
Direct or Indirect ESPT Mechanism in CFP psamFP488? A Theoretical-Computational Investigation



Abstract
1. Introduction
2. Results and Discussion
2.1. Characterization of the Cyan Fluorescent Protein Photocycle
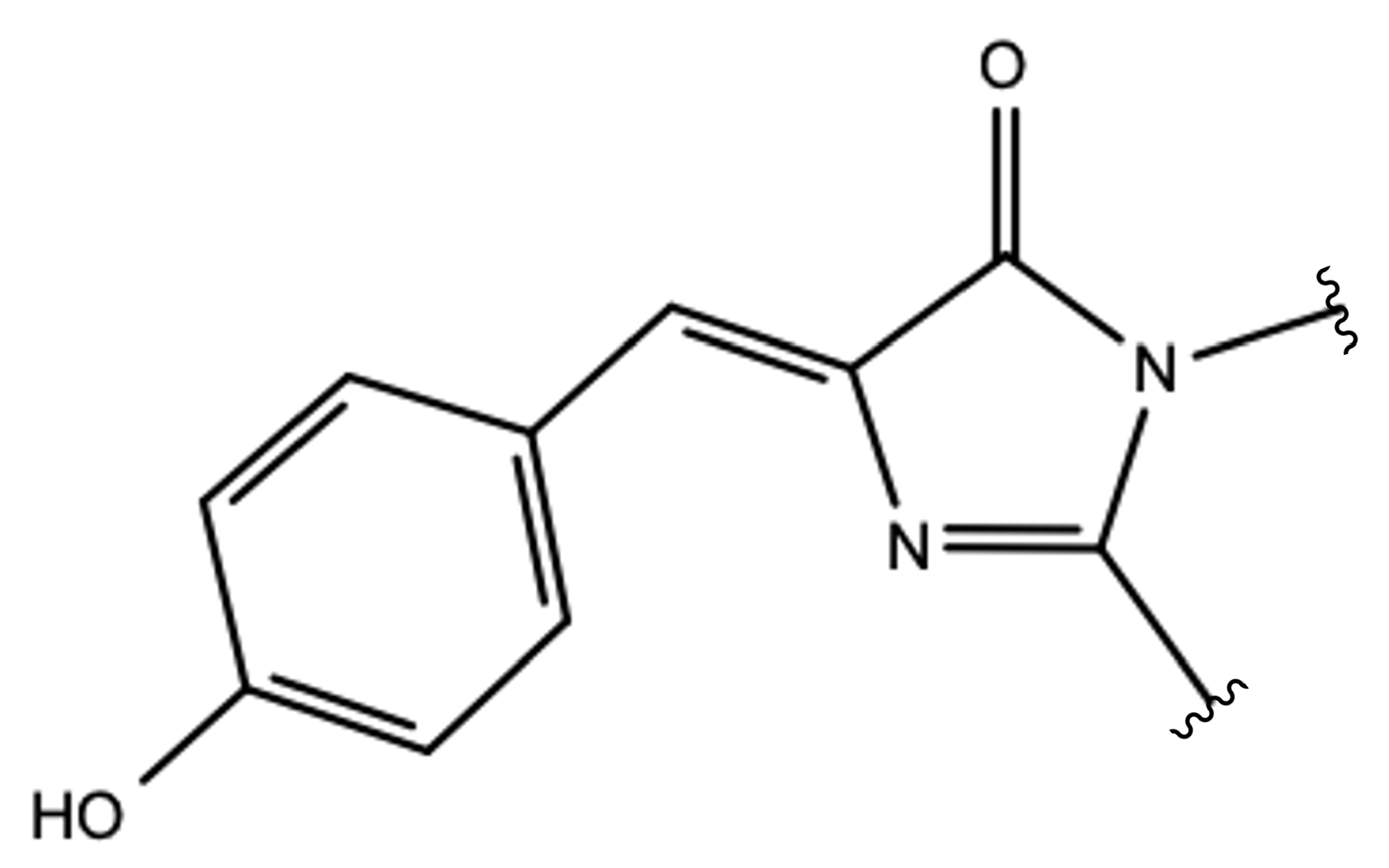
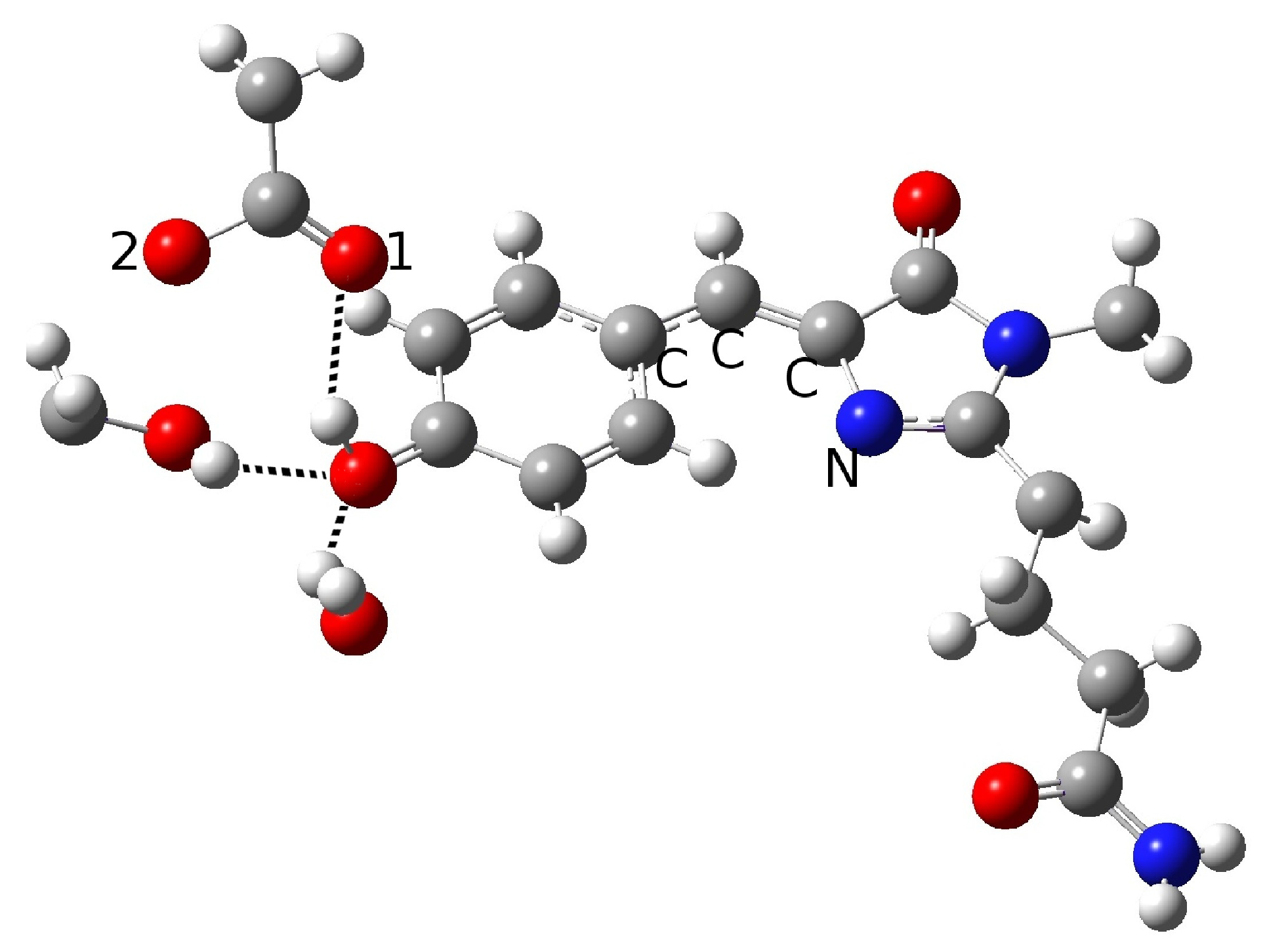
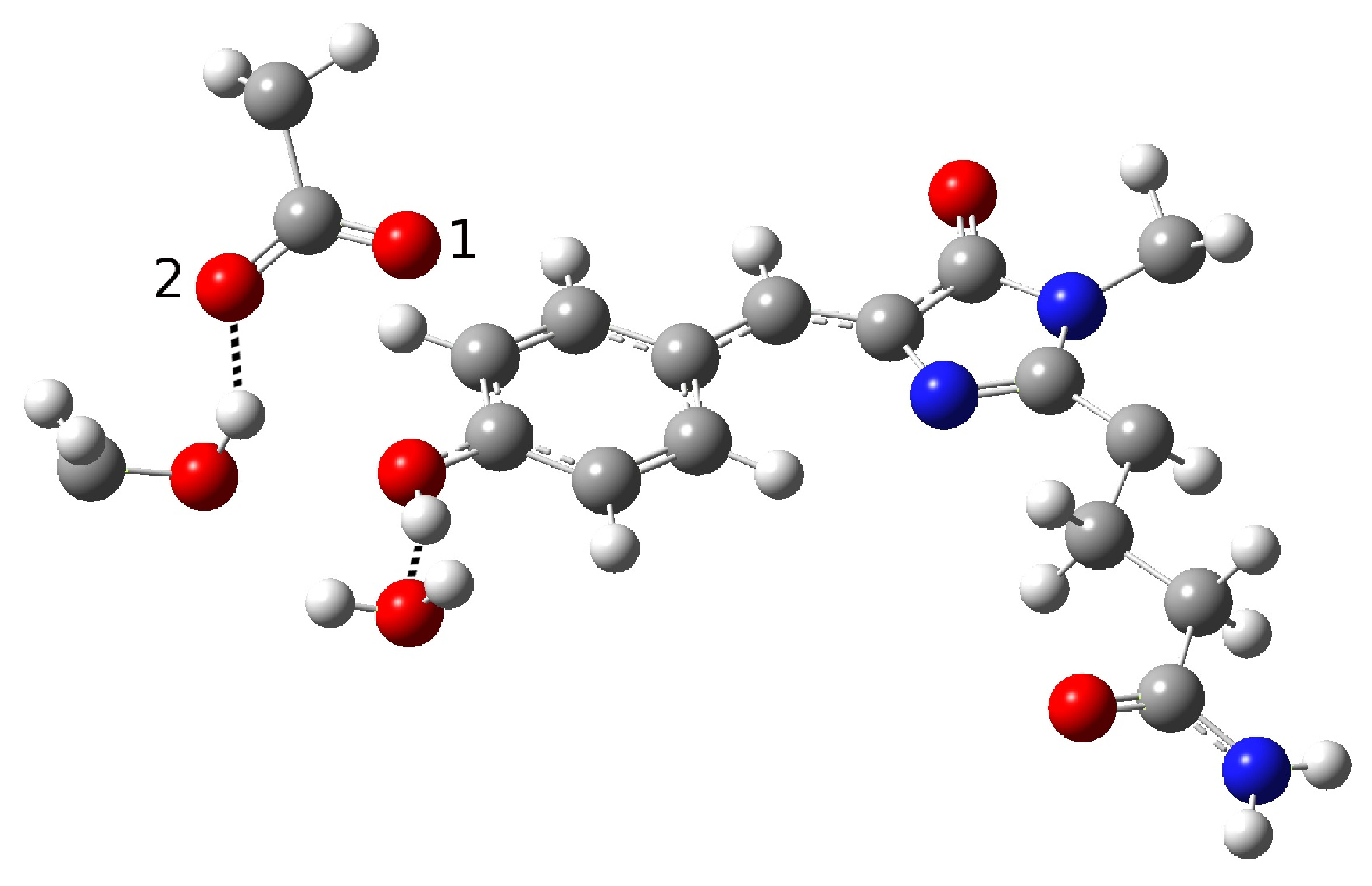
2.1.1. The Chromophore in Its Neutral Form; Part I: The Experimental Model
2.1.2. The Chromophore in Its Neutral Form; Part II: An Alternative Model
2.1.3. Optical Properties of the Neutral CFP
2.1.4. Chromophore Anionic Form Characterization
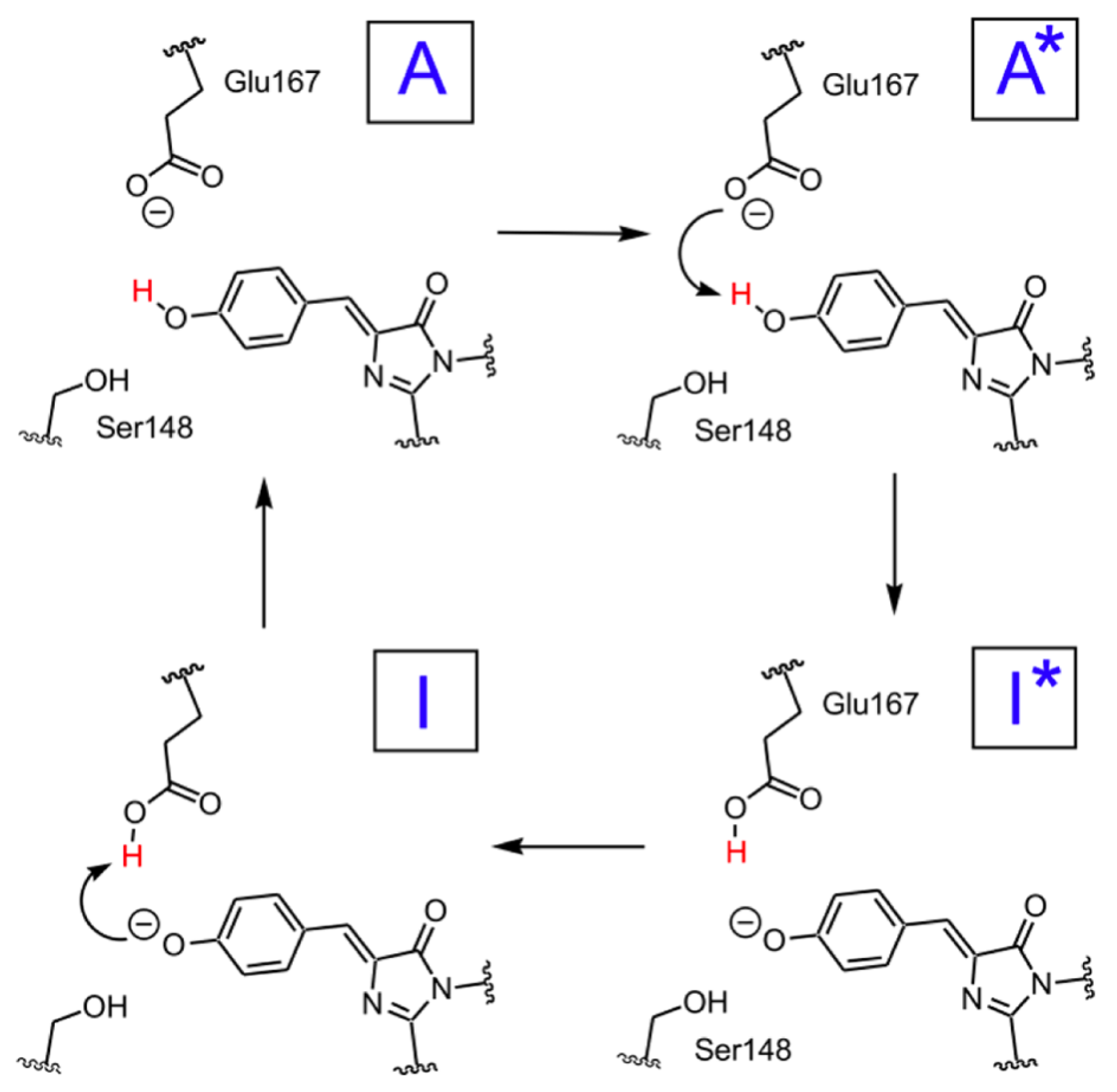
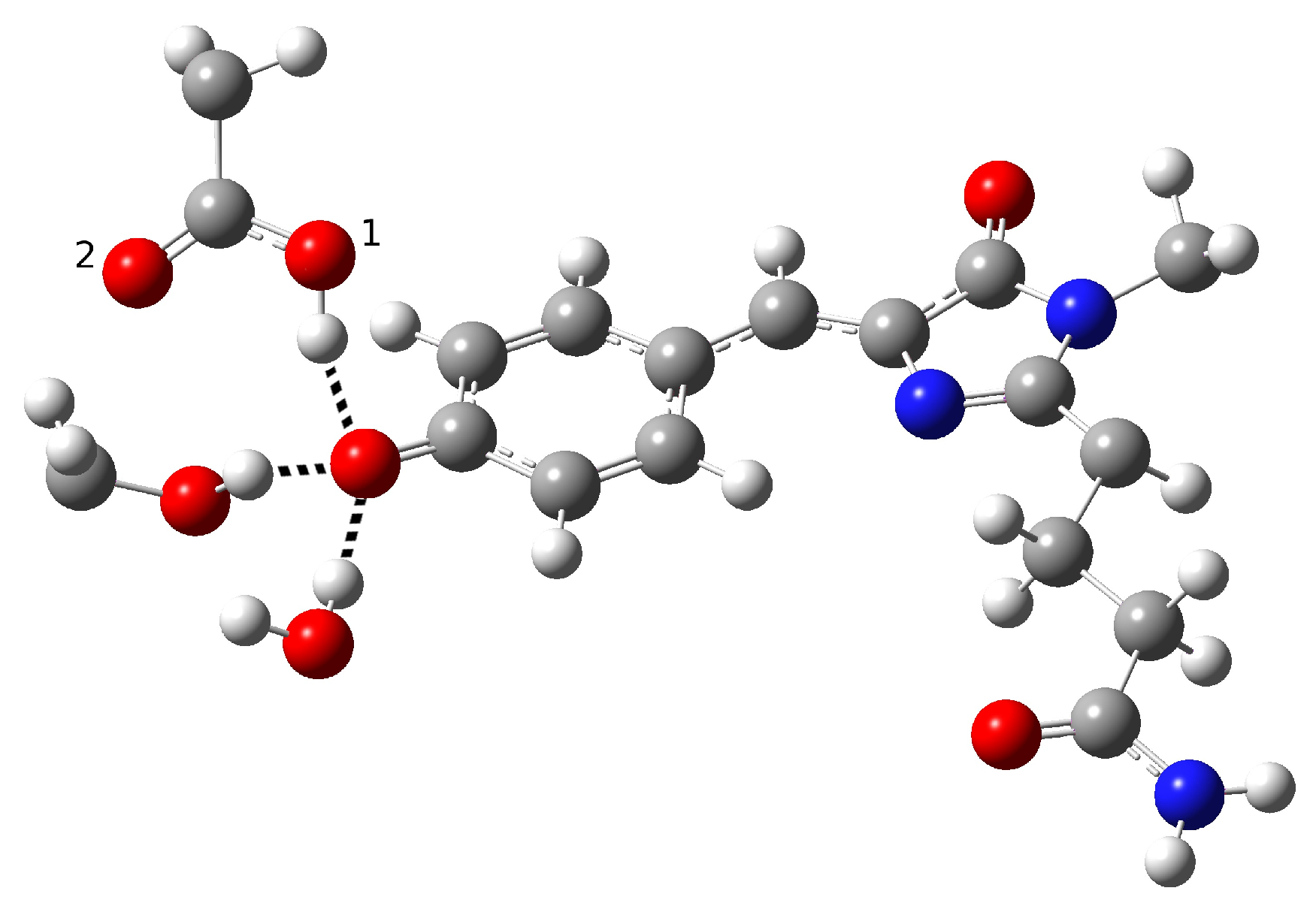
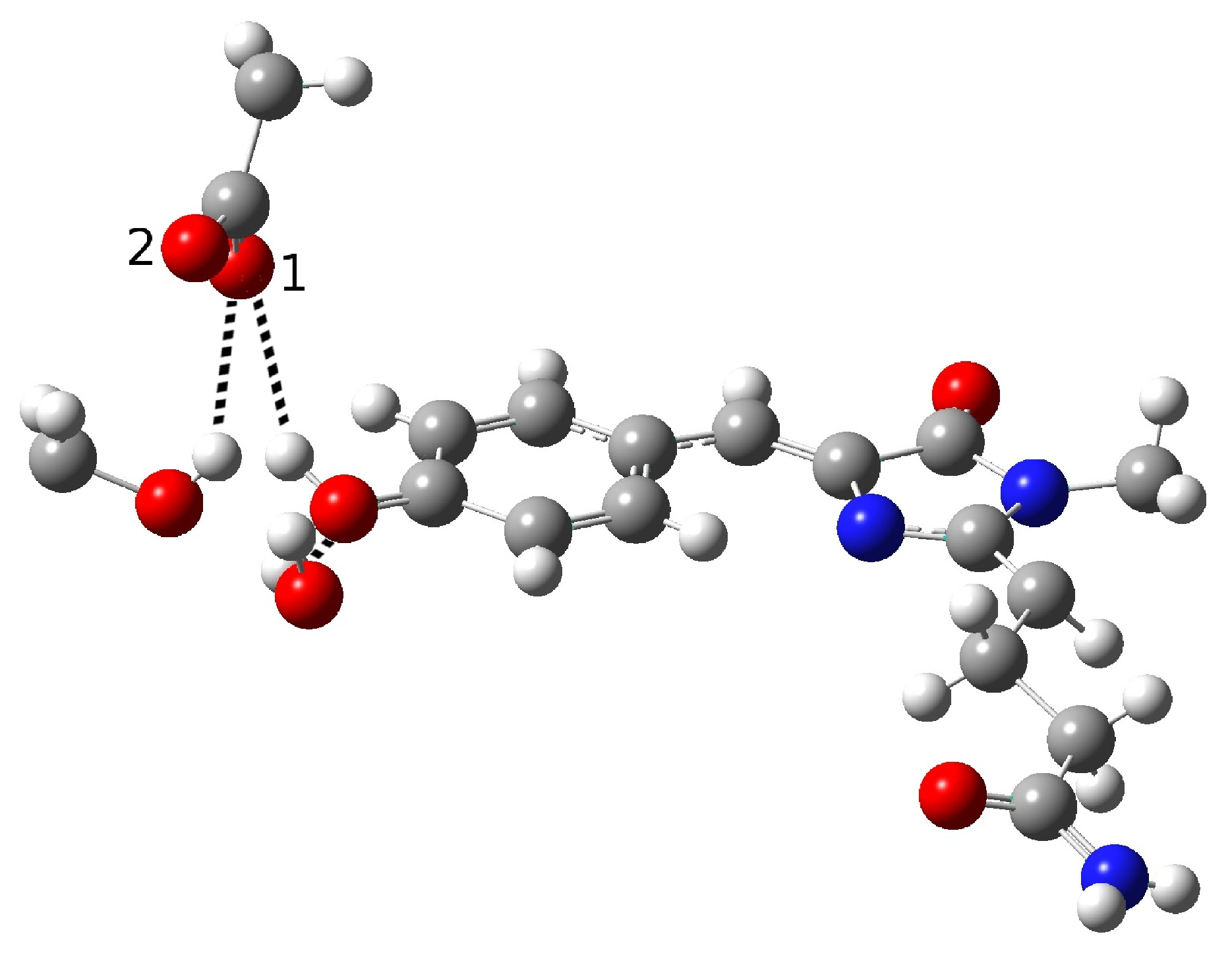
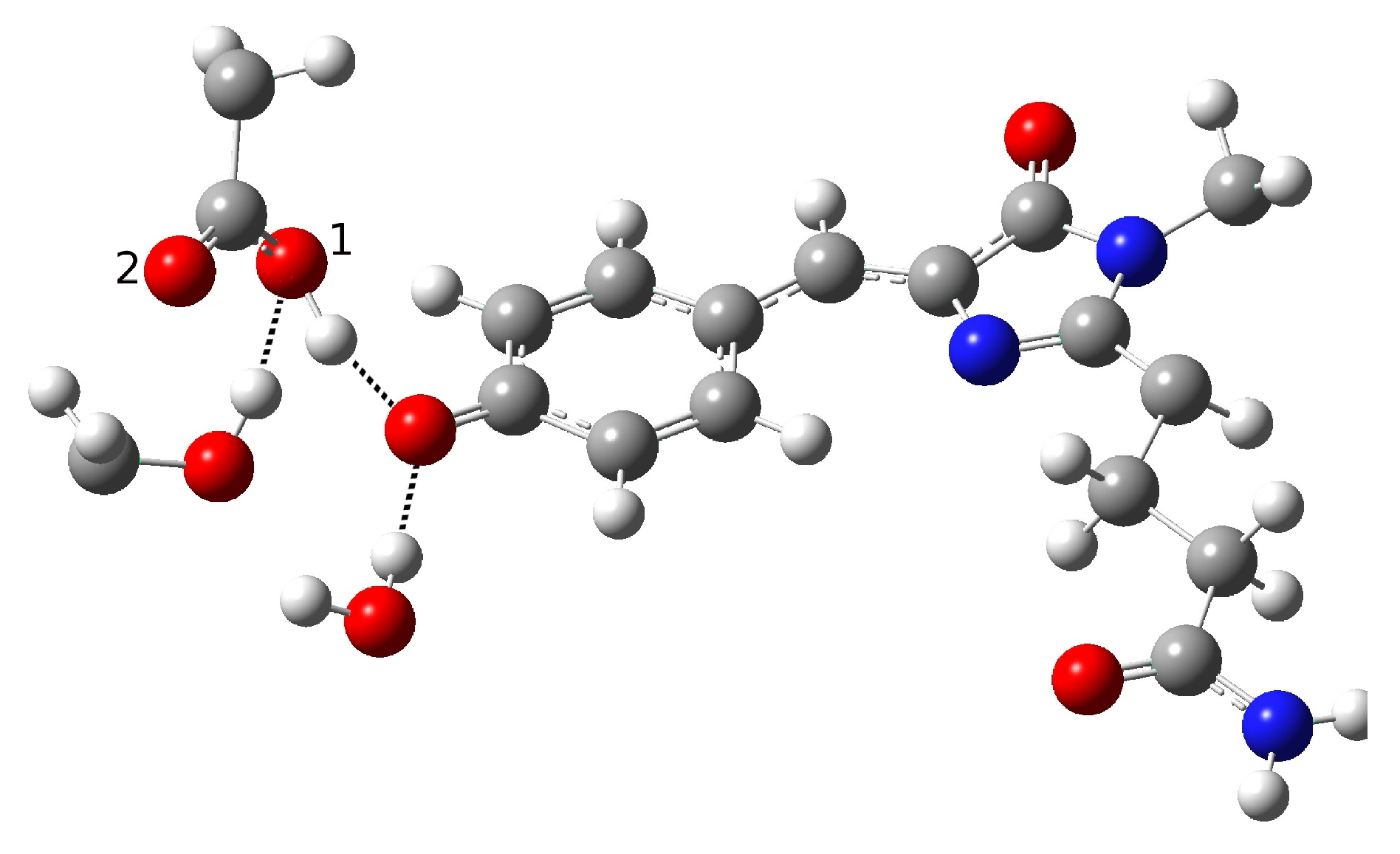
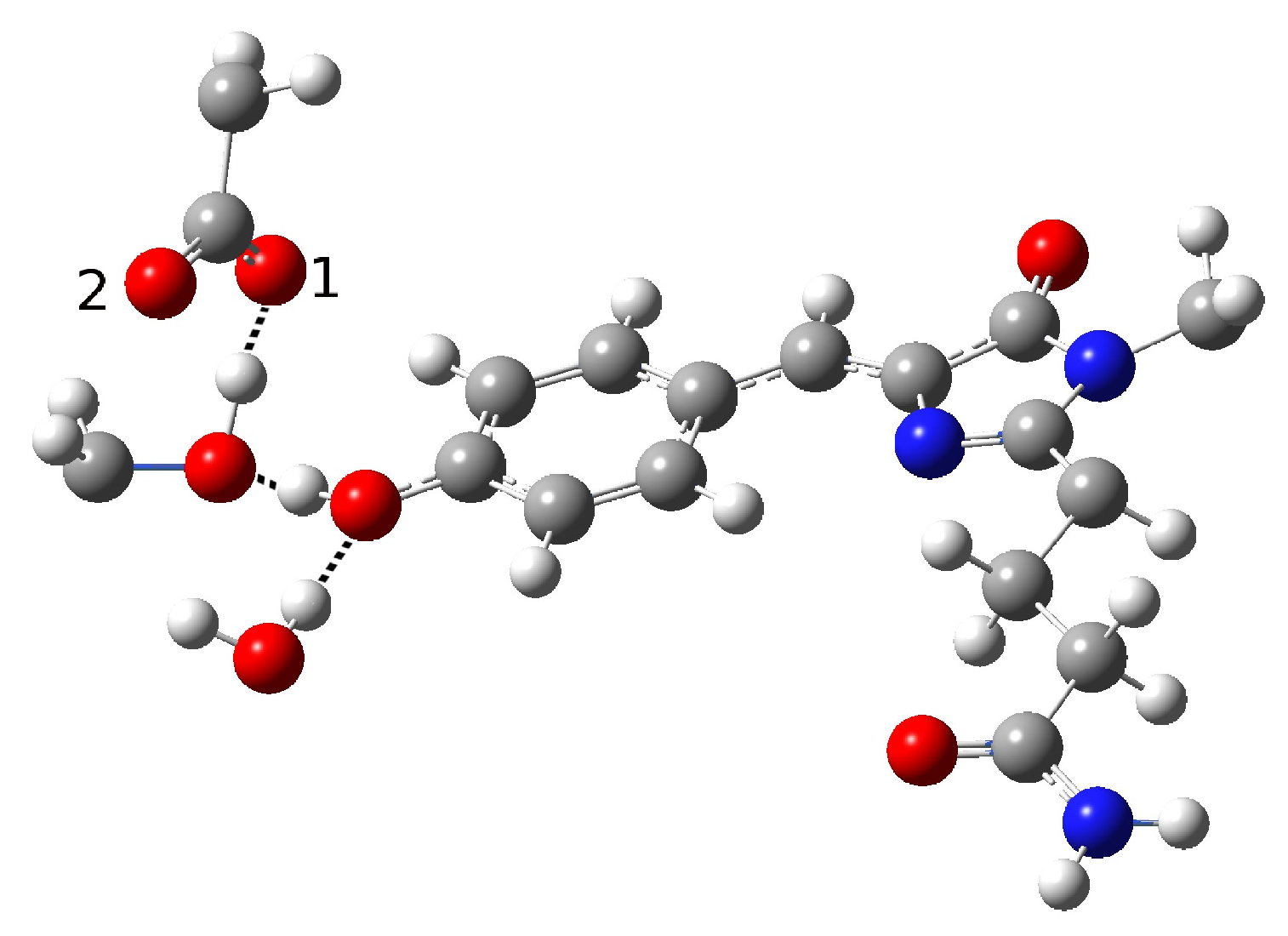
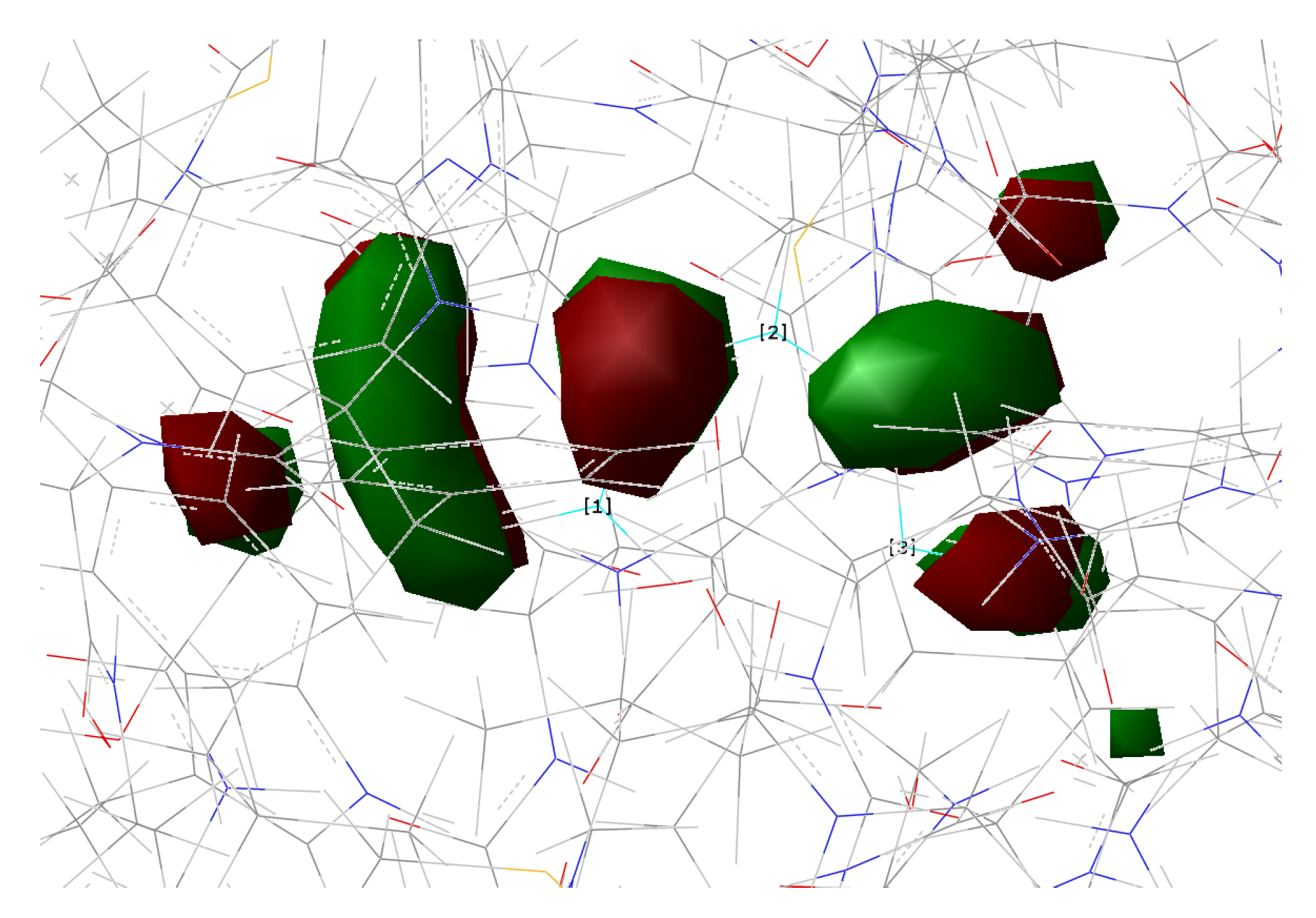
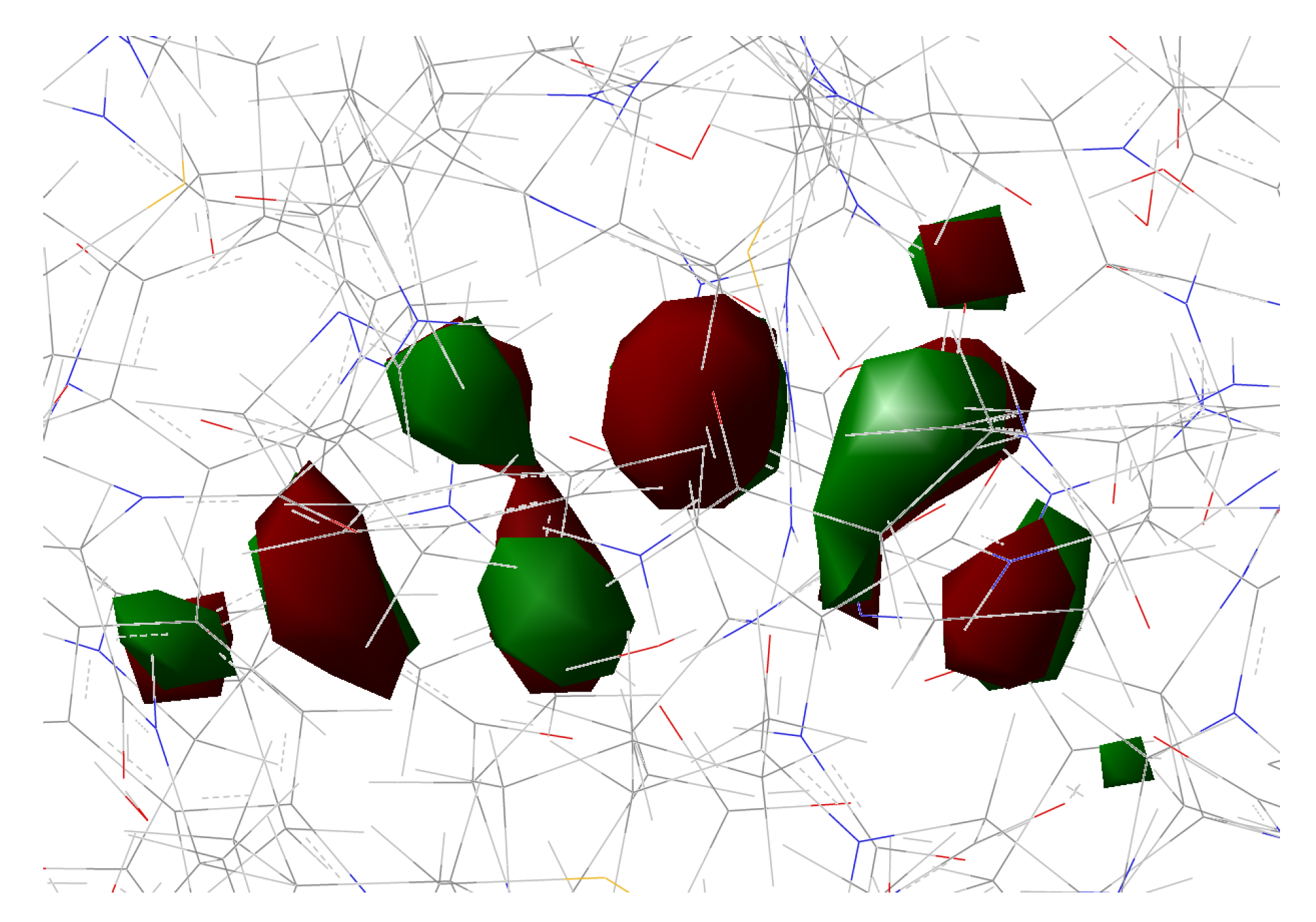
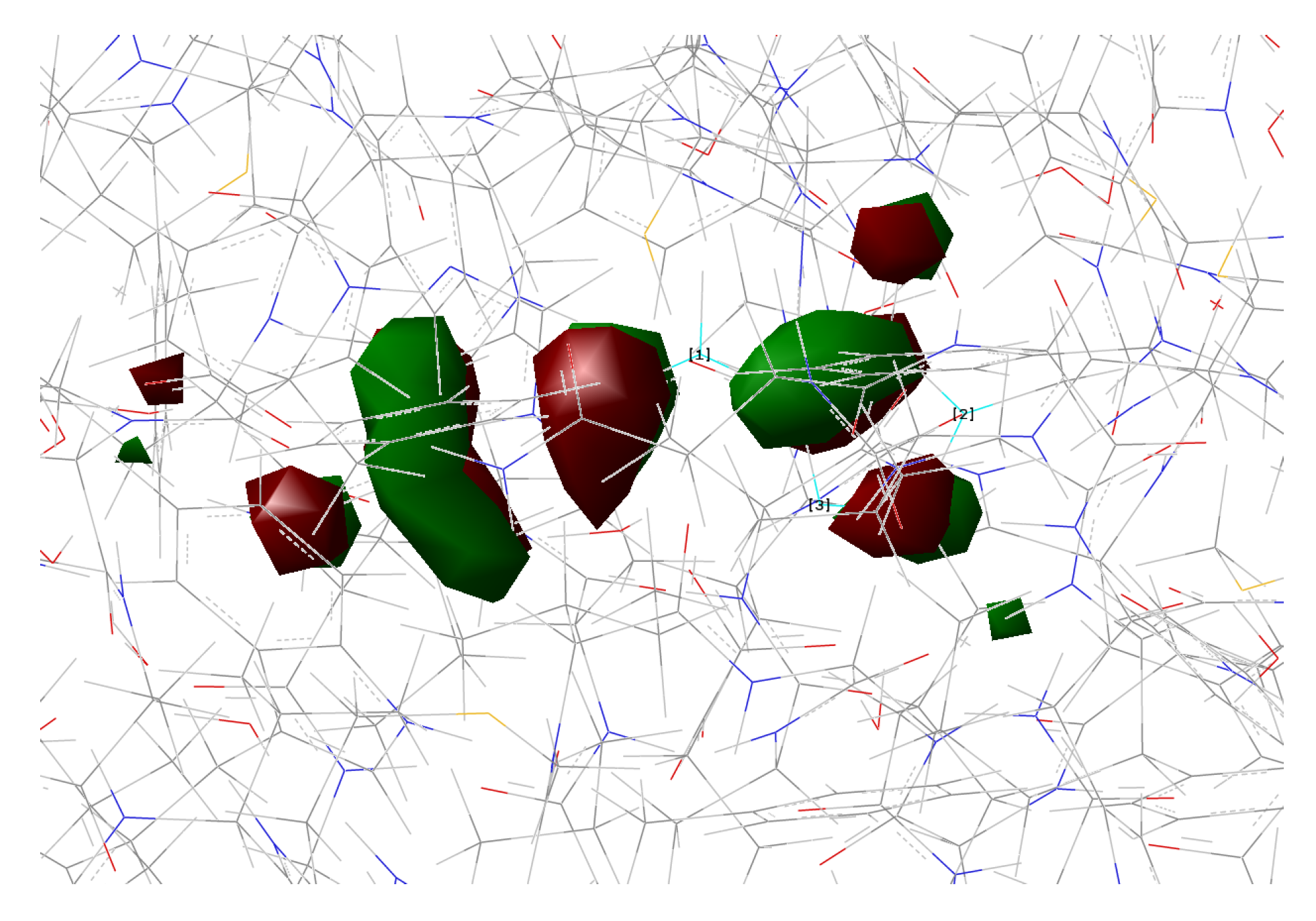
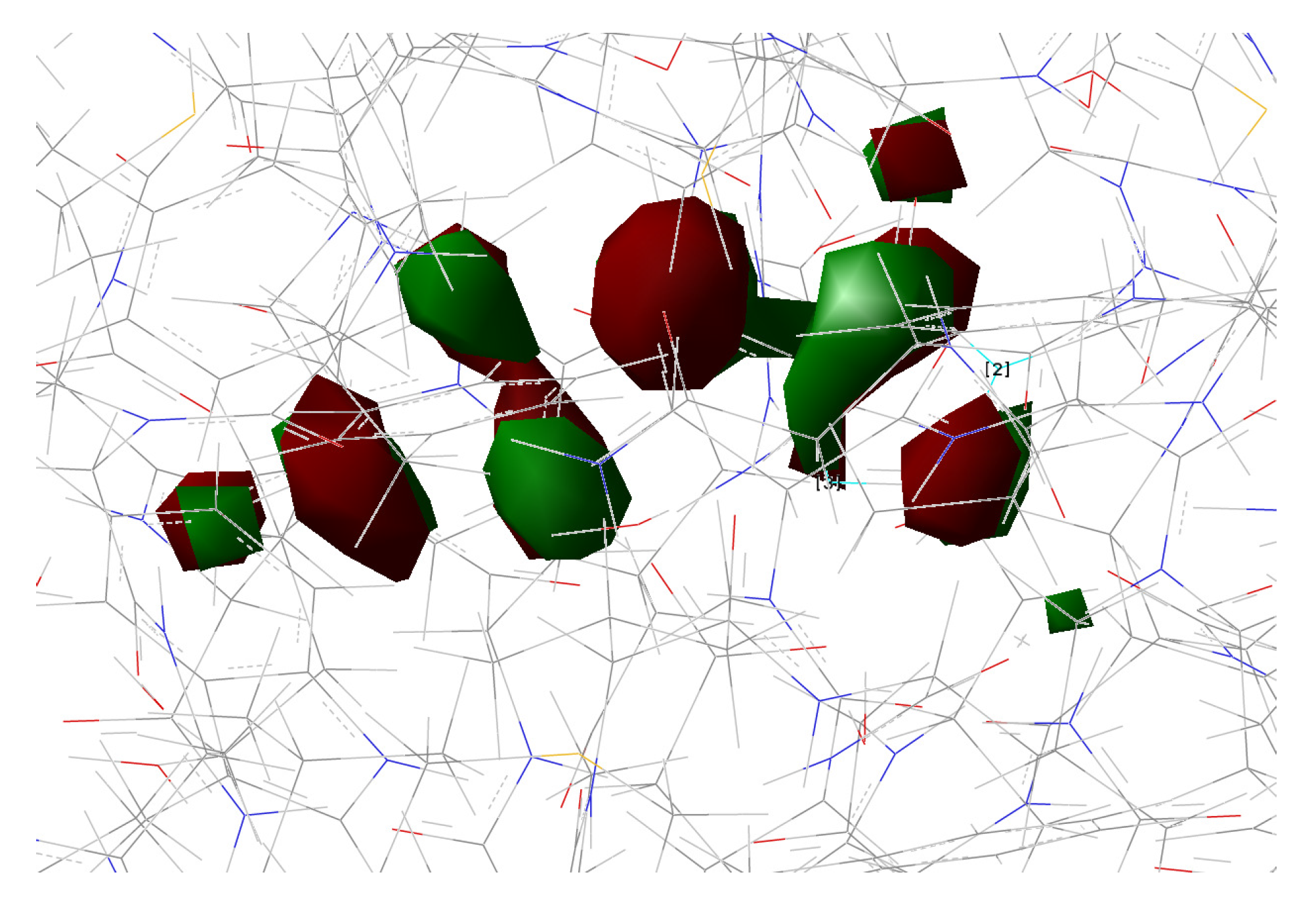
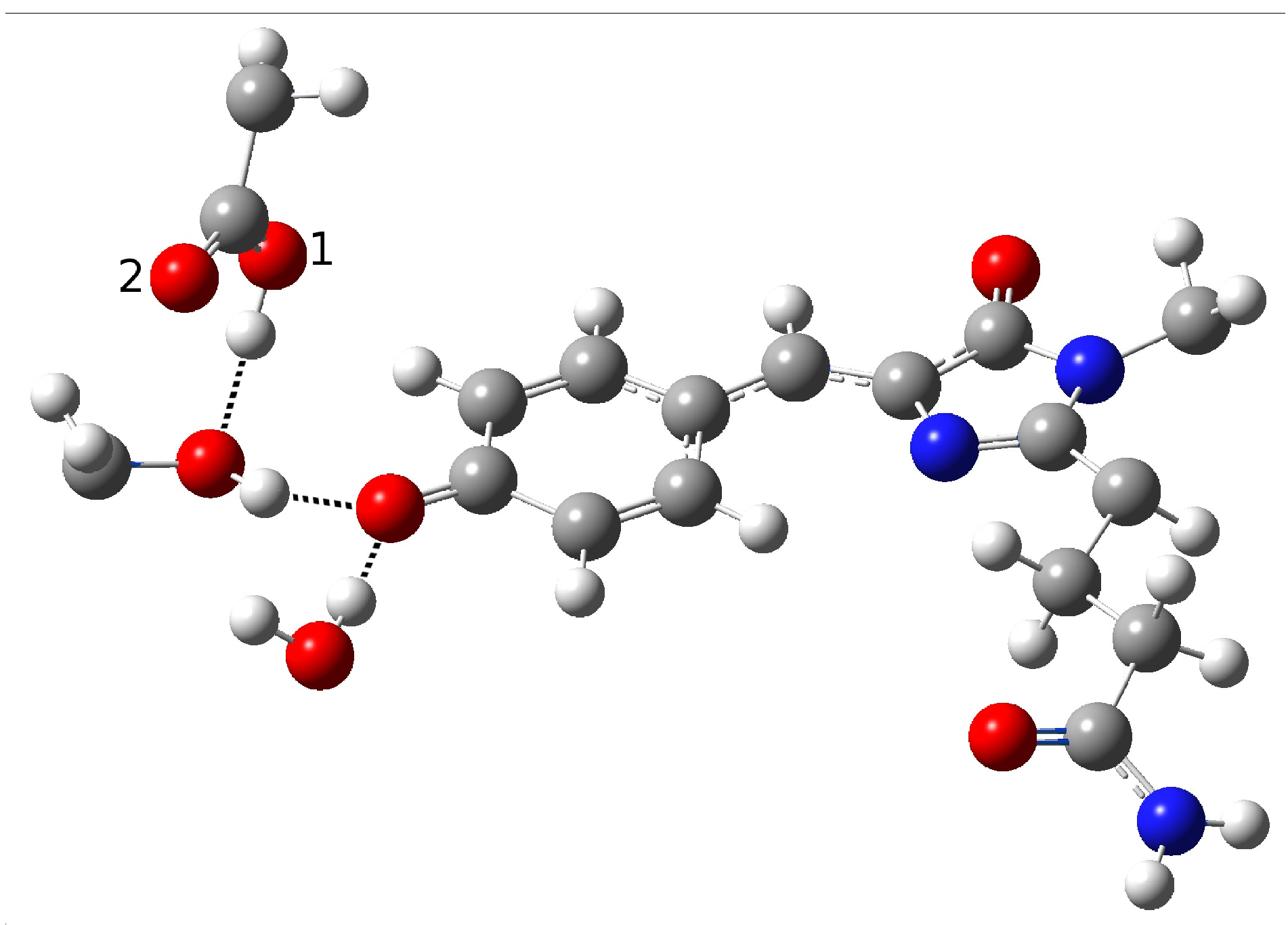
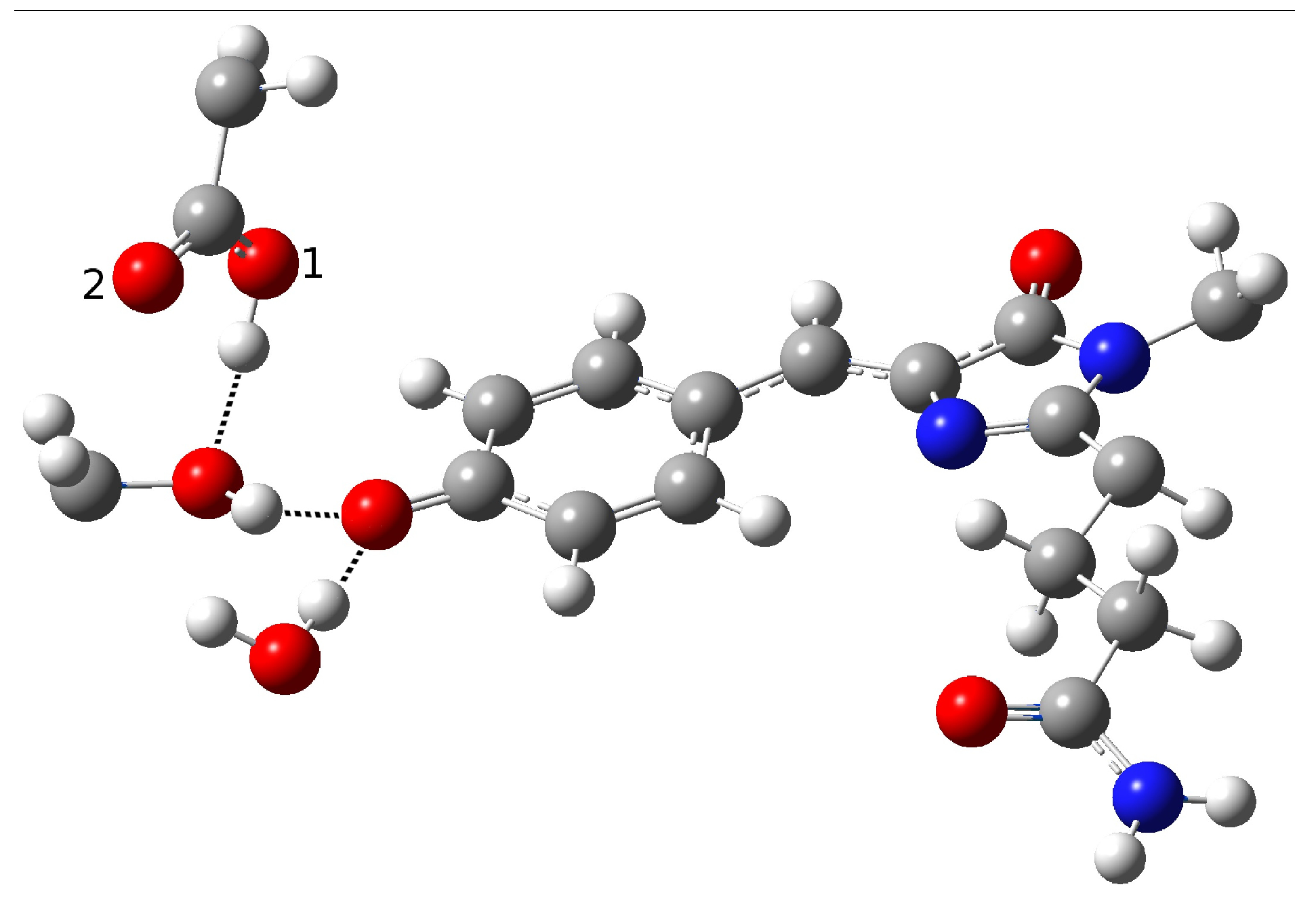
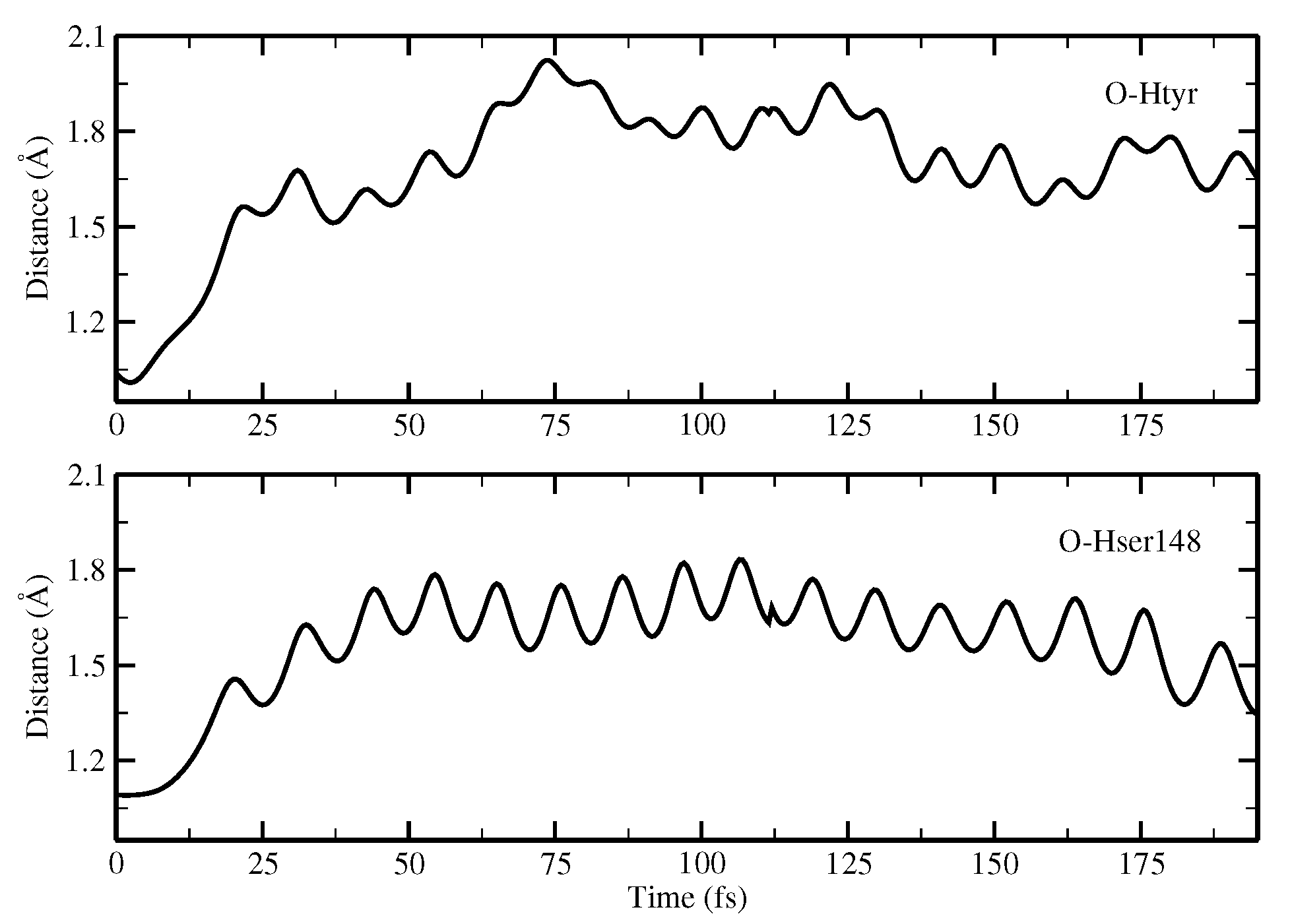
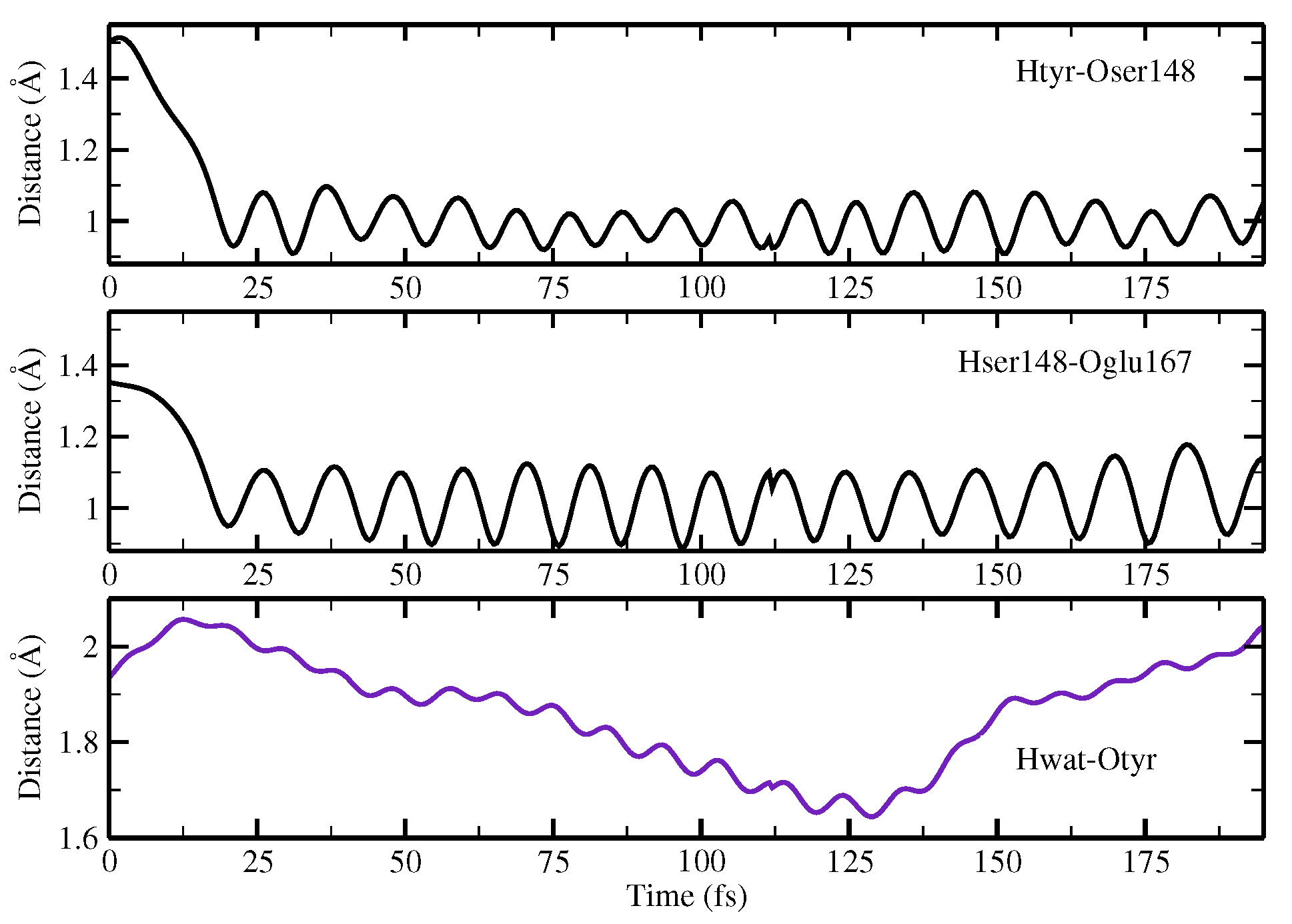
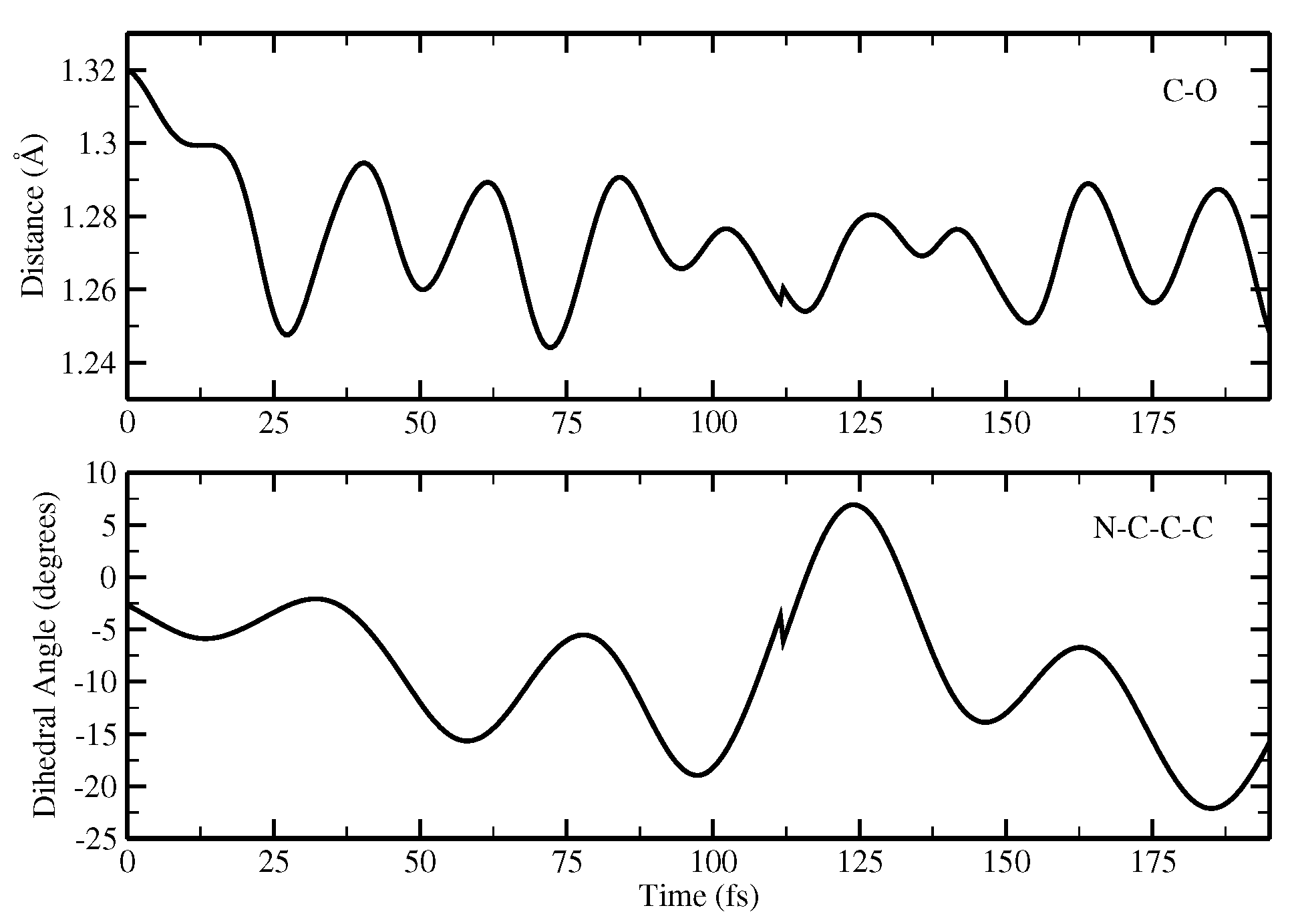
2.1.5. ESPT Reaction Mechanism Investigation
3. Materials and Methods
