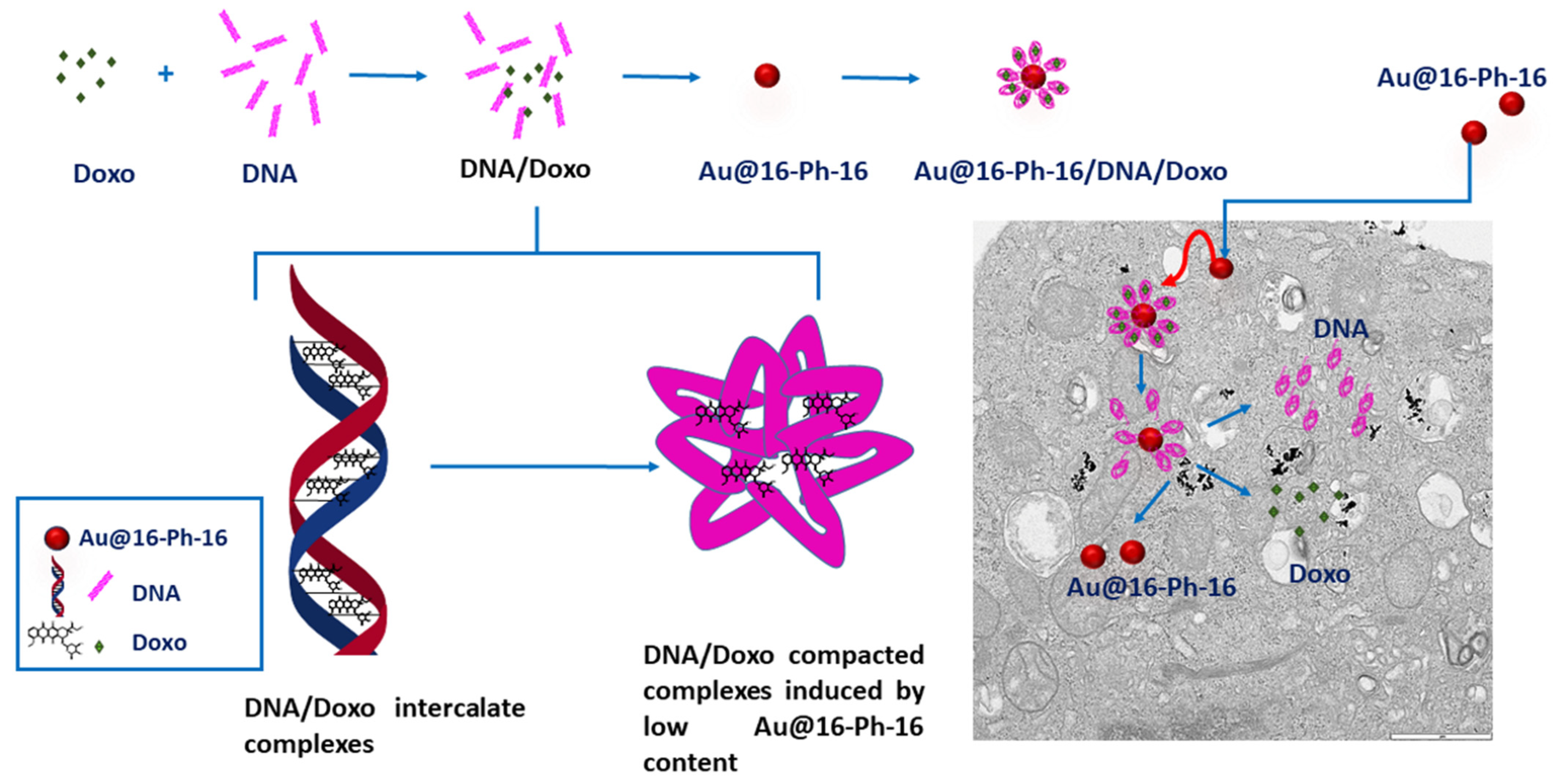
2.4. Internalization of Au@16-Ph-16 and Au@16-Ph-16/DNA–Doxo Nanosystems

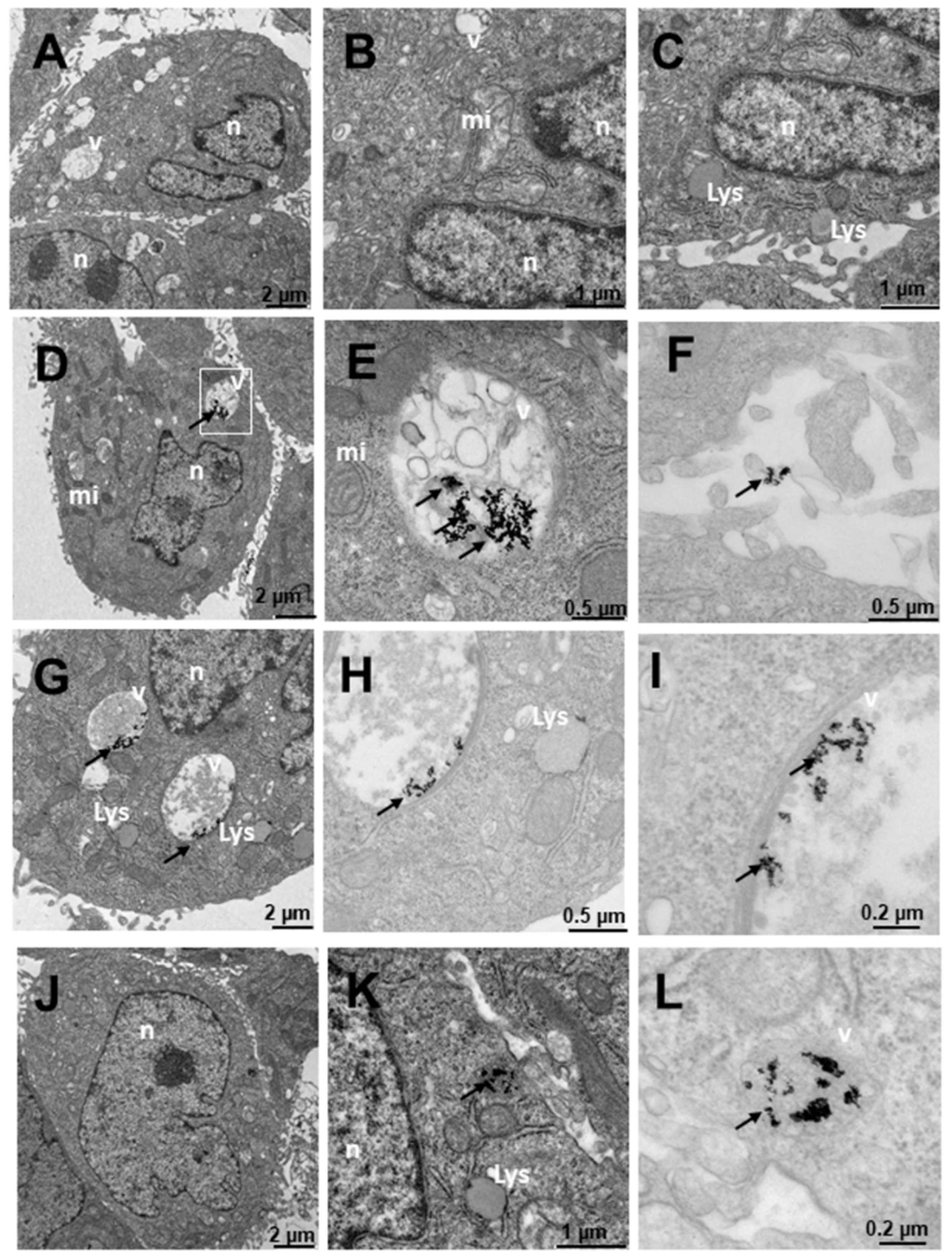

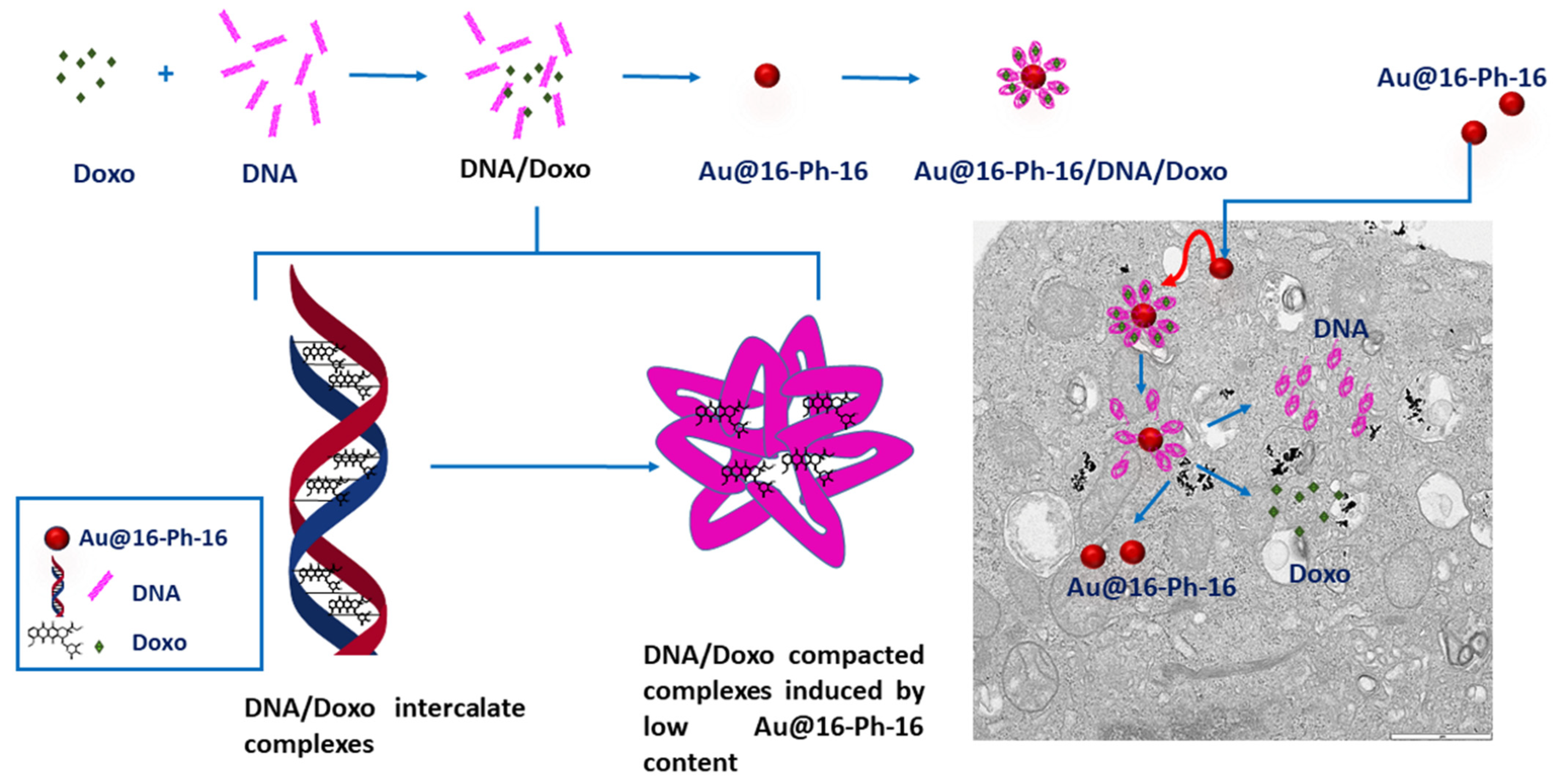
3. Materials and Methods
3.1. Materials
3.1.1. Cell Lines and Culture Conditions
