
Cas9-Mediated Nanopore Sequencing Enables Precise Characterization of Structural Variants in CCM Genes
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
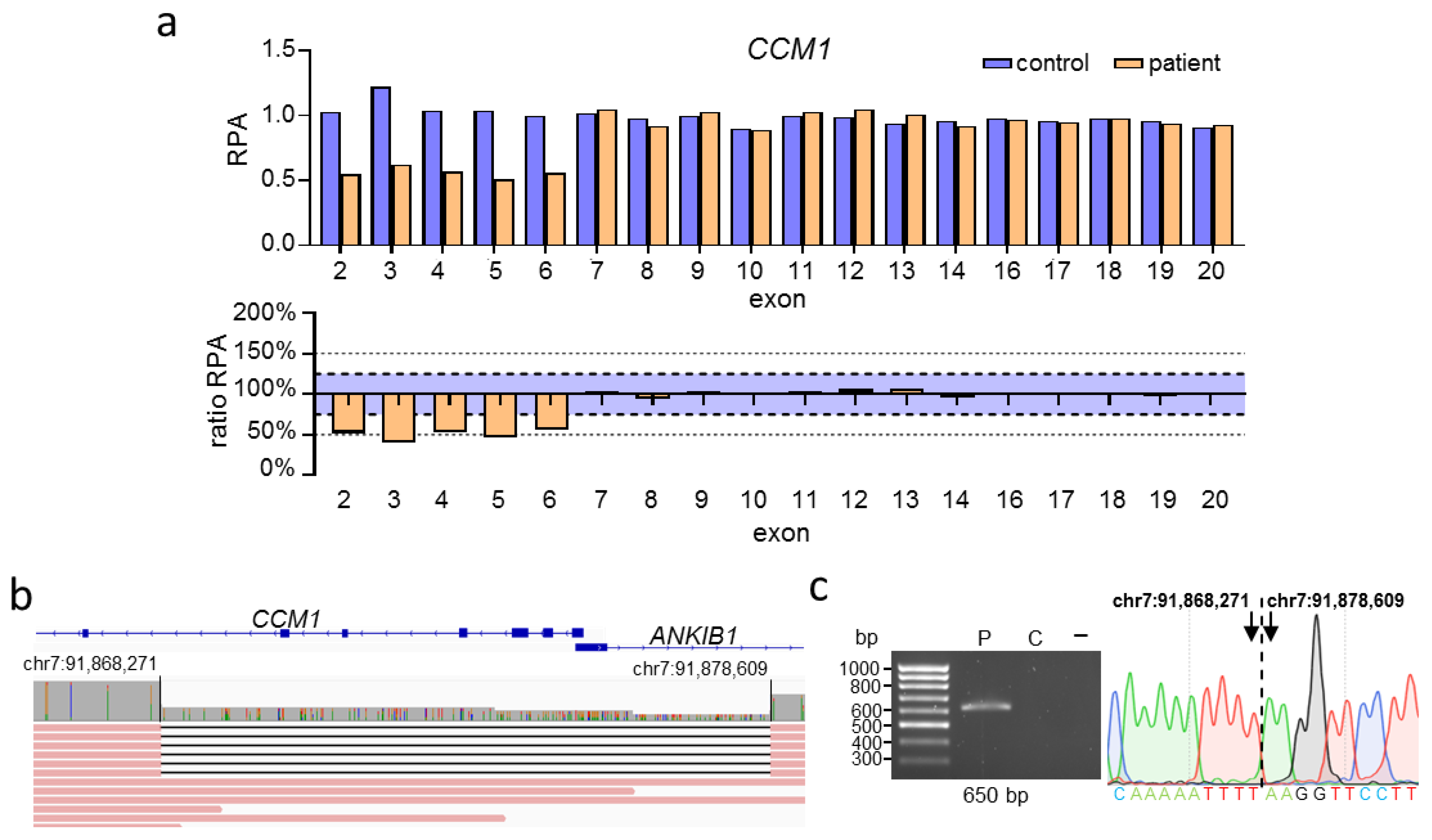
2.1. Cas9-Mediated Nanopore Sequencing Confirmed a 2552 bp Deletion in CCM1
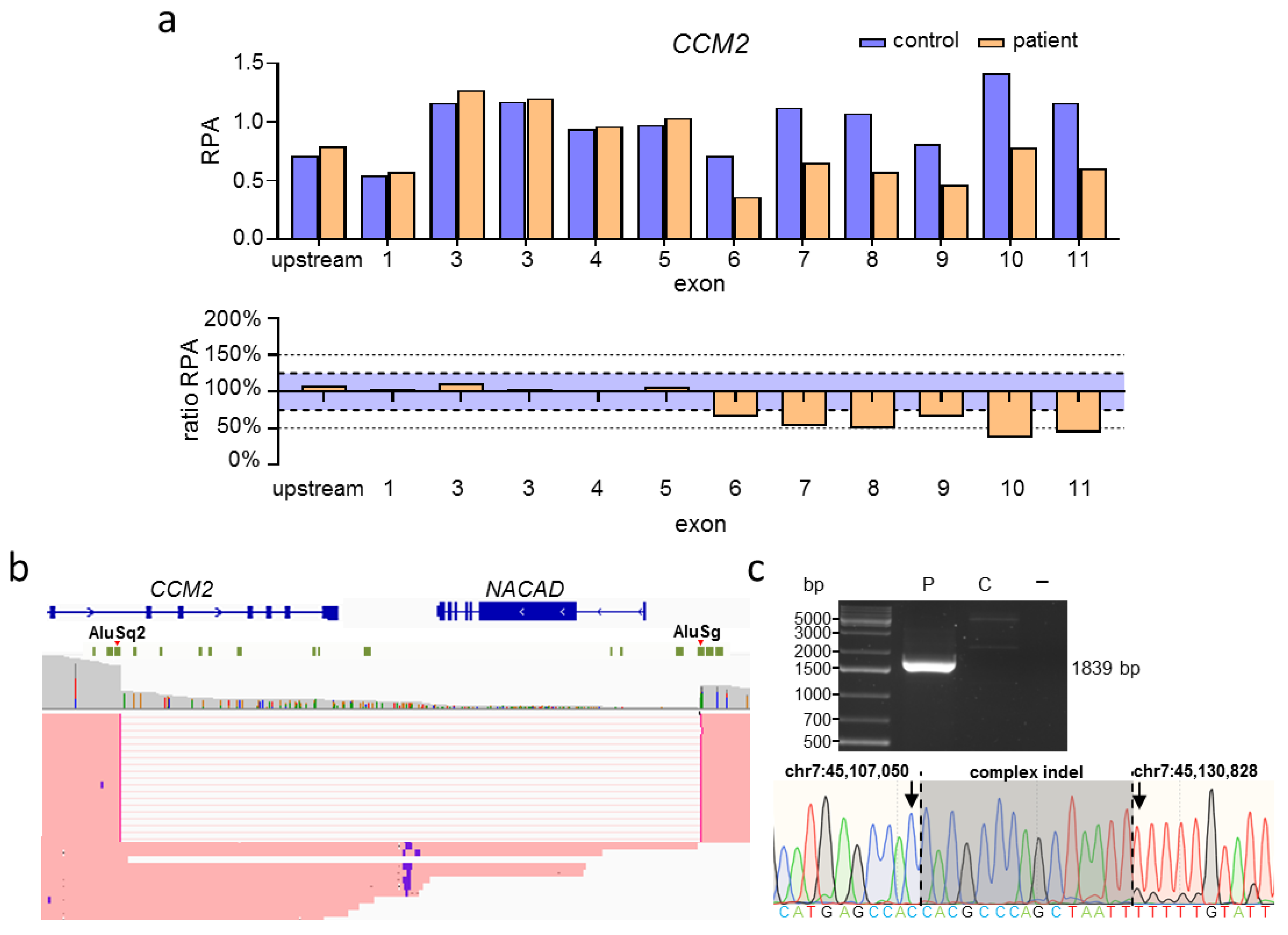
2.2. Targeted Nanopore Sequencing Revealed the Exact Size of Large Deletions in Familial CCM Cases
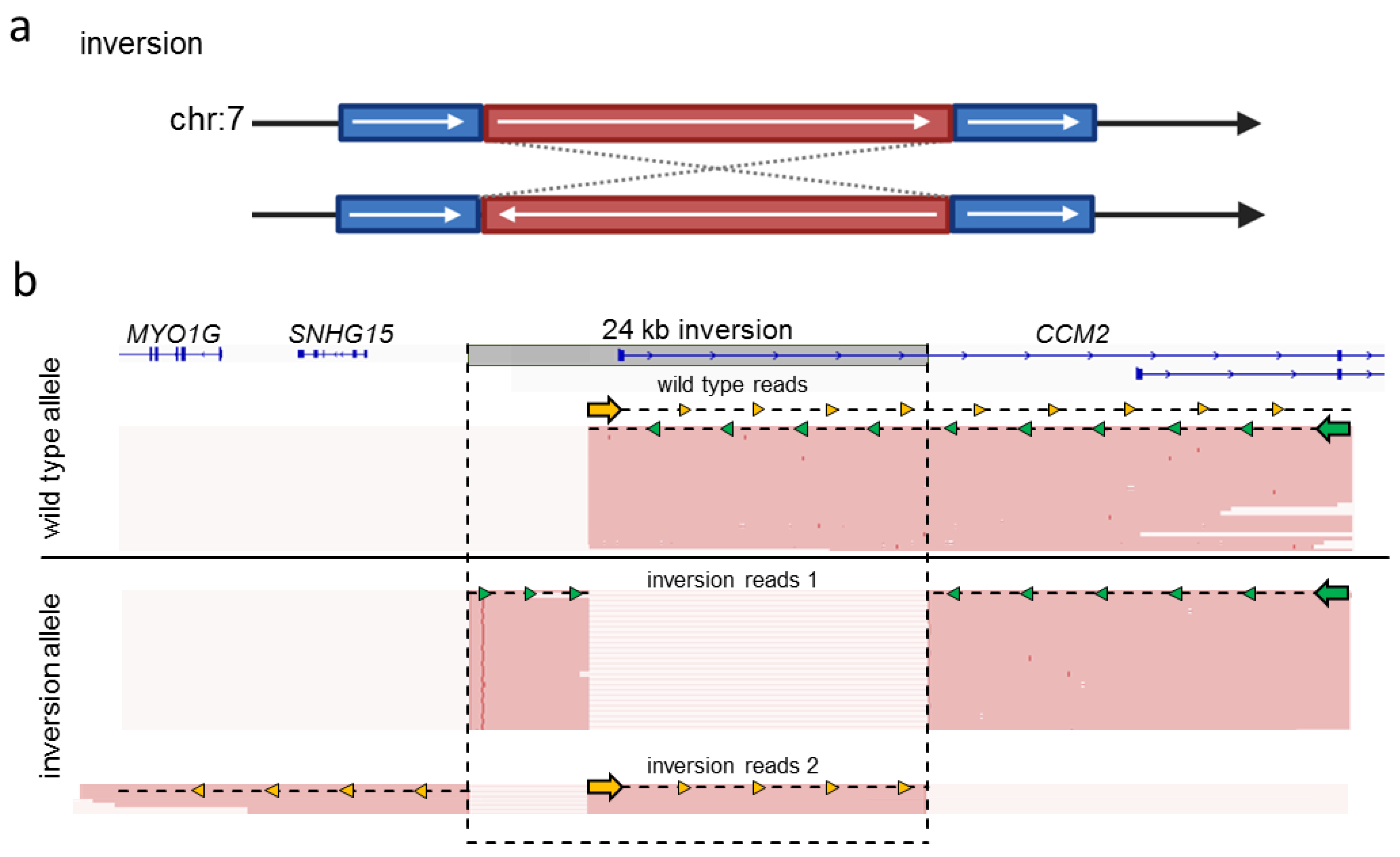
2.3. Targeted Nanopore Sequencing Can Be Used to Identify Complex SVs in CCM
2.4. Targeted Nanopore Sequencing on the Flongle Flow Cell Is Also Able to Determine Variant Breakpoints
3. Discussion
4. Materials and Methods
4.1. Design of crRNAs
4.2. Nanopore Sequencing
4.3. Multiplex Ligation-Dependent Probe Amplification, PCR, and Sanger Sequencing
4.4. Illumina Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiegler, S.; Rath, M.; Paperlein, C.; Felbor, U. Cerebral Cavernous Malformations: An Update on Prevalence, Molecular Genetic Analyses, and Genetic Counselling. Mol. Syndromol. 2018, 9, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Felbor, U.; Gaetzner, S.; Verlaan, D.J.; Vijzelaar, R.; Rouleau, G.A.; Siegel, A.M. Large germline deletions and duplication in isolated cerebral cavernous malformation patients. Neurogenetics 2007, 8, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Much, C.D.; Schwefel, K.; Skowronek, D.; Shoubash, L.; von Podewils, F.; Elbracht, M.; Spiegler, S.; Kurth, I.; Flöel, A.; Schroeder, H.W.S.; et al. Novel Pathogenic Variants in a Cassette Exon of CCM2 in Patients With Cerebral Cavernous Malformations. Front. Neurol. 2019, 10, 1219. [Google Scholar] [CrossRef] [Green Version]
- Liquori, C.L.; Berg, M.J.; Squitieri, F.; Leedom, T.P.; Ptacek, L.; Johnson, E.W.; Marchuk, D.A. Deletions in CCM2 Are a Common Cause of Cerebral Cavernous Malformations. Am. J. Hum. Genet. 2007, 80, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riant, F.; Cecillon, M.; Saugier-Veber, P.; Tournier-Lasserve, E. CCM molecular screening in a diagnosis context: Novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 2013, 14, 133–141. [Google Scholar] [CrossRef]
