
Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences

 and
and
Abstract
1. Introduction
2. Results
2.1. Quality and Lineages of the SARS-CoV-2 Genomes Analyzed
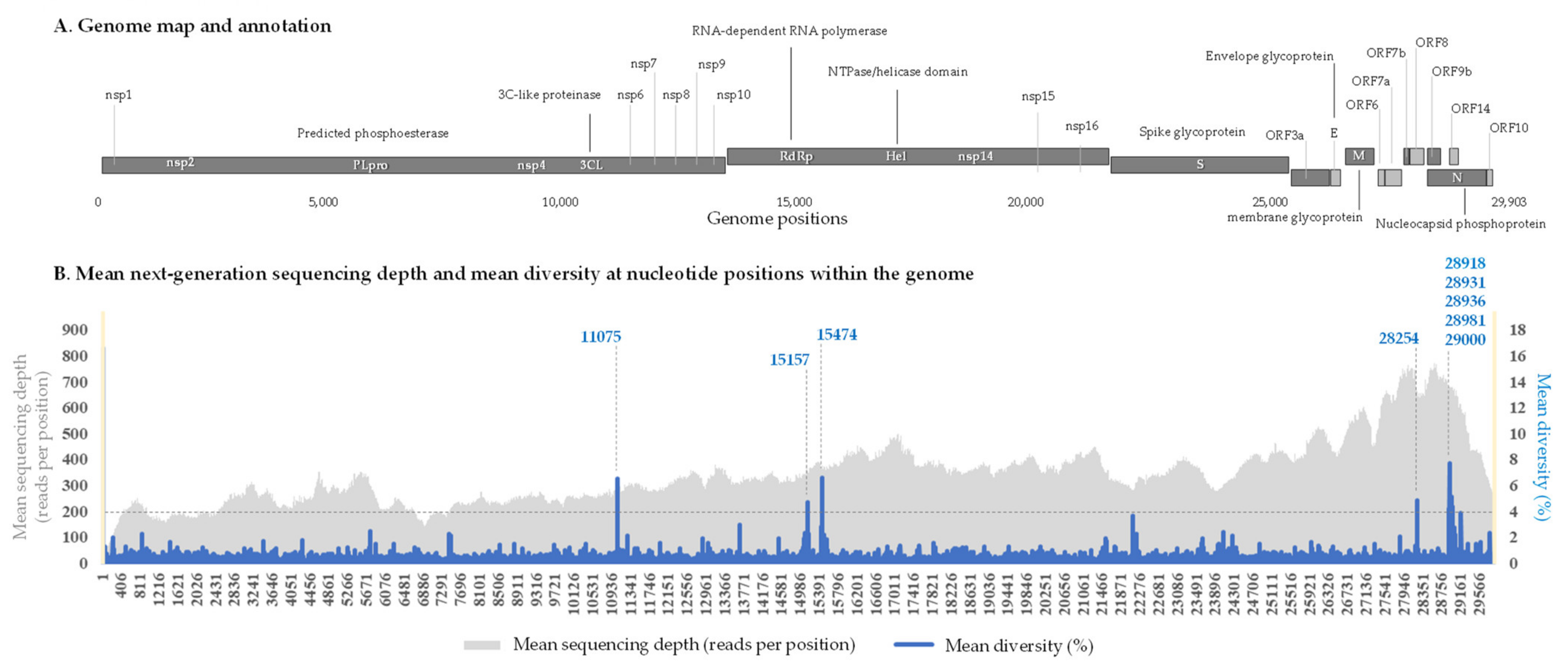
2.2. Nucleotide Diversity in the SARS-CoV-2 Genomes and Genes
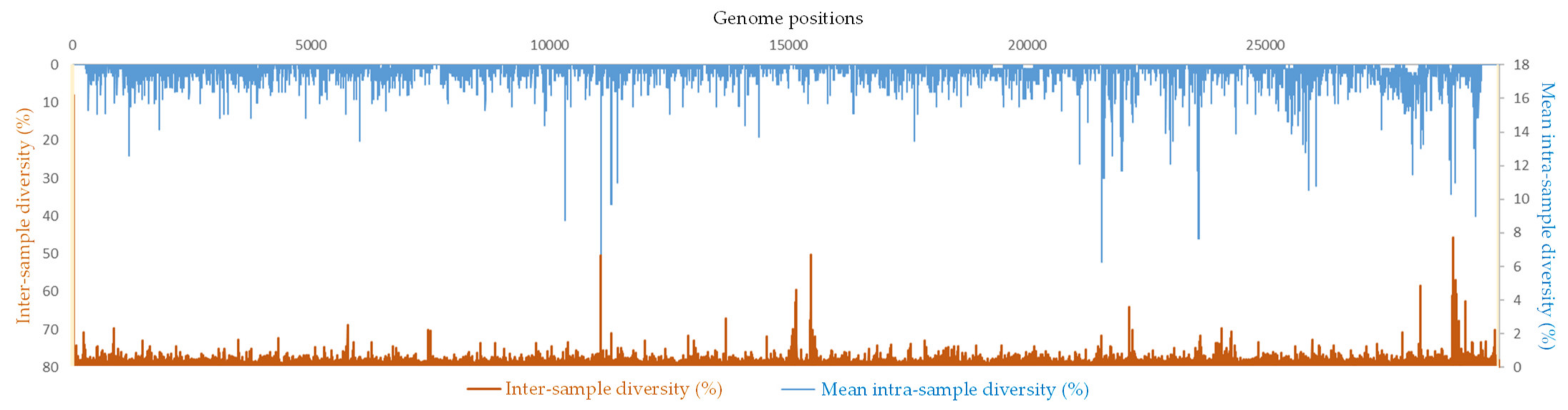
2.3. Hot Spots of Intra-Sample Genetic Diversity
2.4. Correlation between Intra-Sample and Inter-Sample Genetic Diversity in SARS-CoV-2 Genomes
2.5. Presence in Viral Quasispecies of Variant Hallmark Mutations of the Spike Gene
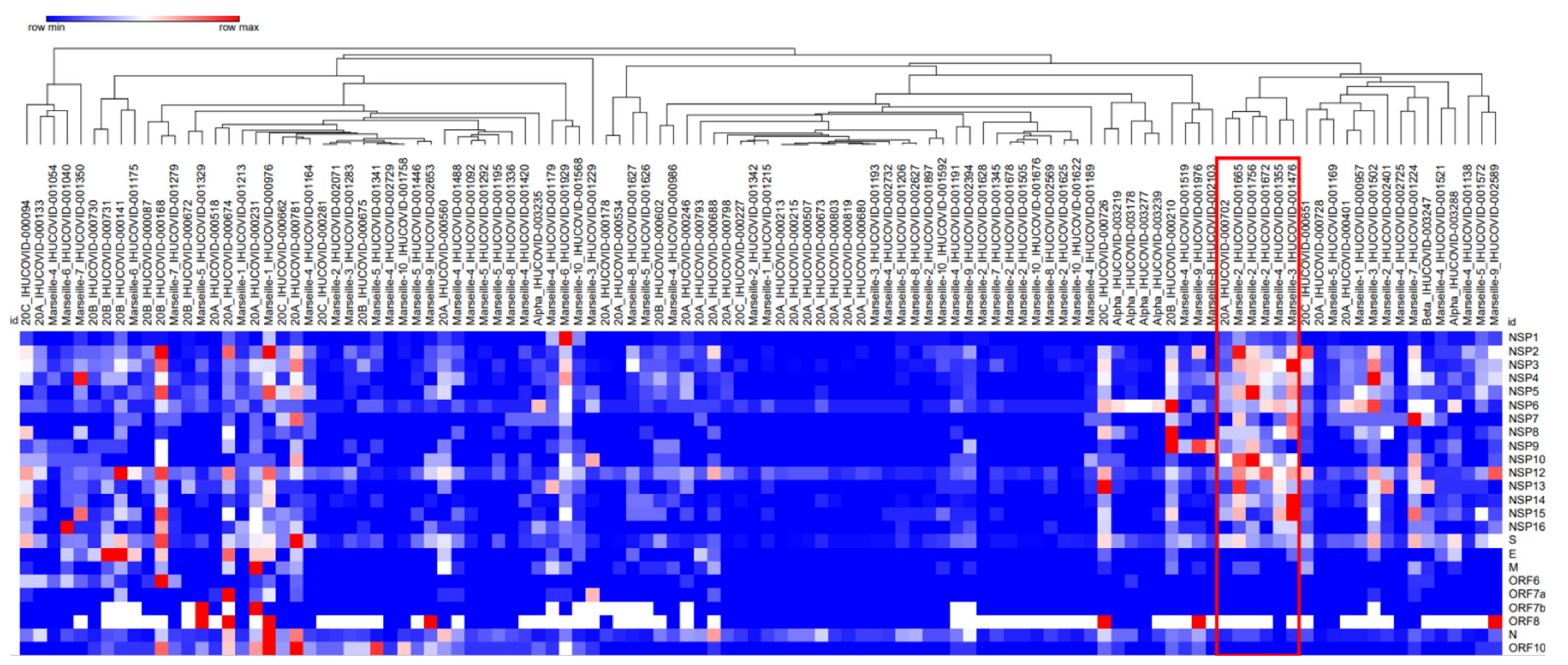
2.6. Intra-Sample Genetic Diversity for the Different SARS-CoV-2 Lineages and Variants
3. Discussion
4. Materials and Methods
4.1. Next-Generation SARS-CoV-2 Genome Sequencing Methods and Data
4.2. Detection and Characterization of Genetic Quasispecies
4.3. Analysis of SARS-CoV-2 Intra-Sample Genetic Diversity at the Genome and Gene Scales
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Prates, E.T.; Garvin, M.R.; Pavicic, M.; Jones, P.; Shah, M.; Demerdash, O.; Amos, B.K.; Geiger, A.; Jacobson, D. Potential Pathogenicity Determinants Identified from Structural Proteomics of SARS-CoV and SARS-CoV-2. Mol. Biol. Evol. 2021, 38, 702–715. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M. Newly Emerged Antiviral Strategies for SARS-CoV-2: From Deciphering Viral Protein Structural Function to the Development of Vaccines, Antibodies and Small Molecules. Int. J. Mol. Sci. 2022, 23, 6083. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, N.; Qi, W.; Li, T.; Zhang, Y.; Zhang, H.; Wu, J.; Zhu, Z.; Ai, J.; Qiu, C.; et al. Intra-host SARS-CoV-2 single-nucleotide variants emerged during the early stage of COVID-19 pandemic forecast population fixing mutations. J. Infect. 2022, 84, 722–746. [Google Scholar] [CrossRef]
- Domingo, E.; Escarmís, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Rambaut, A. Viral evolution and the emergence of SARS coronavirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T. Insertion-and-Deletion Mutations between the Genomes of SARS-CoV, SARS-CoV-2 and Bat Coronavirus RaTG13. Microbiol. Spectr. 2022, 10, e0071622. [Google Scholar] [CrossRef] [PubMed]