3.1.2. Synthesis of N,N’-[1,3-phenylenebis(methylene))bis[N,N’-dimethyl-N-(1-hexadecyl)]-ammonium dibromide, 16-Ph-16
3.1.3. Synthesis of Au@16-Ph-16 Gold Nanoparticles
3.1.4. Synthesis of Au@16-Ph-16/DNA–Doxo Nanocomplexes
3.2. Methods
3.2.1. Cell Viability
3.2.2. UV–Vis Spectroscopy
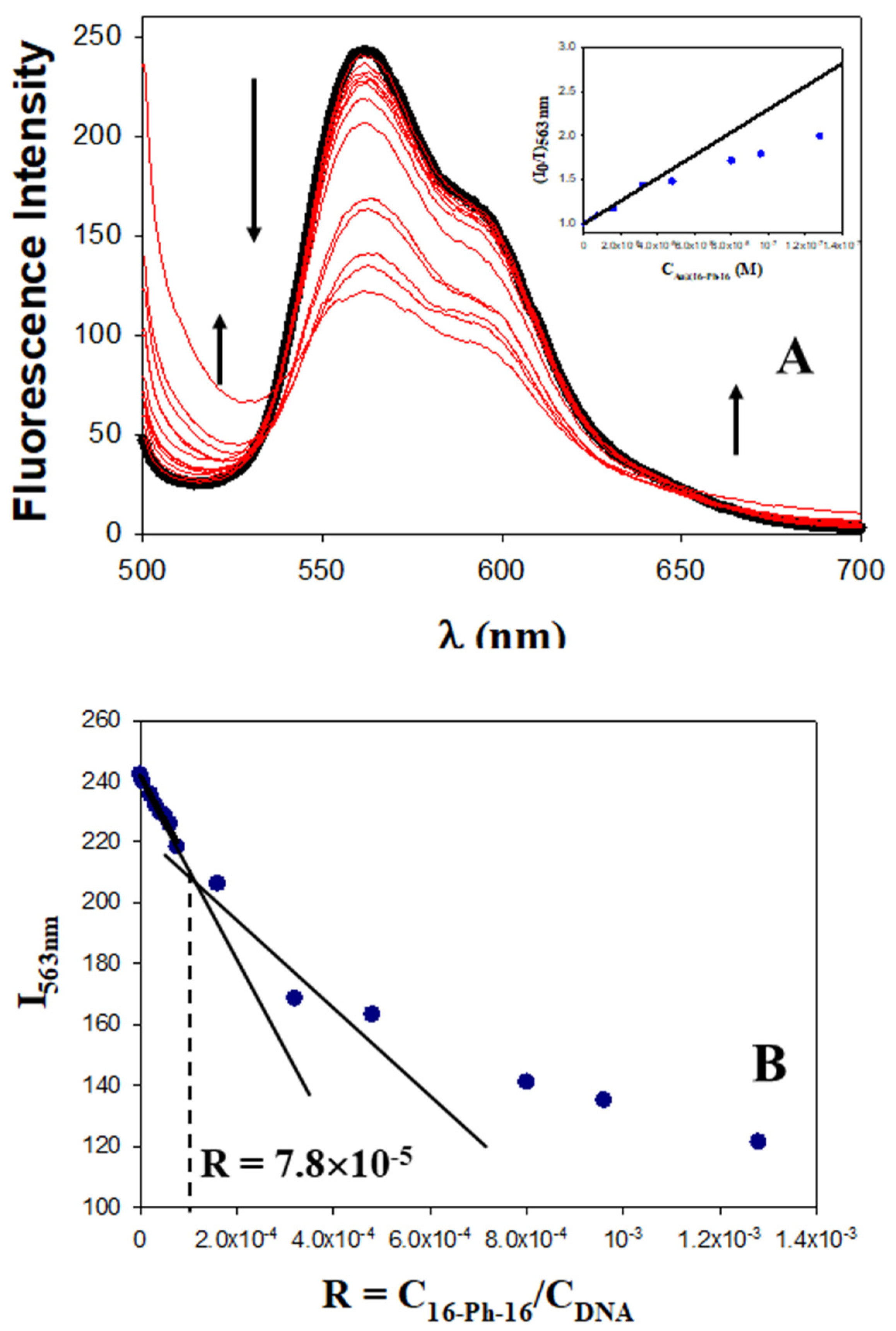
3.2.3. Fluorescence Spectroscopy