- Riant, F.; Odent, S.; Cecillon, M.; Pasquier, L.; de Baracé, C.; Carney, M.P.; Tournier-Lasserve, E. Deep intronic KRIT1 mutation in a family with clinically silent multiple cerebral cavernous malformations. Clin. Genet. 2014, 86, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Pilz, R.A.; Skowronek, D.; Hamed, M.; Weise, A.; Mangold, E.; Radbruch, A.; Pietsch, T.; Felbor, U.; Rath, M. Using CRISPR/Cas9 genome editing in human iPSCs for deciphering the pathogenicity of a novel CCM1 transcription start site deletion. Front. Mol. Biosci. 2022, 9, 953048. [Google Scholar] [CrossRef]
- Spiegler, S.; Rath, M.; Hoffjan, S.; Dammann, P.; Sure, U.; Pagenstecher, A.; Strom, T.; Felbor, U. First large genomic inversion in familial cerebral cavernous malformation identified by whole genome sequencing. Neurogenetics 2018, 19, 55–59. [Google Scholar] [CrossRef]
- Pilz, R.A.; Schwefel, K.; Weise, A.; Liehr, T.; Demmer, P.; Spuler, A.; Spiegler, S.; Gilberg, E.; Hübner, C.A.; Felbor, U.; et al. First interchromosomal insertion in a patient with cerebral and spinal cavernous malformations. Sci. Rep. 2020, 10, 6306. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 246. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, K.; Ishihara, H.; Sakazaki, H.; Suzuki, T.; Horie, M.; Ohno, S. Long-Read Sequence Confirmed a Large Deletion Including MYH6 and MYH7 in an Infant of Atrial Septal Defect and Atrial Arrhythmias. Circ. Genom. Precis. Med. 2021, 14, e003223. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.M.; Bavi, O. DNA sequencing: An overview of solid-state and biological nanopore-based methods. Biophys Rev. 2022, 14, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.M.; Holliday, D.L.; Crinnion, L.A.; Bonthron, D.T. Long-read nanopore DNA sequencing can resolve complex intragenic duplication/deletion variants, providing information to enable preimplantation genetic diagnosis. Prenat. Diagn. 2022, 42, 226–232. [Google Scholar] [CrossRef]