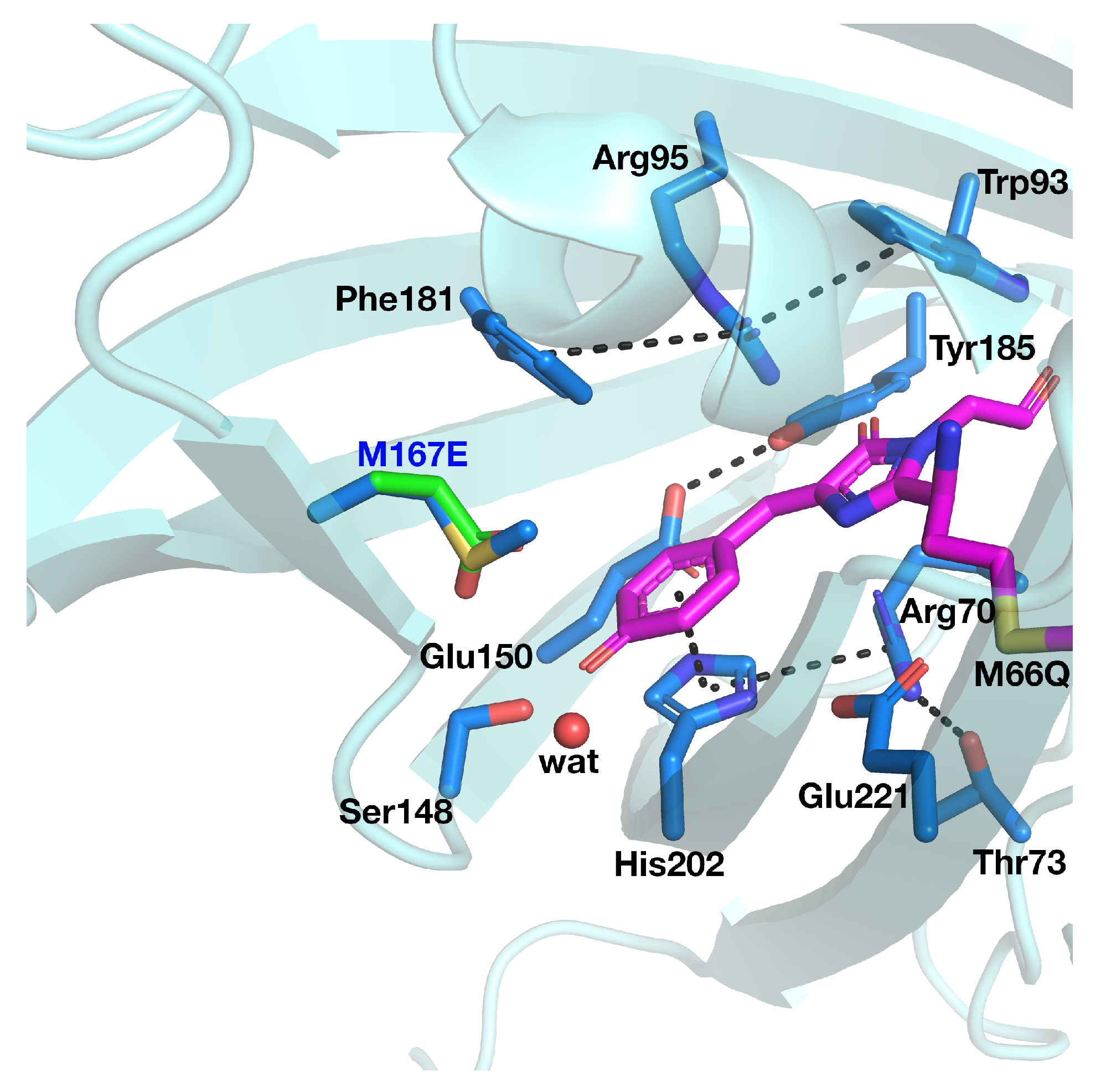
3.1. psammFP488 Model Preparation
3.2. ESPT Network Optimization and Calculation of Optical Properties
3.3. Ab Initio Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 68, 509. [Google Scholar] [CrossRef]
- Remington, S.J. Fluorescent proteins: Maturation, photochemistry and photophysics. Curr. Opin. Struc. Biol. 2006, 16, 714. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Patterson, G.H.; Davidson, M.W. Advances in fluorescent protein technology. J. Cell. Sci. 2007, 120, 4247. [Google Scholar] [CrossRef] [PubMed]
- Tonge, P.; Meech, S.R. Excited state dynamics in the green fluorescent protein. J. Photochem. PhotoBiol. A 2009, 205, 1. [Google Scholar] [CrossRef]
- Beddard, G. Molecular photophysics. Rep. Prog. Phys. 1993, 56, 63. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461. [Google Scholar] [CrossRef]
- Reid, G.D.; Wynne, K. Handbook of Laser Technology and Applications; Taylor and Francis: Milton Park, UK, 2003. [Google Scholar]
- Brejc, K.; Kitts, T.; Kain, S.R.; Tsien, R.Y.; Ormo, M.; Remington, S.J. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2306. [Google Scholar] [CrossRef]