3.2.4. Circular Dichroism (CD) Spectroscopy
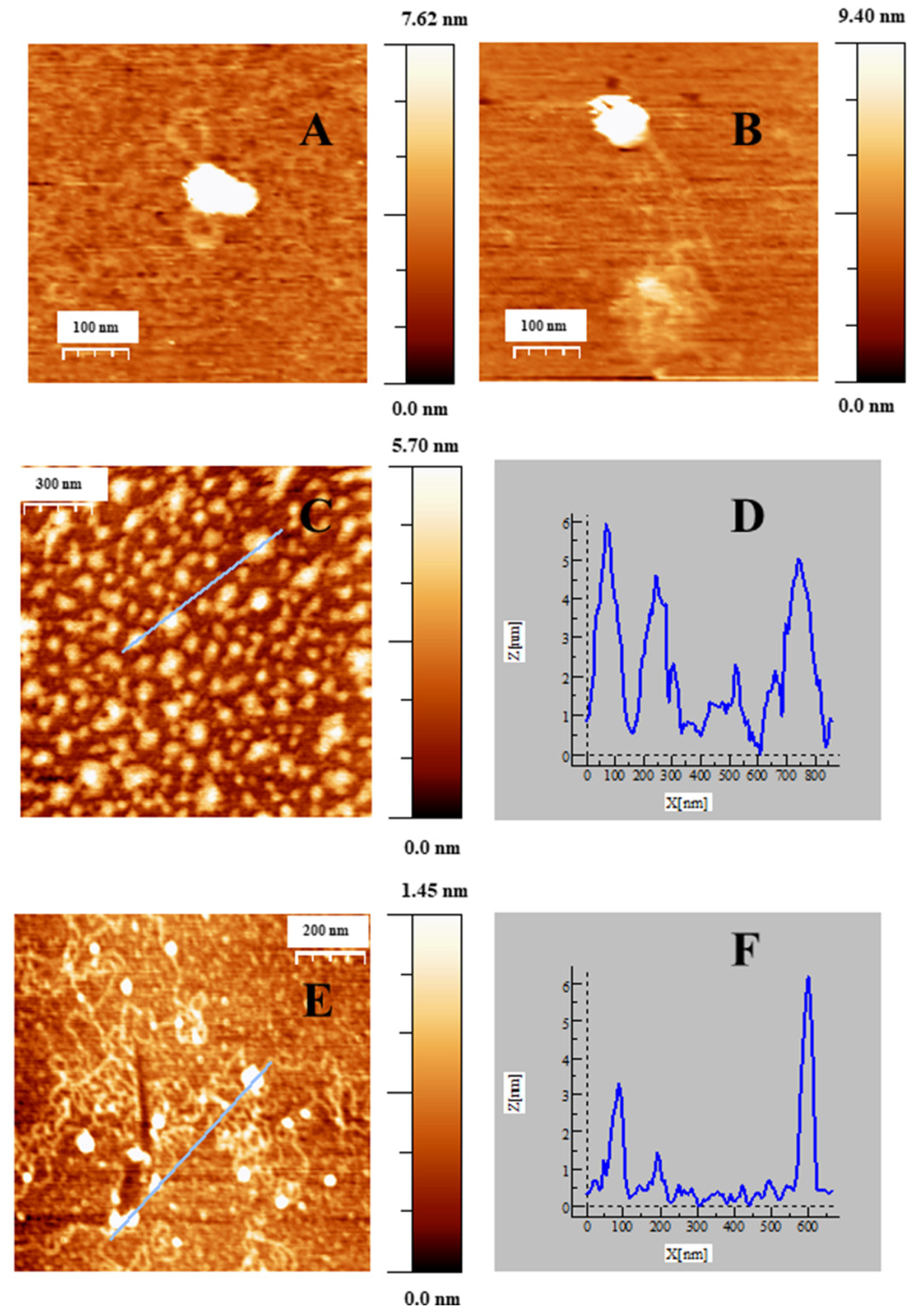
3.2.5. Atomic Force Microscopy Experiments
3.2.6. Dynamic Light Scattering (DLS) and Zeta Potential Measurements
3.2.7. Transmission Electron Microscopy (TEM)
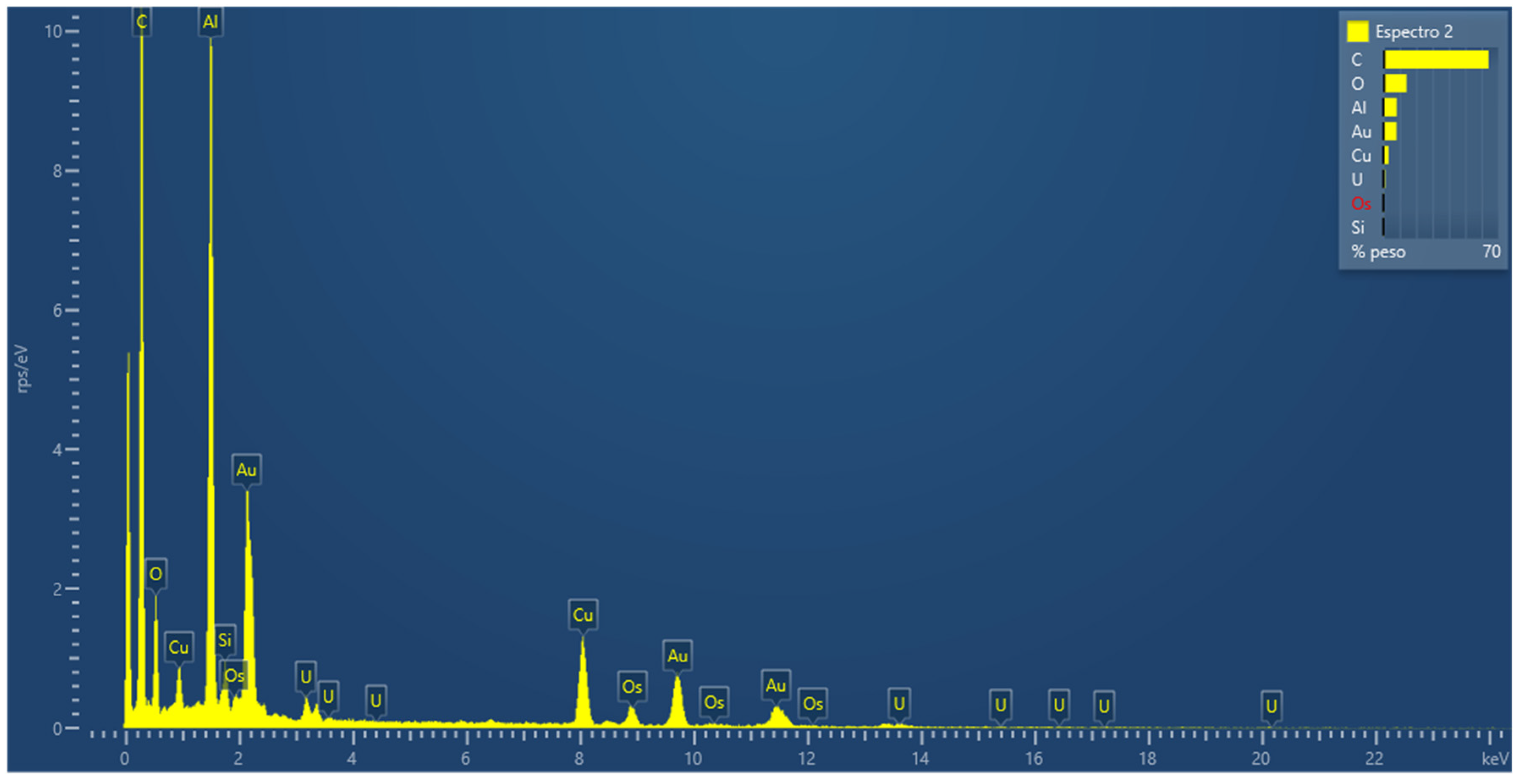
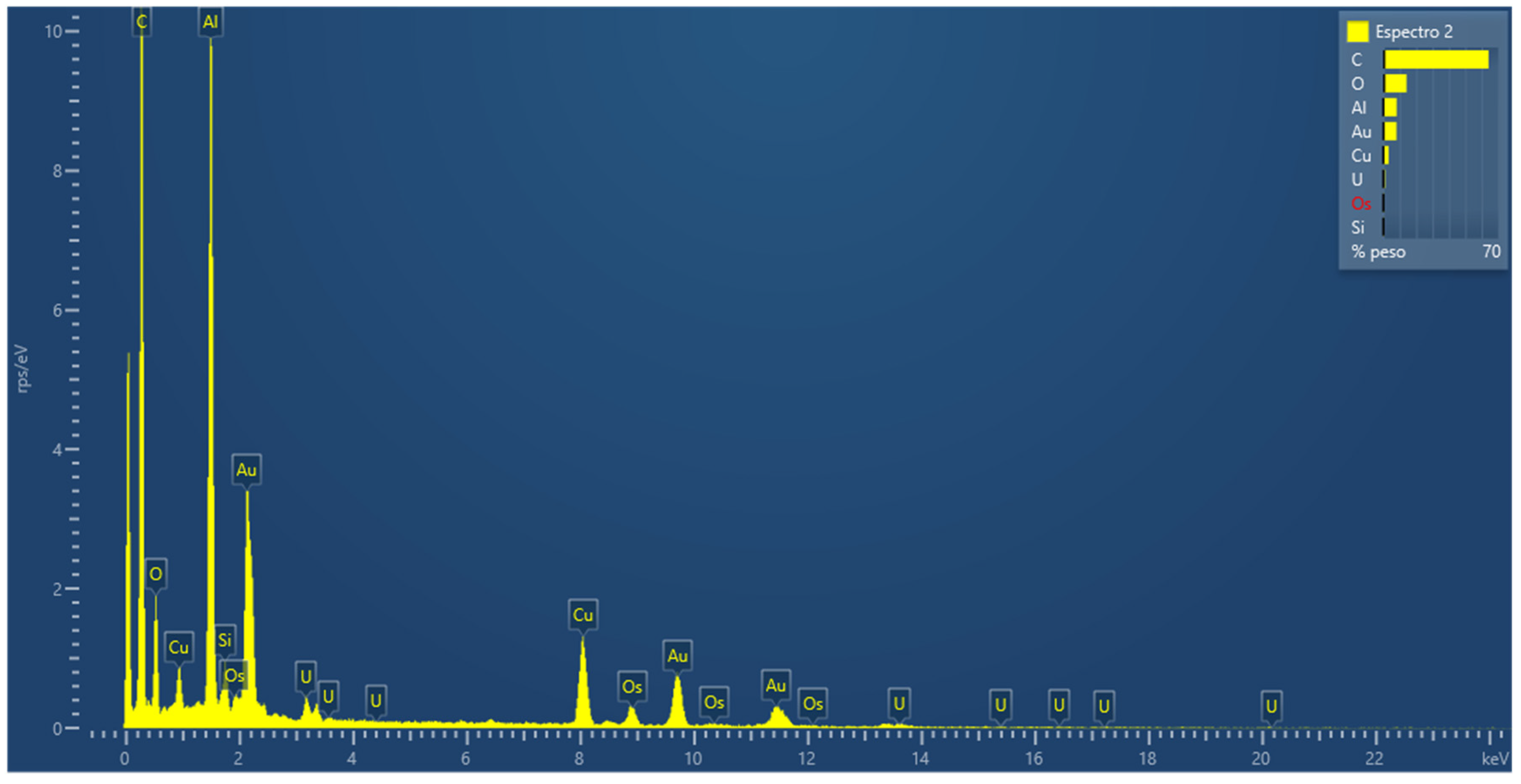