- Steiert, T.A.; Fuß, J.; Juzenas, S.; Wittig, M.; Hoeppner, M.P.; Vollstedt, M.; Varkalaite, G.; ElAbd, H.; Brockmann, C.; Görg, S.; et al. High-throughput method for the hybridisation-based targeted enrichment of long genomic fragments for PacBio third-generation sequencing. NAR Genom. Bioinform. 2022, 4, lqac051. [Google Scholar] [CrossRef]
- Wang, M.; Beck, C.R.; English, A.C.; Meng, Q.; Buhay, C.; Han, Y.; Doddapaneni, H.V.; Yu, F.; Boerwinkle, E.; Lupski, J.R.; et al. PacBio-LITS: A large-insert targeted sequencing method for characterization of human disease-associated chromosomal structural variations. BMC Genomics 2015, 16, 214. [Google Scholar] [CrossRef] [Green Version]
- Madsen, E.B.; Höijer, I.; Kvist, T.; Ameur, A.; Mikkelsen, M.J. Xdrop: Targeted sequencing of long DNA molecules from low input samples using droplet sorting. Hum. Mutat. 2020, 41, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.E.; Sulovari, A.; Wang, T.; Loucks, H.; Hoekzema, K.; Munson, K.M.; Lewis, A.P.; Fuerte, E.P.A.; Paschal, C.R.; Walsh, T.; et al. Targeted long-read sequencing identifies missing disease-causing variation. Am. J. Hum. Genet. 2021, 108, 1436–1449. [Google Scholar] [CrossRef]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Munson, K.M.; Eng, M.; Mandell, J.B.; Gulsuner, S.; King, M.C. CRISPR-Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J. Med. Genet. 2021, 58, 850–852. [Google Scholar] [CrossRef]
- López-Girona, E.; Davy, M.W.; Albert, N.W.; Hilario, E.; Smart, M.E.M.; Kirk, C.; Thomson, S.J.; Chagné, D. CRISPR-Cas9 enrichment and long read sequencing for fine mapping in plants. Plant Methods 2020, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Fiol, A.; Jurado-Ruiz, F.; López-Girona, E.; Aranzana, M.J. An efficient CRISPR-Cas9 enrichment sequencing strategy for characterizing complex and highly duplicated genomic regions. A case study in the Prunus salicina LG3-MYB10 genes cluster. Plant Methods 2022, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; et al. Mutations within the Programmed Cell Death 10 Gene Cause Cerebral Cavernous Malformations. Am. J. Hum. Genet. 