- Ignatieva, A.; Hein, J.; Jenkins, P.A. Ongoing Recombination in SARS-CoV-2 Revealed through Genealogical Reconstruction. Mol. Biol. Evol. 2022, 39, msac028. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e8. [Google Scholar] [CrossRef]
- Colson, P.; Fournier, P.E.; Delerce, J.; Million, M.; Bedotto, M.; Houhamdi, L.; Yahi, N.; Bayette, J.; Levasseur, A.; Fantini, J.; et al. Culture and identification of a “Deltamicron” SARS-CoV-2 in a three cases cluster in southern France. J. Med. Virol. 2022, 94, 3739–3749. [Google Scholar] [CrossRef]
- Andrés, C.; Garcia-Cehic, D.; Gregori, J.; Piñana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally occurring SARS-CoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasispecies of COVID19 patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13, e0297921. [Google Scholar] [CrossRef]
- Colson, P.; Fournier, P.E.; Chaudet, H.; Delerce, J.; Giraud-Gatineau, A.; Houhamdi, L.; Andrieu, C.; Brechard, L.; Bedotto, M.; Prudent, E.; et al. Analysis of SARS-CoV-2 Variants from 24,181 Patients Exemplifies the Role of Globalization and Zoonosis in Pandemics. Front. Microbiol. 2022, 12, 786233. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Molecular self-organization and the early stages of evolution. Q. Rev. Biophys. 1971, 4, 149–212. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. On the nature of virus quasispecies. Trends Microbiol. 1996, 4, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Flavell, R.A.; Weissmann, C. In vitro site-directed mutagenesis: Generation and properties of an infectious extracistronic mutant of bacteriophage Qβ. Gene 1976, 1, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Tamalet, C.; Yahi, N.; Tourrès, C.; Colson, P.; Quinson, A.M.; Poizot-Martin, I.; Dhiver, C.; Fantini, J. Multidrug resistance genotypes (insertions in the beta3-beta4 finger subdomain and MDR mutations) of HIV-1 reverse transcriptase from extensively treated patients: Incidence and association with other resistance mutations. Virology 2000, 270, 310–316. [Google Scholar] [CrossRef][Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Kuipers, J.; Batavia, A.A.; Jablonski, K.P.; Bayer, F.; Borgsmüller, N.; Dondi, A.; Drăgan, M.