3.2.8. Energy Dispersive Spectroscopy (EDS) Measurements
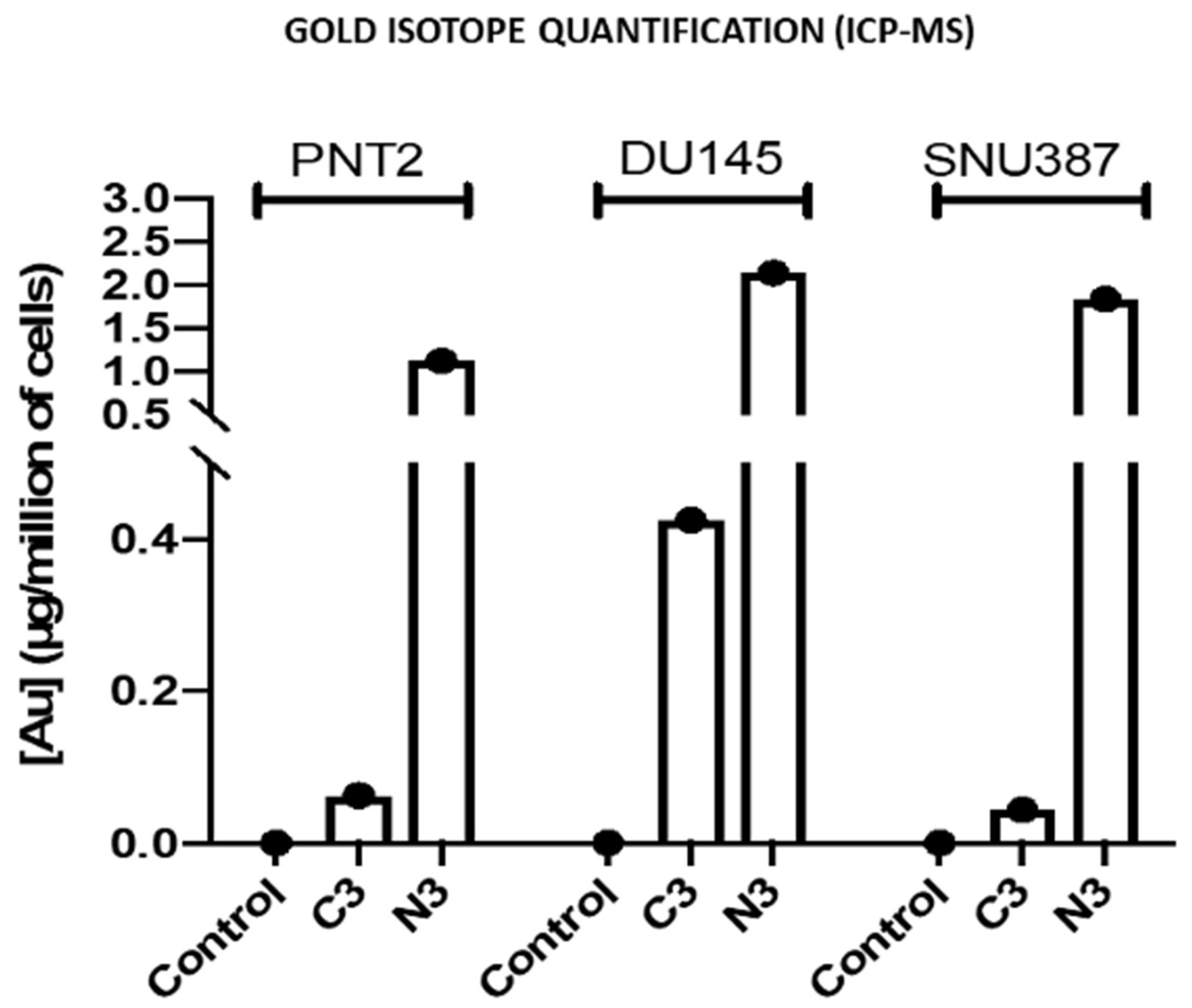
3.2.9. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
3.2.10. Confocal Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Cancer—Key Facts. 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 24 July 2022).