2005, 76, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liquori, C.L.; Penco, S.; Gault, J.; Leedom, T.P.; Tassi, L.; Esposito, T.; Awad, I.A.; Frati, L.; Johnson, E.W.; Squitieri, F.; et al. Different spectra of genomic deletions within the CCM genes between Italian and American CCM patient cohorts. Neurogenetics 2008, 9, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Choe, C.U.; Riant, F.; Gerloff, C.; Tournier-Lasserve, E.; Orth, M. Multiple cerebral cavernous malformations and a novel CCM3 germline deletion in a German family. J. Neurol. 2010, 257, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Cigoli, M.S.; Avemaria, F.; De Benedetti, S.; Gesu, G.P.; Accorsi, L.G.; Parmigiani, S.; Corona, M.F.; Capra, V.; Mosca, A.; Giovannini, S.; et al. PDCD10 Gene Mutations in Multiple Cerebral Cavernous Malformations. PLoS ONE 2014, 9, e110438. [Google Scholar] [CrossRef]
- Nardella, G.; Visci, G.; Guarnieri, V.; Castellana, S.; Biagini, T.; Bisceglia, L.; Palumbo, O.; Trivisano, M.; Vaira, C.; Scerrati, M.; et al. A single-center study on 140 patients with cerebral cavernous malformations: 28 new pathogenic variants and functional characterization of a PDCD10 large deletion. Hum. Mutat. 2018, 39, 1885–1900. [Google Scholar] [CrossRef]
- Fusco, C.; Copetti, M.; Mazza, T.; Amoruso, L.; Mastroianno, S.; Nardella, G.; Guarnieri, V.; Micale, L.; D′Agruma, L.; Castori, M. Molecular diagnostic workflow, clinical interpretation of sequence variants, and data repository procedures in 140 individuals with familial cerebral cavernous malformations. Hum. Mutat. 2019, 40, e24–e36. [Google Scholar] [CrossRef] [Green Version]
- Muscarella, L.A.; Guarnieri, V.; Coco, M.; Belli, S.; Parrella, P.; Pulcrano, G.; Catapano, D.; D′Angelo, V.A.; Zelante, L.; D′Agruma, L. Small Deletion at the 7q21.2 Locus in a CCM Family Detected by Real-Time Quantitative PCR. J. Biomed. Biotechnol. 2010, 2010, 854737. [Google Scholar] [CrossRef]
- Benedetti, V.; Canzoneri, R.; Perrelli, A.; Arduino, C.; Zonta, A.; Brusco, A.; Retta, S.F. Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM). Antioxidants 2022, 11, 1294. [Google Scholar] [CrossRef]