-A.; Ferreira, P.; Jahn, K.; Lamberti, L.; et al. Within-patient genetic diversity of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of Globally Shared SARS-CoV-2 Data to Identify and Characterize New Variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Nour, D.; Rafei, R.; Lamarca, A.P.; de Almeida, L.G.P.; Osman, M.; Ismail, M.B.; Mallat, H.; Berry, A.; Burfin, G.; Semanas, Q.; et al. The Role of Lebanon in the COVID-19 Butterfly Effect: The B.1.398 Example. Viruses 2022, 14, 1640. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L. Lost in deletion: The enigmatic ORF8 protein of SARS-CoV-2. Biochem. Biophys. Res. Commun. 2021, 538, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, S.; Pandey, S.; Puvar, A.; Shah, T.; Joshi, M.; Joshi, C.; Kumar, S. Identification of unique mutations in SARS-CoV-2 strains isolated from India suggests its attenuated pathotype. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dudas, G.; Hong, S.L.; Potter, B.I.; Calvignac-Spencer, S.; Niatou-Singa, F.S.; Tombolomako, T.B.; Fuh-Neba, T.; Vickos, U.; Ulrich, M.; Leendertz, F.H.; et al. Emergence and spread of SARS-CoV-2 lineage B.1.620 with variant of concern-like mutations and deletions. Nat. Commun. 2021, 12, 5769. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, D.; Zhang, L.; Sun, W.; Zhang, Z.; Chen, W.; Zhu, A.; Huang, Y.; Xiao, F.; Yao, J.; et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 2021, 13, 30. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Colson, P.; Levasseur, A.; Gautret, P.; Fenollar, F.; Thuan Hoang, V.; Delerce, J.; Bitam, I.; Saile, R.; Maaloum, M.; Padane, A.; et al. Introduction into the Marseille geographical area of a mild SARS-CoV-2 variant originating from sub-Saharan Africa: An investigational study. Travel. Med. Infect. Dis. 2021, 40, 101980. [Google Scholar] [CrossRef]
- Hodcroft, E. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. 2021. Available online: https://covariants.org/ (accessed on 30 September 2022).
- Fournier, P.E.; Colson, P.; Levasseur, A.; Devaux, C.A.; Gautret, P.; Bedotto, M.; Delerce, J.; Brechard, L.; Pinault, L.; Lagier, J.C.; et al. Emergence and outcomes of the SARS-CoV-2 ‘Marseille-4’ variant. Int. J. Infect. Dis. 2021, 106, 228–236. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Long, S.W.; Christensen, P.A.; Olsen, R.J.; Olson, R.; Shukla, M.; Subedi, S.; Stevens, R.; Musser, J.M. Analysis of the ARTIC Version 3 and Version 4 SARS-CoV-2 Primers and Their Impact on the Detection of the G142D Amino Acid Substitution in the Spike Protein. Microbiol. Spectr. 2021, 9, e0180321. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, M.; Mloka, D.; Tovanabutra, S.; Sanders-Buell, E.; Hoffmann, O.; Maboko, L.; Mmbando, D.; Birx, D.L.; McCutchan, F.E.; Hoelscher, M. In-depth, longitudinal analysis of viral quasispecies from an individual triply infected with late-stage human immunodeficiency virus type 1, using a multiple PCR primer approach. J. Virol. 2005, 79, 8249–8261. [Google Scholar] [CrossRef] [PubMed]
- Bracho, M.A.; García-Robles, I.; Jiménez, N.; Torres-Puente, M.; Moya, A.; González-Candelas, F. Effect of oligonucleotide primers in determining viral variability within hosts. Virol. J. 2004, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Tanaka, R.; Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE 2020, 15, e0239403. [Google Scholar] [CrossRef]
- Gao, R.; Zu, W.; Liu, Y.; Li, J.; Li, Z.; Wen, Y.; Wang, H.; Yuan, J.; Cheng, L.; Zhang, S.; et al. Quasispecies of SARS-CoV-2 revealed by single nucleotide polymorphisms (SNPs) analysis. Virulence 2021, 12, 1209–1226. [Google Scholar] [CrossRef]
- Han, L.; Zheng, Y.; Deng, J.; Nan, M.L.; Xiao, Y.; Zhuang, M.W.; Zhang, J.; Wang, W.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J. Med. Virol. 2022, 94, 5174–5188. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.C. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef]
- Quaranta, E.G.; Fusaro, A.; Giussani, E.; D’Amico, V.; Varotto, M.; Pagliari, M.; Giordani, M.T.; Zoppelletto, M.; Merola, F.; Antico, A.; et al. SARS-CoV-2 intra-host evolution during prolonged infection in an immunocompromised patient. Int. J. Infect. Dis. 2022, 122, 444–448. [Google Scholar] [CrossRef]
- Chaguza, C.; Hahn, A.M.; Petrone, M.E.; Zhou, S.; Ferguson, D.; Breban, M.I.; Pham, K.; Peña-Hernández, M.A.; Castaldi, C.; Hill, V.; et al. Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection. medRxiv 2022. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