- Chattoraj, M.; King, B.A.; Bublitz, G.U.; Boxer, S.G. Ultra-fast excited state dynamics in green fluorescent protein: Multiple states and proton transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 8362. [Google Scholar] [CrossRef]
- Donati, G.; Petrone, A.; Caruso, P.; Rega, N. The mechanism of a green fluorescent protein proton shuttle unveiled in the time-resolved frequency domain by excited state ab initio dynamics. Chem. Sci. 2018, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Petrone, A.; Caruso, P.; Tenuta, S.; Rega, N. On the optical absorption of the anionic GFP chromophore in vacuum, solution, and protein. Phys. Chem. Chem. Phys. 2013, 15, 20536. [Google Scholar] [CrossRef] [PubMed]
- Petrone, A.; Cimino, P.; Donati, G.; Hratchian, H.P.; Frisch, M.; Rega, N. On the driving force of the excited-state proton shuttle in the green fluorescent protein: A time-dependent density functional theory (TD-DFT) study of the intrinsic reaction path. J. Chem. Theory Comput. 2016, 12, 4925. [Google Scholar] [CrossRef] [PubMed]
- Coppola, F.; Perrella, F.; Donati, G.; Petrone, A.; Rega, N. A not obvious correlation between the structure of green fluorescent protein chromophore pocket and hydrogen bond dynamics: A choreography from ab initio molecular dynamics. Front. Mol. Biosci. 