- Mondéjar, R.; Solano, F.; Rubio, R.; Delgado, M.; Pérez-Sempere, Á.; González-Meneses, A.; Vendrell, T.; Izquierdo, G.; Martinez-Mir, A.; Lucas, M. Mutation Prevalence of Cerebral Cavernous Malformation Genes in Spanish Patients. PLoS ONE 2014, 9, e86286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denier, C.; Goutagny, S.; Labauge, P.; Krivosic, V.; Arnoult, M.; Cousin, A.; Benabid, A.L.; Comoy, J.; Frerebeau, P.; Gilbert, B.; et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2004, 74, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Pös, O.; Radvanszky, J.; Styk, J.; Pös, Z.; Buglyó, G.; Kajsik, M.; Budis, J.; Nagy, B.; Szemes, T. Copy Number Variation: Methods and Clinical Applications. Appl. Sci. 2021, 11, 819. [Google Scholar] [CrossRef]
- Coughlin, C.R., 2nd; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Olsen, M.F.; Lavik, L.A.S.; Vold, T.; Drabløs, F.; Sjursen, W. Detecting copy number variation in next generation sequencing data from diagnostic gene panels. BMC Med. Genomics 2021, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- Gaetzner, S.; Stahl, S.; Sürücü, O.; Schaafhausen, A.; Halliger-Keller, B.; Bertalanffy, H.; Sure, U.; Felbor, U. CCM1 gene deletion identified by MLPA in cerebral cavernous malformation. Neurosurg. Rev. 2007, 30, 155–159; discussion 159–160. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.; Ulm, S.L.; Uddin, S.; Förster, S.; Seifert, D.; Oehme, R.; Corty, M.; Schaade, L.; Michel, J.; Nitsche, A. AmpliCoV: Rapid Whole-Genome Sequencing Using Multiplex PCR Amplification and Real-Time Oxford Nanopore MinION Sequencing Enables Rapid Variant Identification of SARS-CoV-2. Front. Microbiol. 2021, 12, 651151. [Google Scholar] [CrossRef]
- Alfano, M.; De Antoni, L.; Centofanti, F.; Visconti, V.V.; Maestri, S.; Degli Esposti, C.; Massa, R.; D′Apice, M.R.; Novelli, G.; Delledonne, M.; et al. Characterization of full-length CNBP expanded alleles in myotonic dystrophy type 2 patients by Cas9-mediated enrichment and nanopore sequencing. elife 2022, 11, e80229. [Google Scholar] [CrossRef] [PubMed]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef]