- Gewirtz, D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yoshida, S. Mechanism of the Inhibition of Calf Thymus DNA Polymerases α and β by Daunomycin and Adriamycin1. J. Biochem. 1980, 87, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Ascensão, A.; Oliveira, P.J.; Magalhães, J. Exercise as a beneficial adjunct therapy during Doxorubicin treatment—Role of mitochondria in cardioprotection. Int. J. Cardiol. 2011, 156, 4–10. [Google Scholar] [CrossRef]
- Meredith, A.-M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharmacol. 2016, 68, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Keizer, H.; Pinedo, H.; Schuurhuis, G.; Joenje, H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol. Ther. 1990, 47, 219–231. [Google Scholar] [CrossRef]
- Ruggiero, A.; De Rosa, G.; Rizzo, D.; Leo, A.; Maurizi, P.; De Nisco, A.; Vendittelli, F.; Zuppi, C.; Mordente, A.; Riccardi, R. Myocardial performance index and biochemical markers for early detection of doxorubicin-induced cardiotoxicity in children with acute lymphoblastic leukaemia. Int. J. Clin. Oncol. 2012, 18, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Barton, J.; Chaikin, P.; Swenson, C.; Welles, L. Myocet (liposome-encapsulated doxorubicin citrate): A new approach in breast cancer therapy. Expert Opin. Pharmacother. 2002, 3, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Marina, N.M.; Cochrane, D.; Harney, E.; Zomorodi, K.; Blaney, S.; Winick, N.; Bernstein, M.; Link, M.P. Dose escalation and pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in children with solid tumors: A pediatric oncology group study. Clin. Cancer Res. 2002, 8, 413–418. [Google Scholar] [PubMed]
- Guo, B.; Zhu, H.-L.; Li, S.-X.; Lu, X.-C.; Fan, H. Individualized Liposomal Doxorubicin-Based Treatment in Elderly Patients with Non-Hodgkin’s Lymphoma. Onkologie 2011, 34, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Weekes, C.D.; Vose, J.M.; Lynch, J.C.; Weisenburger, D.D.; Bierman, P.J.; Greiner, T.; Bociek, G.; Enke, C.; Bast, M.; Chan, W.C.; et al. Hodgkin’s Disease in the Elderly: Improved Treatment Outcome With a Doxorubicin-Containing Regimen. J. Clin. Oncol. 2002, 20, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef]
- Bogner, J.R.; Kronawitter, U.; Rolinski, B.; Truebenbach, K.; Goebel, F.-D. Liposomal Doxorubicin in the Treatment of Advanced AIDS-Related Kaposi Sarcoma. J. Acquir. Immune Defic. Syndr. 1994, 7, 463–468. [Google Scholar] [PubMed]
- Lotem, M.; Hubert, A.; Lyass, O.; Goldenhersh, M.A.; Ingber, A.; Peretz, T.; Gabizon, A. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch. Dermatol. 2000, 136, 1475–1480. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.; Shih, V.; Kian, T.C. Liposomal doxorubicin-associated acute hypersensitivity despite appropriate preventive measures. J. Oncol. Pharm. Pract. 2007, 13, 105–107. [Google Scholar] [CrossRef]
- Stathopoulos, G.P.; Antoniou, D.; Dimitroulis, I.; Michalopoulou, P.; Bastas, A.; Marosis, K.; Stathopoulos, J.; Provata, A.; Yiamboudakis, P.; Veldekis, D.; et al. Liposomal cisplatin combined with paclitaxel versus cisplatin and paclitaxel in non-small-cell lung cancer: A randomized phase III multicenter trial. Ann. Oncol. 2010, 21, 2227–2232. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy caused by amphiphilic drug carriers: The role of lipoproteins. Curr. Drug Deliv. 2005, 2, 443–449. [Google Scholar] [CrossRef]
- Boulikas, T.; Mylonakis, N.I.; Athanasiou, A.; Kosmas, C.; Angel, J. Liposomally-encapsulated cisplatin (Lipoplatin) plus gemcitabine in NSCLC: Preliminary results of a phase II trial and its antiangiogenesis potential. J. Clin. Oncol. 2008, 26, 19024. [Google Scholar] [CrossRef]
- Boulikas, T.; Mylonakis, N.; Sarikos, G.; Angel, J.; Athanasiou, A.; Politis, G.; Rapti, A.; Rassidakis, A.; Karabatzaki, M.; Anyfantis, N. Lipoplatin plus gemcitabine versus cisplatin plus gemcitabine in NSCLC: Preliminary results of a phase III trial. J. Clin. Oncol. 2007, 25, 18028. [Google Scholar] [CrossRef]
- Veal, G.J.; Griffin, M.J.; Price, E.; Parry, A.; Dick, G.S.; Little, M.A.; Yule, S.M.; Morland, B.; Estlin, E.J.; Hale, J.P.; et al. A phase I study in paediatric patients to evaluate the safety and pharmacokinetics of SPI-77, a liposome encapsulated formulation of cisplatin. Br. J. Cancer 2001, 84, 1029–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.C.; Lorigan, P.; Margison, G.P.; Margison, J.M.; Martin, F.; Thatcher, N.; Anderson, H.; Ranson, M. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br. J. Cancer 2006, 95, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, A.; Karamouzis, M.V.; Stavrinides, H.; Ardavanis, A.; Kandilis, K.; Stavrakakis, J.; Georganta, C.; Rigatos, G. Phase II study of pegylated liposomal doxorubicin (Caelyx®) and docetaxel as first-line treatment in metastatic breast cancer. Ann. Oncol. 2004, 15, 891–895. [Google Scholar] [CrossRef]
- Katsaros, D.; Oletti, M.V.; de la Longrais, I.A.R.; Ferrero, A.; Celano, A.; Fracchioli, S.; Donadio, M.; Passera, R.; Cattel, L.; Bumma, C. Clinical and pharmacokinetic phase II study of pegylated liposomal doxorubicin and vinorelbine in heavily pretreated recurrent ovarian carcinoma. Ann. Oncol. 2005, 16, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, N.; Fujiwara, Y.; Kamura, T.; Nakanishi, T.; Hatae, M.; Aoki, D.; Tanaka, K.; Tsuda, H.; Kamiura, S.; Takehara, K.; et al. Phase II Clinical Trial of Pegylated Liposomal Doxorubicin (JNS002) in Japanese Patients with Mullerian Carcinoma (Epithelial Ovarian Carcinoma, Primary Carcinoma of Fallopian Tube, Peritoneal Carcinoma) Having a Therapeutic History of Platinum-based Chemotherapy: A Phase II Study of the Japanese Gynecologic Oncology Group. Jpn. J. Clin. Oncol. 2008, 38, 777–785. [Google Scholar] [CrossRef]
- Hrelia, S.; Fiorentini, D.; Maraldi, T.; Angeloni, C.; Bordoni, A.; Biagi, P.L.; Hakim, G. Doxorubicin induces early lipid peroxidation associated with changes in glucose transport in cultured cardiomyocytes. Biochim. Biophys. Acta (BBA)-Biomembr. 2002, 1567, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Thandavarayan, R.A.; Giridharan, V.V.; Arumugam, S.; Suzuki, K.; Ko, K.M.; Krishnamurthy, P.; Watanabe, K.; Konishi, T. Schisandrin B Prevents Doxorubicin Induced Cardiac Dysfunction by Modulation of DNA Damage, Oxidative Stress and Inflammation through Inhibition of MAPK/p53 Signaling. PLoS ONE 2015, 10, e0119214. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Xu, T.; Wang, C.; Li, Y.; Lin, Z.; Zhao, M.; Zhu, B. Novel functionalized nanoparticles for tumor-targeting co-delivery of doxorubicin and siRNA to enhance cancer therapy. Int. J. Nanomed. 2017, 13, 143–159. [Google Scholar] [CrossRef]