2020, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Alieva, N.O.; Konzen, K.A.; Field, S.F.; Meleshkevitch, E.A.; Hunt, M.E.; Beltran-Ramirez, V.; Miller, D.J.; Wiedenmann, J.; Salih, A.; Matz, V. Diversity and evolution of coral fluorescent proteins. PLoS ONE 2008, 3, e2680. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.N.; Remington, S.J. Crystal structures and mutational analysis of amFP486, a cyan fluorescent protein from Anemonia majano. Proc. Natl. Acad. Sci. USA 2005, 102, 12712. [Google Scholar] [CrossRef] [PubMed]
- Remington, S.J.; Wachter, R.M.; Yarbrough, D.K.; Branchaud, B.; Anderson, D.C.; Kallio, K.; Lukyanov, K.A. zFP538, a yellow-fluorescent protein from Zoanthus, contains a novel three-ring chromophore. Biochemistry 2005, 44, 202. [Google Scholar] [CrossRef]
- Gross, L.A.; Baird, G.S.; Hoffman, R.C.; Baldridge, K.K.; Tsien, R.Y. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 87, 11990. [Google Scholar] [CrossRef]
- Kennis, J.T.M.; van Stokkum, I.H.M.; Peterson, D.S.; Pandit, A.; Wachter, R.M. Ultrafast Proton Shuttling in Psammocora Cyan Fluorescent Protein. J. Phys. Chem. B 2013, 117, 11134. [Google Scholar] [CrossRef]