- Iyer, S.V.; Kramer, M.; Goodwin, S.; McCombie, W.R. ACME: An Affinity-based Cas9 Mediated Enrichment method for targeted nanopore sequencing. bioRxiv 2022, preprint. [Google Scholar]
- Mizuguchi, T.; Toyota, T.; Miyatake, S.; Mitsuhashi, S.; Doi, H.; Kudo, Y.; Kishida, H.; Hayashi, N.; Tsuburaya, R.S.; Kinoshita, M.; et al. Complete sequencing of expanded SAMD12 repeats by long-read sequencing and Cas9-mediated enrichment. Brain 2021, 144, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Stangl, C.; de Blank, S.; Renkens, I.; Westera, L.; Verbeek, T.; Valle-Inclan, J.E.; González, R.C.; Henssen, A.G.; van Roosmalen, M.J.; Stam, R.W.; et al. Partner independent fusion gene detection by multiplexed CRISPR-Cas9 enrichment and long read nanopore sequencing. Nat. Commun. 2020, 11, 2861. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.D.; Sasani, T.A.; Swanier, J.; Gates, B.L.; Greenland, J.; Pedersen, B.S.; Varley, K.E.; Quinlan, A.R. CaBagE: A Cas9-based Background Elimination strategy for targeted, long-read DNA sequencing. PLoS ONE 2021, 16, e0241253. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentsch, M.; Kaczmarczyk, A.; van Leeuwen, K.; de Boer, M.; Kaus-Drobek, M.; Dagher, M.C.; Kaiser, P.; Arkwright, P.D.; Gahr, M.; Rösen-Wolff, A.; et al. Alu-repeat-induced deletions within the NCF2 gene causing p67-phox-deficient chronic granulomatous disease (CGD). Hum. Mutat. 2010, 31, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Pluta, N.; Hoffjan, S.; Zimmer, F.; Köhler, C.; Lücke, T.; Mohr, J.; Vorgerd, M.; Nguyen, H.H.P.; Atlan, D.; Wolf, B.; et al. Homozygous Inversion on Chromosome 13 Involving SGCG Detected by Short Read Whole Genome Sequencing in a Patient Suffering from Limb-Girdle Muscular Dystrophy. Genes 2022, 13, 1752. [Google Scholar] [CrossRef] [PubMed]
- Schilter, K.F.; Reis, L.M.; Sorokina, E.A.; Semina, E.V. Identification of an Alu-repeat-mediated deletion of OPTN upstream region in a patient with a complex ocular phenotype. Mol. Genet. Genomic. Med. 2015, 3, 490–499. [Google Scholar] [CrossRef]