- Vellas, C.; Del Bello, A.; Debard, A.; Steinmeyer, Z.; Tribaudeau, L.; Ranger, N.; Jeanne, N.; Martin-Blondel, G.; Delobel, P.; Kamar, N.; et al. Influence of treatment with neutralizing monoclonal antibodies on the SARS-CoV-2 nasopharyngeal load and quasispecies. Clin. Microbiol. Infect. 2022, 28, 139.e5–139.e8. [Google Scholar] [CrossRef] [PubMed]
- Pondé, R.A.A. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience 2022, 25, 103589. [Google Scholar] [CrossRef]
- Van Cleemput, J.; Van Snippenberg, W.; Lambrechts, L.; Dendooven, A.; D’Onofrio, V.; Couck, L.; Trypsteen, W.; Vanrusselt, J.; Theuns, S.; Vereecke, N.; et al. Organ-specific genome diversity of replication-competent SARS-CoV-2. Nat. Commun. 2021, 12, 6612. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Weissgerber, T.L.; Winham, S.J.; Heinzen, E.P.; Milin-Lazovic, J.S.; Garcia-Valencia, O.; Bukumiric, Z.; Savic, M.D.; Garovic, V.D.; Milic, N.M. Reveal, Don’t Conceal: Transforming Data Visualization to Improve Transparency. Circulation 2019, 140, 1506–1518. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Coordinates on the Genome GenBank Accession no. NC_045512.2 | Mean Diversity (%) | Number of Gene Positions Exhibiting a Significant (>4%) Diversity in Any of the 110 Samples | Number of Positions per 100 Nucleotides |
|---|---|---|---|---|
| ORF1ab | 266..21555 | 0.2 | 3123 | 0.15 |
| S | 21563..25384 | 0.22 | 1281 | 0.34 |
| ORF3a | 25393..26220 | 0.2 | 105 | 0.13 |
| E | 26245..26472 | 0.2 | 31 | 0.14 |
| M | 26523..27191 | 0.21 | 68 | 0.10 |
| ORF6 | 27202..27387 | 0.17 | 26 | 0.14 |
| ORF7a | 27394..27759 | 0.2 | 28 | 0.08 |
| ORF7b | 27756..27887 | 0.19 | 4 | 0.03 |
| ORF8 | 27894..28259 | 0.21 | 14 | 0.04 |
| N | 28274..29533 | 0.34 | 301 | 0.24 |
| ORF10 | 29558..29674 | 0.2 | 65 | 0.56 |
| Coordinates on the Genome GenBank Accession no. NC_045512.2 | Gene_Codon | Inter-Patient Diversity (%) | Mean Intra-Sample Diversity (%) | Nucleotide Position in Codon |
|---|---|---|---|---|
| 516 | ORF1a_84 | 11 | 1.21 | 1 |
| 517 | 11 | 1.06 | 2 | |
| 518 | ORF1a_85 | 13 | 1.25 | 1 |
| 519 | 13 | 1.11 | 2 | |
| 520 | 13 | 1.15 | 3 | |
| 521 | ORF1a_86 | 6 | 1.03 | 1 |
| 522 | 6 | 1.27 | 2 | |
| 867 | ORF1a_201 | 1 | 1.01 | 2 |
| 868 | 1 | 2.33 | 3 | |
| 873 | ORF1a_203 | 4 | 1.41 | 2 |
| 963 | ORF1a_233 | 1 | 1.12 | 2 |
| 1465 | ORF1a_400 | 3 | 1.61 | 3 |
| 1600 | ORF1a_445 | 7 | 1.05 | 3 |
| 3067 | ORF1a_934 | 1 | 1.12 | 3 |