- Malo, G.D.; Wang, M.; Wu, D.; Stelling, A.L.; Tonge, P.J.; Wachter, R.M. Crystal structure and Raman studies of dsFP483, a cyan fluorescent protein from Discosoma striata. J. Mol. Biol. 2008, 378, 871. [Google Scholar] [CrossRef]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200–204. [Google Scholar] [CrossRef]
- Adams, D.P.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Dapprich, S.; Byun, K.S.; Komaromi, I.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. 1999, 462, 1. [Google Scholar] [CrossRef]
- Vreven, T.; Byun, K.; Komaromi, I.; Dapprich, S.; Morokuma, K.; Frisch, M.J. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput. 2006, 2, 815. [Google Scholar] [CrossRef] [PubMed]
- Vreven, T.; Morokuma, K. Chapter 3 Hybrid Methods: ONIOM(QM:MM) and QM/MM. Annu. Rep. Comput. Chem. 2006, 2, 35–51. [Google Scholar]
- Clemente, F.; Vreven, T.; Frisch, M.J. Quantum Biochemistry; Matta, C., Ed.; Wiley VCH: Weinheim, Germany, 2010; pp. 61–84. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179. [Google Scholar] [CrossRef]
- Reuter, N.; Lin, H.; Thiel, H.L. Green fluorescent proteins: Empirical force field for the neutral and deprotonated forms of the chromophore. Molecular dynamics simulations of the wild type and S65T mutant. J. Phys. Chem. B 2002, 106, 6310. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamarsky, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218. [Google Scholar] [CrossRef]