- Stephens, Z.; Wang, C.; Iyer, R.K.; Kocher, J.P. Detection and visualization of complex structural variants from long reads. BMC Bioinformatics 2018, 19, 508. [Google Scholar] [CrossRef] [Green Version]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Mai, Y.; Liu, D.; He, W.; Lin, X.; Xu, C.; Zhang, L.; Meng, X.; Mafofo, J.; Zaher, W.A.; et al. Fast-bonito: A faster deep learning based basecaller for nanopore sequencing. Artif. Intell. Life Sci. 2021, 1, 100011. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, Y.; Jiang, Y.; Li, J.; Gao, Y.; Cui, Z.; Liu, Y.; Liu, B.; Wang, Y. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skowronek, D.; Pilz, R.A.; Bonde, L.; Schamuhn, O.J.; Feldmann, J.L.; Hoffjan, S.; Much, C.D.; Felbor, U.; Rath, M. Cas9-Mediated Nanopore Sequencing Enables Precise Characterization of Structural Variants in CCM Genes. Int. J. Mol. Sci. 2022, 23, 15639. https://doi.org/10.3390/ijms232415639
Skowronek D, Pilz RA, Bonde L, Schamuhn OJ, Feldmann JL, Hoffjan S, Much CD, Felbor U, Rath M. Cas9-Mediated Nanopore Sequencing Enables Precise Characterization of Structural Variants in CCM Genes. International Journal of Molecular Sciences. 2022; 23(24):15639. https://doi.org/10.3390/ijms232415639
Chicago/Turabian StyleSkowronek, Dariush, Robin A. Pilz, Loisa Bonde, Ole J. Schamuhn, Janne L. Feldmann, Sabine Hoffjan, Christiane D. Much, Ute Felbor, and Matthias Rath. 2022. "Cas9-Mediated Nanopore Sequencing Enables Precise Characterization of Structural Variants in CCM Genes" International Journal of Molecular Sciences 23, no. 24: 15639. https://doi.org/10.3390/ijms232415639
APA StyleSkowronek, D., Pilz, R. A., Bonde, L., Schamuhn, O. J., Feldmann, J. L., Hoffjan, S., Much, C. D., Felbor, U., & Rath, M. (2022). Cas9-Mediated Nanopore Sequencing Enables Precise Characterization of Structural Variants in CCM Genes. International Journal of Molecular Sciences, 23(24), 15639. https://doi.org/10.3390/ijms232415639