| 3468 | ORF1a_1068 | 2 | 1.65 | 2 |
| 4318 | ORF1a_1351 | 1 | 1.76 | 3 |
| 5434 | ORF1a_1723 | 1 | 1.03 | 3 |
| 5743 | ORF1a_1826 | 1 | 1.24 | 3 |
| 5886 | ORF1a_1874 | 1 | 1.50 | 2 |
| 6268 | ORF1a_2001 | 2 | 1.51 | 3 |
| 6713 | ORF1a_2149 | 2 | 1.24 | 3 |
| 9044 | ORF1a_2927 | 1 | 1.10 | 1 |
| 9714 | ORF1a_3150 | 1 | 1.47 | 2 |
| 10037 | ORF1a_3258 | 3 | 1.23 | 1 |
| 11075 | ORF1a_3604 | 1 | 6.64 | 1 |
| 11117 | ORF1a_3618 | 5 | 1.02 | 1 |
| 11289 | ORF1a_3675 | 32 | 1.63 | 2 |
| 11290 | 32 | 1.57 | 3 | |
| 11291 | ORF1a_3676 | 37 | 1.57 | 1 |
| 11292 | 37 | 1.68 | 2 | |
| 11293 | 37 | 2.03 | 3 | |
| 11294 | ORF1a_3677 | 37 | 1.73 | 1 |
| 11295 | 37 | 1.67 | 2 | |
| 11296 | 37 | 1.72 | 3 | |
| 11997 | ORF1a_3911 | 1 | 1.57 | 2 |
| 15156 | ORF1b_572 | 1 | 1.14 | 2 |
| 15157 | 1 | 3.87 | 3 | |
| 15173 | ORF1b_578 | 1 | 2.18 | 1 |
| 15491 | ORF1b_684 | 1 | 1.52 | 1 |
| 15492 | 1 | 1.01 | 2 | |
| 15500 | ORF1b_687 | 1 | 1.15 | 1 |
| 15501 | 1 | 1.11 | 2 | |
| 15576 | ORF1b_712 | 1 | 1.11 | 2 |
| 17152 | ORF1b_1237 | 1 | 1.34 | 3 |
| 17514 | ORF1b_1358 | 1 | 1.01 | 2 |
| 18314 | ORF1b_1625 | 1 | 1.14 | 1 |
| 18354 | ORF1b_1638 | 2 | 1.21 | 2 |
| 18482 | ORF1b_1681 | 6 | 1.25 | 1 |
| 19477 | ORF1b_2012 | 2 | 1.31 | 3 |
| 21243 | ORF1b_2601 | 1 | 1.05 | 2 |
| 21492 | ORF1b_2684 | 1 | 1.23 | 2 |
| 21765 | S_68 | 4 | 1.19 | 2 |
| 22214 | S_218 | 3 | 2.25 | 1 |
| 22216 | 3 | 1.13 | 3 | |
| 22218 | S_219 | 1 | 1.38 | 2 |
| 22219 | 1 | 1.15 | 3 | |
| 22645 | S_361 | 1 | 1.03 | 3 |
| 23531 | S_657 | 3 | 1.18 | 1 |
| 23534 | S_658 | 1 | 1.09 | 1 |
| 23622 | S_687 | 2 | 1.16 | 2 |
| 23642 | S_694 | 3 | 1.88 | 1 |
| 23652 | S_697 | 1 | 1.14 | 2 |
| 24038 | S_826 | 2 | 1.20 | 1 |
| 24089 | S_843 | 1 | 2.30 | 1 |
| 24365 | S_935 | 1 | 1.29 | 1 |
| 25620 | ORF3a_76 | 2 | 1.26 | 3 |
| 25979 | ORF3a_196 | 3 | 1.66 | 2 |
| 26390 | E_49 | 2 | 1.01 | 2 |
| 26433 | E_63 | 1 | 1.31 | 3 |
| 27870 | ORF7b_37 | 5 | 2.10 | 3 |
| 28215 | ORF8_108 | 1 | 1.38 | 1 |
| 28251 | ORF8_120 | 11 | 1.15 | 1 |
| 28252 | ORF8_120 | 11 | 1.01 | 2 |
| 28253 | 11 | 1.17 | 3 | |
| 28254 | ORF8_121 | 22 | 4.84 | 1 |
| 20918 | N_215 | 4 | 4.27 | 3 |
| 28920 | N_216 | 2 | 2.70 | 2 |
| 28921 | 2 | 1.24 | 3 | |
| 28922 | N_217 | 2 | 1.10 | 1 |
| 28923 | 2 | 1.69 | 2 | |