- Caillie, C.V.; Amos, R.D. Geometric derivatives of excitation energies using SCF and DFT. Chem. Phys. Lett. 1999, 308, 249. [Google Scholar] [CrossRef]
- Brancato, K.; Barone, V.; Rega, N. Theoretical modeling of spectroscopic properties of molecules in solution: Toward an effective dynamical discrete/continuum approach. Theor. Chem. Acc. 2007, 117, 1001. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance (Å) | |
| O(water)-O(tyr) | 2.894 |
| O(tyr)-O(ser) | 2.794 |
| O(water)-O(ser) | 4.390 |
| O(tyr)-O1(glu) | 2.661 |
| O(tyr)-O2(glu) | 3.500 |
| O(ser)-O1(glu) | 4.282 |
| O(ser)-O2(glu) | 3.509 |
| C−O(tyr) | 1.282 |
| O-H(glu) | 0.994 |
| H(water)-O(tyr) | 1.917 |
| H(ser)-O(tyr) | 1.842 |
| Angle (degrees) | |
| H-O(water)-O(tyr) | 2.73 |
| H-O(ser)-O(tyr) | 10.25 |
| H-O(glu)-O(tyr) | 13.16 |
| Distance (Å) | |
| O(water)-O(tyr) | 2.909 |
| O(tyr)-O(ser) | 3.096 |
| O(water)-O(ser) | 4.617 |
| O(tyr)-O1(glu) | 2.479 |
| O(tyr)-O2(glu) | 3.602 |
| O(ser)-O1(glu) | 2.944 |
| O(ser)-O2(glu) | 4.072 |
| C-O(tyr) | 1.277 |
| O-H(glu) | 1.036 |
| H(water)-O(tyr) | 1.933 |
| H(ser)-O1(glu) | 2.082 |