- Khutale, G.V.; Casey, A. Synthesis and characterization of a multifunctional gold-doxorubicin nanoparticle system for pH triggered intracellular anticancer drug release. Eur. J. Pharm. Biopharm. 2017, 119, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.; Zhao, Z.-Z.; Kang, J.; Lin, L.-R.; Chen, W.-T.; Liu, J.-X.; Wu, X.-L.; Lu, T.-L. Cinnamaldehyde and Doxorubicin Co-Loaded Graphene Oxide Wrapped Mesoporous Silica Nanoparticles for Enhanced MCF-7 Cell Apoptosis. Int. J. Nanomed. 2020, 15, 10285–10304. [Google Scholar] [CrossRef]
- Csikós, Z.; Kerekes, K.; Fazekas, E.; Kun, S.; Borbély, J. Biopolymer based nanosystem for doxorubicin targeted delivery. Am. J. Cancer Res. 2017, 7, 715–726. [Google Scholar]
- Szuplewska, A.; Joniec, A.R.; Pocztańska, E.; Krysiński, P.; Dybko, A.; Chudy, M. Magnetic field-assisted selective delivery of doxorubicin to cancer cells using magnetoliposomes as drug nanocarriers. Nanotechnology 2019, 30, 315101. [Google Scholar] [CrossRef]
- Lee, C.-S.; Kim, T.; Oh, D.; Bae, S.; Ryu, J.; Kong, H.; Jeon, H.; Seo, H.; Jeon, S.; Kim, T. In Vivo and In Vitro Anticancer Activity of Doxorubicin-loaded DNA-AuNP Nanocarrier for the Ovarian Cancer Treatment. Cancers 2020, 12, 634. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.M.; Shrivastav, A.; Shrivastav, B.R. Biogenic gold nanoparticles: As a potential candidate for brain tumor directed drug delivery. Artif. Cells Nanomed. Biotechnol. 2013, 43, 311–317. [Google Scholar] [CrossRef]
- Giráldez-Pérez, R.; Grueso, E.; Domínguez, I.; Pastor, N.; Kuliszewska, E.; Prado-Gotor, R.; Requena-Domenech, F. Biocompatible DNA/5-Fluorouracil-Gemini Surfactant-Functionalized Gold Nanoparticles as Promising Vectors in Lung Cancer Therapy. Pharmaceutics 2021, 13, 423. [Google Scholar] [CrossRef]
- Grueso, E.; Roldan, E.; Perez-Tejeda, P.; Kuliszewska, E.; Molero, B.; Brecker, L.; Giráldez-Pérez, R.M. Reversible DNA compaction induced by partial intercalation of 16-Ph-16 gemini surfactants: Evidence of triple helix formation. Phys. Chem. Chem. Phys. 2018, 20, 24902–24914. [Google Scholar] [CrossRef] [PubMed]
- Giráldez-Pérez, R.M.; Grueso, E.; Lhamyani, S.; Perez-Tejeda, P.; Gentile, A.-M.; Kuliszewska, E.; Roman-Perez, J.; El Bekay, R. miR-21/Gemini surfactant-capped gold nanoparticles as potential therapeutic complexes: Synthesis, characterization and in vivo nanotoxicity probes. J. Mol. Liq. 2020, 313, 113577. [Google Scholar] [CrossRef]
- Thorek, D.L.; Tsourkas, A. Size, charge and concentration dependent uptake of iron oxide particles by non-phagocytic cells. Biomaterials 2008, 29, 3583–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouton, C.W.; Seymour, L.W. Key issues in non-viral gene delivery. Adv. Drug Deliv. Rev. 2001, 46, 187–203. [Google Scholar] [CrossRef]
- Nayerossadat, N.; Ali, P.A.; Maedeh, T. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef]
- González-Pérez, A. Reversible DNA Compaction. Curr. Top. Med. Chem. 2014, 14, 766–773. [Google Scholar] [CrossRef]
- Emerson, M.; Renwick, L.; Tate, S.; Rhind, S.; Milne, E.; Painter, H.; Boyd, A.C.; McLachlan, G.; Griesenbach, U.; Cheng, S.H.; et al. Transfection efficiency and toxicity following delivery of naked plasmid DNA and cationic lipid–DNA complexes to ovine lung segments. Mol. Ther. 2003, 8, 646–653. [Google Scholar] [CrossRef]
- Estévez-Torres, A.; Baigl, D. DNA compaction: Fundamentals and applications. Soft Matter 2011, 7, 6746–6756. [Google Scholar] [CrossRef]
- Hirano, K.; Ichikawa, M.; Ishido, T.; Ishikawa, M.; Baba, Y.; Yoshikawa, K. How environmental solution conditions determine the compaction velocity of single DNA molecules. Nucleic Acids Res. 2011, 40, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Gawęda, S.; Morán, M.C.; Pais, A.A.; Dias, R.; Schillén, K.; Lindman, B.; Miguel, M. Cationic agents for DNA compaction. J. Colloid Interface Sci. 2008, 323, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Grueso, E.; Kuliszewska, E.; Prado-Gotor, R.; Perez-Tejeda, P.; Roldan, E. Improving the understanding of DNA–propanediyl-1,3-bis(dodecyldimethylammonium) dibromide interaction using thermodynamic, structural and kinetic approaches. Phys. Chem. Chem. Phys. 2013, 15, 20064–20074. [Google Scholar] [CrossRef]
- Grueso, E.; Kuliszewska, E.; Roldan, E.; Perez-Tejeda, P.; Prado-Gotor, R.; Brecker, L. DNA conformational changes induced by cationic gemini surfactants: The key to switching DNA compact structures into elongated forms. RSC Adv. 2015, 5, 29433–29446. [Google Scholar] [CrossRef]
- Lin, J.; Miao, L.; Zhong, G.; Lin, C.-H.; Dargazangy, R.; Alexander-Katz, A. Understanding the synergistic effect of physicochemical properties of nanoparticles and their cellular entry pathways. Commun. Biol. 2020, 3, 205. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Wu, D.-C.; Li, Z.-J.; Chen, G.-Q. Chpater 7—Polymer NanoparticlesPolymer Nanoparticles. Prog. Mol. Biol. Transl. Sci. 2011, 104, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef]
- Gatoo, M.A.; Naseem, S.; Arfat, M.Y.; Dar, A.M.; Qasim, K.; Zubair, S. Physicochemical Properties of Nanomaterials: Implication in Associated Toxic Manifestations. BioMed Res. Int. 2014, 2014, 498420. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, T.; Naqvi, K. Isosbestic points in emission spectra. Chem. Phys. Lett. 1968, 2, 374–378. [Google Scholar] [CrossRef]
- Neidle, S. Nucleic Acid Structure and Recognition, 1st ed.; Oxford University Press: New York, NY, USA, 2002; ISBN 0-19-850635-X. [Google Scholar]
- Garbett, N.C.; Ragazzon, P.; Chaires, J.B. Circular dichroism to determine binding mode and affinity of ligand–DNA interactions. Nat. Protoc. 2007, 2, 3166–3172. [Google Scholar] [CrossRef]
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular Dichroism: Principles and Applications, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000; ISBN 978-0-471-33003-5. [Google Scholar]
- Monnot, M.; Mauffret, O.; Lescot, E.; Fermandjian, S. Probing intercalation and conformational effects of the anticancer drug 2-methyl-9-hydroxyellipticinium acetate in DNA fragments with circular dichroism. Eur. J. Biochem. 1992, 204, 1035–1039. [Google Scholar] [CrossRef]
- Prado-Gotor, R.; Grueso, E. A kinetic study of the interaction of DNA with gold nanoparticles: Mechanistic aspects of the interaction. Phys. Chem. Chem. Phys. 2010, 13, 1479–1489. [Google Scholar] [CrossRef]
- Grueso, E.; Cerrillos, C.; Hidalgo, J.; Cornejo, P.L. Compaction and Decompaction of DNA Induced by the Cationic Surfactant CTAB. Langmuir 2012, 28, 10968–10979. [Google Scholar] [CrossRef]