| 28924 | 2 | 2.42 | 3 | |
| 28926 | N_218 | 1 | 1.52 | 2 |
| 28927 | 1 | 3.04 | 3 | |
| 28929 | N_219 | 1 | 1.87 | 2 |
| 28931 | N_220 | 5 | 4.20 | 1 |
| 28933 | 5 | 1.23 | 3 | |
| 28949 | N_226 | 1 | 1.12 | 1 |
| 28952 | N_227 | 1 | 1.56 | 1 |
| 28954 | 1 | 2.84 | 3 | |
| 28959 | N_229 | 3 | 1.49 | 2 |
| 28962 | N_230 | 1 | 1.77 | 2 |
| 28967 | N_232 | 5 | 1.31 | 1 |
| 28974 | N_234 | 31 | 1.92 | 2 |
| 28976 | N_235 | 6 | 1.07 | 1 |
| 28979 | N_236 | 1 | 1.33 | 1 |
| 28981 | 1 | 5.20 | 3 | |
| 28985 | N_238 | 7 | 2.52 | 1 |
| 28987 | 7 | 1.46 | 3 | |
| 28989 | N_239 | 1 | 2.27 | 2 |
| 28994 | N_241 | 1 | 1.96 | 1 |
| 28997 | N_242 | 1 | 2.36 | 1 |
| 29000 | N_243 | 3 | 4.36 | 3 |
| 29004 | N_244 | 3 | 1.45 | 2 |
| 29010 | N_246 | 1 | 1.11 | 2 |
| 29014 | N_247 | 1 | 1.08 | 3 |
| 29021 | N_250 | 2 | 1.62 | 1 |
| 29024 | N_251 | 1 | 1.12 | 1 |
| 29029 | N_252 | 4 | 1.93 | 3 |
| 29035 | N_254 | 2 | 1.42 | 3 |
| 29039 | N_256 | 2 | 1.69 | 1 |
| 29041 | 2 | 2.43 | 3 | |
| 29049 | N_259 | 1 | 2.77 | 2 |
| 29057 | N_262 | 1 | 1.55 | 1 |
| 29059 | 1 | 2.23 | 3 | |
| 29072 | N_267 | 1 | 1.13 | 1 |
| 29325 | N_351 | 1 | 1.49 | 2 |
| 29336 | N_355 | 1 | 1.37 | 1 |
| 29514 | N_414 | 7 | 1.25 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, W.; Delerce, J.; Aherfi, S.; La Scola, B.; Colson, P. Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences. Int. J. Mol. Sci. 2022, 23, 15658. https://doi.org/10.3390/ijms232415658
Bader W, Delerce J, Aherfi S, La Scola B, Colson P. Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences. International Journal of Molecular Sciences. 2022; 23(24):15658. https://doi.org/10.3390/ijms232415658
Chicago/Turabian StyleBader, Wahiba, Jeremy Delerce, Sarah Aherfi, Bernard La Scola, and Philippe Colson. 2022. "Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences" International Journal of Molecular Sciences 23, no. 24: 15658. https://doi.org/10.3390/ijms232415658
APA StyleBader, W., Delerce, J., Aherfi, S., La Scola, B., & Colson, P. (2022). Quasispecies Analysis of SARS-CoV-2 of 15 Different Lineages during the First Year of the Pandemic Prompts Scratching under the Surface of Consensus Genome Sequences. International Journal of Molecular Sciences, 23(24), 15658. https://doi.org/10.3390/ijms232415658