| Angle (degrees) | |
| H-O(water)-O(tyr) | 2.19 |
| H-O(ser)-O(glu) | 22.32 |
| H-O(glu)-O(tyr) | 10.69 |
| Distance (Å) | |
| O(water)-O(tyr) | 2.901 |
| O(tyr)-O(ser) | 2.538 |
| O(water)-O(ser) | 4.464 |
| O(tyr)-O1(glu) | 3.196 |
| O(tyr)-O2(glu) | 3.567 |
| O(ser)-O1(glu) | 2.442 |
| O(ser)-O2(glu) | 3.326 |
| C-O(tyr) | 1.320 |
| H(water)-O(tyr) | 1.934 |
| H(tyr)-O(ser) | 1.500 |
| H(ser)-O1(glu) | 1.352 |
| Angle (degrees) | |
| H-O(water)-O(tyr) | 4.81 |
| H-O(tyr)-O(ser) | 2.91 |
| H-O(ser)-O(glu) | 2.77 |
| Distance (Å) | |
| O(water)-O(tyr) | 2.810 |
| O(tyr)-O(ser) | 2.728 |
| O(water)-O(ser) | 4.452 |
| O(tyr)-O1(glu) | 2.926 |
| O(tyr)-O2(glu) | 3.173 |
| O(ser)-O1(glu) | 4.250 |
| O(ser)-O2(glu) | 2.765 |
| C-O(tyr) | 1.311 |
| H(tyr)-O(water) | 1.853 |
| H(ser)-O2(glu) | 1.866 |
| Angle (degrees) | |
| H-O(tyr)-O(water) | 10.89 |
| H-O(ser)-O(glu) | 19.91 |
| eV | nm | |
|---|---|---|
| Network1 | 3.13 | 396.59 |
| Network2 | 3.11 | 398.49 |
| Ground State | Excited State | |
|---|---|---|
| Distance (Å) | ||
| O(water)-O(tyr) | 2.778 | 2.774 |
| O(tyr)-O(ser) | 2.679 | 2.680 |
| O(water)-O(ser) | 4.352 | 4.347 |
| O(tyr)-O1(glu) | 3.511 | 3.486 |
| O(tyr)-O2(glu) | 3.834 | 3.835 |
| O(ser)-O1(glu) | 2.648 | 2.653 |