- Lebrón, J.; López-Cornejo, P.; García-Dionisio, E.; Huertas, P.; García-Calderón, M.; Moyá, M.; Ostos, F.; López-López, M. Cationic Single-Chained Surfactants with a Functional Group at the End of the Hydrophobic Tail DNA Compacting Efficiency. Pharmaceutics 2021, 13, 589. [Google Scholar] [CrossRef]
- Cho, E.J.; Holback, H.; Liu, K.C.; Abouelmagd, S.A.; Park, J.; Yeo, Y. Nanoparticle Characterization: State of the Art, Challenges, and Emerging Technologies. Mol. Pharm. 2013, 10, 2093–2110. [Google Scholar] [CrossRef] [Green Version]
- Dams, E.T.; Oyen, W.J.; Boerman, O.C.; Storm, G.; Laverman, P.; Kok, P.J.; Buijs, W.C.; Bakker, H.; van der Meer, J.W.; Cor-stens, F.H. 99mTc-PEG Liposomes for the Scintigraphic Detection of Infection and Inflammation: Clinical Evaluation. J. Nucl. Med. 2000, 41, 622–630. [Google Scholar]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Freitas, C.; Müller, R.H. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int. J. Pharm. 1998, 168, 221–229. [Google Scholar] [CrossRef]
- Sultana, S.; Alzahrani, N.; Alzahrani, R.; Alshamrani, W.; Aloufi, W.; Ali, A.; Najib, S.; Siddiqui, N.A. Stability issues and approaches to stabilised nanoparticles based drug delivery system. J. Drug Target. 2020, 28, 468–486. [Google Scholar] [CrossRef]
- Ganta, S.; Singh, A.; Coleman, T.P.; Williams, D.; Amiji, M. Pharmaceutical Nanotechnology: Overcoming Drug Delivery Challenges in Contemporary Medicine. In Nanomedicine. Nanostructure Science and Technology; Ge, Y., Li, S., Wang, S., Moore, R., Eds.; Springer: New York, NY, USA, 2014; pp. 191–236. [Google Scholar] [CrossRef]
- González, V.; Kharisov, B.; Gómez, I. Preparation, optical characterization and stability of gold nanoparticles by facile methods. Rev. Mex. Fís. 2019, 65, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Ke, F.; Luu, Y.K.; Hadjiargyrou, M.; Liang, D. Characterizing DNA Condensation and Conformational Changes in Organic Solvents. PLoS ONE 2010, 5, e13308. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.; Köper, I. Polyelectrolyte-Coated Gold Nanoparticles: The Effect of Salt and Polyelectrolyte Concentration on Colloidal Stability. Polymers 2018, 10, 1336. [Google Scholar] [CrossRef] [Green Version]
- Agyapong, D.A.Y.; Zhao, R.-L.; Li, C.-X.; Du, W.-J.; Zeng, H.-J. Novel Mercaptan Derivatives Nanogold Particles with Hy-perchromic and Fluorescence Sensitization Effects. In Proceedings of the 2016 International Conference on Biological Sciences and Technology; Ding, H.B., Yuan, Z., Eds.; Atlantis Press: Paris, France, 2016; pp. 143–148. [Google Scholar]
- Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, M.A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. Condens. Matter 2017, 29, 203002. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C. Development of High-Drug-Loading Nanoparticles. Chempluschem 2020, 85, 2143–2157. [Google Scholar] [CrossRef]
- Grozescu, T.; Popa, F. Prostate cancer between prognosis and adequate/proper therapy. J. Med. Life 2017, 10, 5–12. [Google Scholar]
- Rekasi, Z.; Schally, A.V.; Plonowski, A.; Czompoly, T.; Csernus, B.; Varga, J.L. Regulation of prostate-specific antigen (PSA) gene expression and release in LNCaP prostate cancer by antagonists of growth hormone-releasing hormone and vasoactive intestinal peptide. Prostate 2001, 48, 188–199. [Google Scholar] [CrossRef]
- Nadendla, S.K.; Hazan, A.; Ward, M.; Harper, L.J.; Moutasim, K.; Bianchi, L.S.; Naase, M.; Ghali, L.; Thomas, G.J.; Prowse, D.M.; et al. GLI1 Confers Profound Phenotypic Changes upon LNCaP Prostate Cancer Cells That Include the Acquisition of a Hormone Independent State. PLoS ONE 2011, 6, e20271. [Google Scholar] [CrossRef]
- Appukuttan, A.; Flacke, J.-P.; Flacke, H.; Posadowsky, A.; Reusch, H.P.; Ladilov, Y. Inhibition of soluble adenylyl cyclase increases the radiosensitivity of prostate cancer cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 2656–2663. [Google Scholar] [CrossRef] [Green Version]
- Sarmento-Cabral, A.; L-López, F.; Gahete, M.D.; Castaño, J.P.; Luque, R.M. Metformin Reduces Prostate Tumor Growth, in a Diet-Dependent Manner, by Modulating Multiple Signaling Pathways. Mol. Cancer Res. 2017, 15, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Kytölä, S.; Farnebo, F.; Wang, N.; Lui, W.-O.; Nupponen, N.; Isola, J.; Visakorpi, T.; Bergerheim, U.S.R.; Larsson, C. Characterization of chromosomal abnormalities in prostate cancer cell lines by spectral karyotyping. Cytogenet. Cell Genet. 1999, 87, 225–232. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Broke-Smith, T.P.; Palmer, C.P.; Beckerman, R.; Nastos, T.; Erguler, K.; Djamgoz, M.B. Citrate enhances in vitro metastatic behaviours of PC-3M human prostate cancer cells: Status of endogenous citrate and dependence on aconitase and fatty acid synthase. Int. J. Biochem. Cell Biol. 2006, 38, 1766–1777. [Google Scholar] [CrossRef]
- López-Cánovas, J.L.; del Rio-Moreno, M.; García-Fernandez, H.; Jiménez-Vacas, J.M.; Moreno-Montilla, M.; Sánchez-Frias, M.E.; Amado, V.; L-López, F.; Fondevila, M.F.; Ciria, R.; et al. Splicing factor SF3B1 is overexpressed and implicated in the aggressiveness and survival of hepatocellular carcinoma. Cancer Lett. 2020, 496, 72–83. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, W.D.; Kim, S.K.; Moon, D.H.; Lee, S.J. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 406. [Google Scholar] [CrossRef]
- Rashed, W.M.; Kandeil, M.A.; Mahmoud, M.O.; Maher, D.; Ezzat, S.; Abdel-Rahman, M.H. MET canonical transcript expression is a predictive biomarker for chemo-sensitivity to MET-inhibitors in hepatocellular carcinoma cell lines. J. Cancer Res. Clin. Oncol. 2020, 147, 167–175. [Google Scholar] [CrossRef]
- Son, Y.; Shin, N.-R.; Kim, S.-H.; Park, S.-C.; Lee, H.-J. Fibrinogen-Like Protein 1 Modulates Sorafenib Resistance in Human Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 5330. [Google Scholar] [CrossRef]
- Wani, F.A.; Behera, K.; Padder, R.A.; Husain, M.; Malik, M.A.; Al-Thabaiti, N.S.; Ahmad, R.; Patel, R. Micellization, anti-proliferative activity and binding study of cationic gemini surfactants with calf thymus DNA. Colloids Interface Sci. Commun. 2019, 34, 100221. [Google Scholar] [CrossRef]
- Li, W.; Shi, J.; Zhang, C.; Li, M.; Gan, L.; Xu, H.; Yang, X. Co-delivery of thioredoxin 1 shRNA and doxorubicin by folate-targeted gemini surfactant-based cationic liposomes to sensitize hepatocellular carcinoma cells. J. Mater. Chem. B 2014, 2, 4901–4910. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell1, 6th ed.; Wilson, J., Hunt, T., Eds.; W.W. Norton & Company: New York, NY, USA, 2017; ISBN 9781315735368. [Google Scholar]