| O(ser)-O2(glu) | 3.301 | 3.321 |
| C-O(tyr) | 1.269 | 1.280 |
| H(wat)-O(tyr) | 1.806 | 1.800 |
| H(ser)-O(tyr) | 1.734 | 1.736 |
| H(glu)-O(ser) | 1.648 | 1.652 |
| Angle (degrees) | ||
| H-O(wat)-O(tyr) | 5.98 | 6.15 |
| H-O(ser)-O(tyr) | 13.87 | 14.17 |
| H-O(glu)-O(ser) | 4.20 | 1.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donati, G.; Rega, N. Direct or Indirect ESPT Mechanism in CFP psamFP488? A Theoretical-Computational Investigation. Int. J. Mol. Sci. 2022, 23, 15640. https://doi.org/10.3390/ijms232415640
Donati G, Rega N. Direct or Indirect ESPT Mechanism in CFP psamFP488? A Theoretical-Computational Investigation. International Journal of Molecular Sciences. 2022; 23(24):15640. https://doi.org/10.3390/ijms232415640
Chicago/Turabian StyleDonati, Greta, and Nadia Rega. 2022. "Direct or Indirect ESPT Mechanism in CFP psamFP488? A Theoretical-Computational Investigation" International Journal of Molecular Sciences 23, no. 24: 15640. https://doi.org/10.3390/ijms232415640
APA StyleDonati, G., & Rega, N. (2022). Direct or Indirect ESPT Mechanism in CFP psamFP488? A Theoretical-Computational Investigation. International Journal of Molecular Sciences, 23(24), 15640. https://doi.org/10.3390/ijms232415640