- Oh, N.; Park, J.-H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9 (Suppl. S1), 51–63. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Yang, Y.; Niu, H.; Luo, M.; Wu, Z.-S. Design and application of DNA nanostructures for organelle-targeted delivery of anticancer drugs. Expert Opin. Drug Deliv. 2022, 19, 707–723. [Google Scholar] [CrossRef]
- Jiang, S.; Ge, Z.; Mou, S.; Yan, H.; Fan, C. Designer DNA nanostructures for therapeutics. Chem 2020, 7, 1156–1179. [Google Scholar] [CrossRef]
- Kumar, V.; Palazzolo, S.; Bayda, S.; Corona, G.; Toffoli, G.; Rizzolio, F. DNA Nanotechnology for Cancer Therapeutics. Theranostics 2016, 6, 710–725. [Google Scholar] [CrossRef]
- Li, J.; Fan, C.; Pei, H.; Shi, J.; Huang, Q. Smart Drug Delivery Nanocarriers with Self-Assembled DNA Nanostructures. Adv. Mater. 2013, 25, 4386–4396. [Google Scholar] [CrossRef]
- Chao, J.; Liu, H.; Su, S.; Wang, L.; Huang, W.; Fan, C. Structural DNA Nanotechnology for Intelligent Drug Delivery. Small 2014, 10, 4626–4635. [Google Scholar] [CrossRef]
- Li, J.; Pei, H.; Zhu, B.; Liang, L.; Wei, M.; He, Y.; Chen, N.; Li, D.; Huang, Q.; Fan, C. Self-Assembled Multivalent DNA Nanostructures for Noninvasive Intracellular Delivery of Immunostimulatory CpG Oligonucleotides. ACS Nano 2011, 5, 8783–8789. [Google Scholar] [CrossRef]
- Jiang, Q.; Song, C.; Nangreave, J.; Liu, X.; Lin, L.; Qiu, D.; Wang, Z.-G.; Zou, G.; Liang, X.; Yan, H.; et al. DNA Origami as a Carrier for Circumvention of Drug Resistance. J. Am. Chem. Soc. 2012, 134, 13396–13403. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, Q.; Li, N.; Dai, L.; Liu, Q.; Song, L.; Wang, J.; Li, Y.; Tian, J.; Ding, B.; et al. DNA Origami as an In Vivo Drug Delivery Vehicle for Cancer Therapy. ACS Nano 2014, 8, 6633–6643. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Shaw, A.; Zeng, X.; Benson, E.; Nyström, A.M.; Högberg, B. DNA Origami Delivery System for Cancer Therapy with Tunable Release Properties. ACS Nano 2012, 6, 8684–8691. [Google Scholar] [CrossRef] [Green Version]
- Perrault, S.D.; Shih, W.M. Virus-Inspired Membrane Encapsulation of DNA Nanostructures To Achieve In Vivo Stability. ACS Nano 2014, 8, 5132–5140. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; ISBN 0-87969-309-6. [Google Scholar]
- Felsenfeld, G.; Hirschman, S.Z. A neighbor-interaction analysis of the hypochromism and spectra of DNA. J. Mol. Biol. 1965, 13, 407–427. [Google Scholar] [CrossRef]
- Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Montero-Hidalgo, A.J.; Gómez-Gómez, E.; Fuentes-Fayos, A.C.; León-González, A.J.; Sáez-Martínez, P.; Alors-Pérez, E.; Pedraza-Arévalo, S.; González-Serrano, T.; et al. Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. eBioMedicine 2020, 51, 102547. [Google Scholar] [CrossRef] [Green Version]
- Hormaechea-Agulla, D.; Jiménez-Vacas, J.M.; Gómez-Gómez, E.; López, F.L.; Carrasco-Valiente, J.; Valero-Rosa, J.; Moreno, M.M.; Sánchez-Sánchez, R.; Ortega-Salas, R.; Gracia-Navarro, F.; et al. The oncogenic role of the spliced somatostatin receptor sst5TMD4 variant in prostate cancer. FASEB J. 2017, 31, 4682–4696. [Google Scholar] [CrossRef] [Green Version]
- Lunkenheimer, K.; Rakotoaly, R.H.; Wattebled, L. Spacer effects in dimeric cationic surfactants. Colloid Polym. Sci. 2004, 283, 469–479. [Google Scholar] [CrossRef]
- Kuliszewska, E.; Brecker, L. Gemini Surfactants Foam Formation Ability and Foam Stability Depends on Spacer Length. J. Surfactants Deterg. 2014, 17, 951–957. [Google Scholar] [CrossRef]
- Pérez-Arnaiz, C.; Busto, N.; Leal, J.M.; Garcia, B. New Insights into the Mechanism of the DNA/Doxorubicin Interaction. J. Phys. Chem. B 2014, 118, 1288–1295. [Google Scholar] [CrossRef]
- Karukstis, K.K.; Thompson, E.H.Z.; Whiles, J.A.; Rosenfeld, R.J. Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys. Chem. 1998, 73, 249–263. [Google Scholar] [CrossRef]
- Horcas, I.; Fernández, R.; Gómez-Rodriguez, J.M.; Colchero, J.; Gomez-Herrero, J.; Baro, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 13705. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Zeta Potential/mV in Water | Zeta Potential/mV in PBS 0.1X |
|---|---|---|
| N1 | (30 ± 3) | (18.1 ± 1.7) |
| N2 | (34 ± 3) | (28 ± 3) |
| N3 | (37.0 ± 0.9) | (40.4 ± 0.7) |
| C1 | (−49 ± 6) | (−28 ± 5) |
| C2 | (−49 ± 3) | (−44 ± 5) |
| C3 | (−35.5 ± 1.2) | (−30 ± 3) |
| System | Diameter/nm in Water | Diameter/nm in PBS 0.1X |
|---|---|---|
| C1 | (33 ± 7) | (67 ± 11) |
| C2 | (57 ± 11) | (71 ± 11) |
| C3 | (44 ± 7) | (64 ± 9) |
| Treatment | PNT2 | LNCaP | DU145 | HepG2 | SNU387 |
|---|---|---|---|---|---|
| Au@16-Ph-16 (N3) | 5.9 ± 1.1 | 6.2 ± 1.6 | 5.3 ± 1.4 | 5.6 ± 1.0 | 5.8 ± 1.0 |
| Au@16-Ph-16/DNA/Doxo (C3) | 5.8 ± 1.4 | 6.4 ± 1.3 | 5.78 ± 1.4 | 5.9 ± 1.0 | 5.9 ± 0.9 |
| Au@16-Ph-16/DNA/Doxo + Au@16-Ph-16 (C3 + N3) | 6.7 ± 1.1 | 6.3 ± 1.5 | 5.9 ± 0.8 | 6.1 ± 0.9 | 6.,6 ± 1.1 |
| Treatment | PNT2 | LNCaP | DU145 | HepG2 | SNU387 |
|---|---|---|---|---|---|
| N3 | 415 ± 50 | 947 ± 49 | 968 ± 67 | 678 ± 52 | 761 ± 44 |
| C3 | 87 ± 25 | 605 ± 15 | 212 ± 8 | 48 ± 5 | 63 ± 8 |
| C3 + N3 | 214 ± 8 | 790 ± 30 | 802 ± 15 | 662 ± 84 | 671 ± 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giráldez-Pérez, R.M.; Grueso, E.; Montero-Hidalgo, A.J.; Luque, R.M.; Carnerero, J.M.; Kuliszewska, E.; Prado-Gotor, R. Gold Nanosystems Covered with Doxorubicin/DNA Complexes: A Therapeutic Target for Prostate and Liver Cancer. Int. J. Mol. Sci. 2022, 23, 15575. https://doi.org/10.3390/ijms232415575
Giráldez-Pérez RM, Grueso E, Montero-Hidalgo AJ, Luque RM, Carnerero JM, Kuliszewska E, Prado-Gotor R. Gold Nanosystems Covered with Doxorubicin/DNA Complexes: A Therapeutic Target for Prostate and Liver Cancer. International Journal of Molecular Sciences. 2022; 23(24):15575. https://doi.org/10.3390/ijms232415575
Chicago/Turabian StyleGiráldez-Pérez, Rosa M., Elia Grueso, Antonio J. Montero-Hidalgo, Raúl M. Luque, José M. Carnerero, Edyta Kuliszewska, and Rafael Prado-Gotor. 2022. "Gold Nanosystems Covered with Doxorubicin/DNA Complexes: A Therapeutic Target for Prostate and Liver Cancer" International Journal of Molecular Sciences 23, no. 24: 15575. https://doi.org/10.3390/ijms232415575
APA StyleGiráldez-Pérez, R. M., Grueso, E., Montero-Hidalgo, A. J., Luque, R. M., Carnerero, J. M., Kuliszewska, E., & Prado-Gotor, R. (2022). Gold Nanosystems Covered with Doxorubicin/DNA Complexes: A Therapeutic Target for Prostate and Liver Cancer. International Journal of Molecular Sciences, 23(24), 15575. https://doi.org/10.3390/ijms232415575